一种巯基化透明质酸衍生物中二硫苏糖醇残留的检测方法与流程
1.本发明涉及二硫苏糖醇检测技术领域,特别涉及一种巯基化透明质酸衍生物中的二硫苏糖醇残留的检测方法。
背景技术:
2.透明质酸(hyaluronic acid,简称ha),又名“玻璃酸”,广泛分布于动物和人体的细胞外基质中,是细胞基质和多种组织的重要组成成分,具有多种重要的生理学功能。采用含有二硫键的氨基试剂与透明质酸的侧链羧基进行偶联反应;偶联反应结束后,二硫键可经简单的还原过程还原为自由巯基,即可得到巯基化透明质酸衍生物,这是当前巯基化透明质酸衍生物最为常用的制备方法;在上述二硫键还原为自由巯基的过程中,目前最为广泛使用的还原剂是二硫苏糖醇。
3.二硫苏糖醇(dithiothreitol,简称为dtt)是一种小分子有机还原剂,化学式为c4h
10
o2s2。其还原状态下为线性分子,被氧化后变为包含二硫键的六元环状结构。dtt可以将巯基保持在还原态,常用于还原蛋白分子和多肽的二硫键,更普遍用以防止蛋白半胱氨酸残基形成分子内和分子间的二硫键,通常作为蛋白质巯基保护剂。dtt进入体内可能会破坏蛋白质结构,从而引起酶失活等现象,dtt的残余可能导致潜在的安全性隐患,因此需要严格监控过程中样品以及终产品中的还原剂dtt的残留量。
4.根据目前报道,采用传统分光光度计法进行测定时,dtt极容易被氧化,响应不稳定,灵敏度低,不能满足检测要求。还有一种基于液相色谱的测试方法是用单溴二胺(mbbr)这种荧光探针对dtt进行衍生,衍生后利用反相液相色谱和荧光检测器对dtt进行定量分析,这种方法灵敏度高,但是样品前处理过程复杂,对反应环境要求严格,不适用于工业生产过程中对dtt残留量的检测。中国专利cn112526028a公开了采用液相色谱串级质谱(lc-ms/ms)法同时对二硫苏糖醇还原态和氧化态两种共存形式进行定量分析的液相色谱法,但是这种方法需要同时生成两种物质的标准曲线,增加检测工作量。还有报道利用钴离子反滴定的方法测定dtt含量,但是dtt容易被氧化,而且这种方法灵敏度也不高。中国专利cn107255660b公开了利用石墨烯量子点电极来检测溶液中二硫苏糖醇的方法,这种方法操作简单,灵敏度高,但是限于其制备工艺高昂的价格,工业生产中应用较少。
5.目前也有用hplc对二硫苏糖醇(dtt)残留量进行定量的报道,例如:高效液相色谱法测定药品中的二硫苏糖醇和氧化型二硫苏糖醇(分析科学学报 2016, 32:537-540)采用hplc测定了工程菌代谢产物(一种活性蛋白)中dtt的含量,采用了waters x-bridge c18色谱柱,流动相为30:70(v/v)的甲醇-水;反相高效液相色谱法检测重组类病毒颗粒疫苗原液中二硫苏糖醇的残留量(中国生物制品学杂志2015, 28:1316-1319)采用反相hplc测定了重组hpv疫苗原液中dtt的残留量,采用了dikma bio-bond c18色谱柱,流动相为90:10(v/v)水-乙腈(0~10 min)、100%乙腈(10~15 min)和90:10(v/v)水-乙腈(15~20 min)。但上述的活性蛋白和重组hpv疫苗均具有明确的单一分子量蛋白结构,而本技术涉及的巯基化透明质酸衍生物为具有分散性的大分子多糖衍生物混合体,研究表明上述两种检测方法很
难满足巯基化透明质酸衍生物中微量dtt残留分离检测的实际需求。
6.在巯基化透明质酸衍生物的制备中,二硫苏糖醇的残留定量检测已成为一个亟待解决的问题,现今尚没有巯基化透明质酸衍生物中dtt微量残留的检测方法,这为其在药物或者医疗器械的广泛应用带来了限制,建立一种有效的巯基化透明质酸衍生物中dtt微量残留的检测方法具有重要意义。
技术实现要素:
7.本发明要解决的技术问题是提供一种精确度较高、能够满足实际需求的巯基化透明质酸衍生物中二硫苏糖醇残留的检测方法,一种二硫苏糖醇残留的凝胶渗透色谱检测方法。
8.为解决上述技术问题,本发明提供一种巯基化透明质酸衍生物中二硫苏糖醇残留的检测方法,包括以下步骤:步骤(1)标准品工作液:精密称取一定量的dtt标准品,以ph 3.5~4.5的磷酸盐缓冲液为稀释剂 ,配制成不少于4个浓度梯度、范围为1~20 ppm的dtt标准品工作液;步骤(2)待测样品工作液:精密称取一定量的巯基化透明质酸衍生物,以ph 3.5~4.5的磷酸盐缓冲液为稀释剂,配制浓度范围为3~20mg/ml巯基化透明质酸衍生物溶液;步骤(3)标准曲线:步骤(1)的标准品工作液依次经凝胶渗透色谱检测,分别获得对应的二硫苏糖醇的峰面积,并与标准品工作液浓度进行线性回归产生标准曲线,或由软件系统自动生成标准曲线;步骤(4)待测样品测定:步骤(2)的待测样品工作液经凝胶渗透色谱检测,获得样品的二硫苏糖醇的峰面积,代入至步骤(3)的标准曲线,计算得到待测样品中二硫苏糖醇的残留含量;步骤(2)、步骤(4)所述的凝胶渗透色谱检测采用的是water 2695-2998液相色谱仪;水相凝胶渗透色谱柱的型号是ultrahydrogel
tm 120,7.8
×
300mm,填料粒径 6
µ
m;柱温:30℃。
9.具体的,所述凝胶渗透色谱的进样量为10~100
ꢀµ
l;流速为0.5~1.0 ml/min。
10.具体的,所述凝胶渗透色谱的流动相为ph 3.5~4.5的磷酸缓冲盐与乙腈的混合溶液。
11.具体的,所述磷酸盐缓冲液和乙腈的体积比为85:15~95:5。
12.具体的,所述流速为0.7 ml/min。
13.具体的,所述进样量为40
µ
l。
14.具体的,所述凝胶渗透色谱的检测器是光电二极管阵列检测器。
15.具体的,所述凝胶渗透色谱的检测波长是210nm。
16.具体的,所述标准品工作液的浓度梯度分别为1、2、5、10和20ppm。
17.具体的,所述巯基化透明质酸衍生物的分子量大于120 kda。
18.凝胶渗透色谱(gpc)仪的组成:泵系统、(自动)进样系统、凝胶色谱柱、检测系统和数据采集与处理系统。与高效液相色谱法(hplc)检测类似,gpc检测方法具有定量准确、检测周期短等优点,是较为适于微量检测的方法之一。hplc通过色谱柱进行样品分离,色谱柱中充满非常微小的凝胶颗粒填料,基于被分离物质中各种分子与凝胶颗粒填料之间的亲和力而实现分离。而gpc的分离原理是体积排阻理论,其凝胶渗透色谱柱中所填装的多孔性填料表面及内部有各种各样大小不一的多孔通道,基于被分离物质中各种分子的分子尺寸差异而实现分离。
19.要实现dtt的定量检测就必须首先实现dtt与巯基化透明质酸衍生物及其它可能物质之间的良好分离,参照《中华人民共和国药典(2020版四部)》通则0512《高效液相色谱法》的要求,分离度需要≥1.5。然而,巯基化透明质酸衍生物是一种大分子天然多糖的衍生物,其分子量具有一定的分散性,是不同大分子量的大分子混合体,在hplc所常用的色谱柱(如c18硅胶基色谱柱等)中较难实现与dtt的良好分离。因此在本发明中采用gpc凝胶渗透色谱柱来实现巯基化透明质酸衍生物中dtt微量残留的分离检测。
20.经研究发现,巯基化透明质酸衍生物与dtt的分离度和流动相的组份、流动相的ph值、流动相配比等多种因素相关。综合性考察各方面因素后得出上述本发明的检测方法,能够获得良好的分析结果。
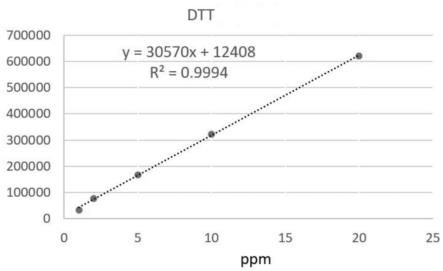
21.本发明具有的有益效果是:本发明提供的巯基化透明质酸衍生物中微量dtt残留检测方法,样品无需处理,操作方法简单易行, dtt在1~20 ppm范围内有良好的线性范围(其线性相关系数(r2)为0.9994),最低检测限可达到1 ppm(信噪比3.67),定量限可达到2.72 ppm(信噪比10),准确度高(回收率(r)在105.6%~111.9%之间(rsd为3%)),能够满足实际检测中的需要。
附图说明
22.为了更清楚地说明本发明的技术方案,下面对本发明所需要使用的附图作简单的介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
23.图1是根据本发明的检测方法提供的dtt色谱图(不同有机相的分离度);图2是根据本发明的检测方法提供的dtt色谱图(不同流动相ph);图3是根据本发明的检测方法提供的dtt色谱图(不同流动相配比);图4是根据本发明的检测方法提供的dtt标准曲线。
具体实施方式
24.下面将结合附图,对本发明中的技术方案进行清楚、完整的描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其它实施例,都属于本发明保护的范围。
25.以下实施例中的设备信息如下:water 2695-2998液相色谱仪(pda检测器);水相凝胶渗透色谱柱(ultrahydrogel
tm 120),7.8
×
300mm,填料粒径 6
µ
m;柱温:30℃。
26.实施例1. 检测条件的考察1.1 dtt试验液的配置精密称取0.100g dtt标准品,用纯水溶解并定容至100ml,配置成浓度为1000ppm的标准储备液;移取一定体积的标准储备液,用纯水稀释成1、2、5、10、20ppm的标准工作液。
27.1.2 巯基化透明质酸衍生物巯基化透明质酸衍生物(mw 120 kda、200 kda、500 kda) 水溶液(10 mg/ml)(ph 3.5)。
28.1.3 检测波长的确定
dtt试验液在紫外可见分光光度计上进行全波长扫描。结果表明,dtt从约260nm处出现吸收,至200~210nm处达到最大,在200nm存在边缘吸收,因此选择210nm作为gpc的检测波长。
29.1.4流动相的的选择在流动相中,有机相和水相、缓冲物质及ph对样品的分离检测都存在影响。
30.水相为磷酸盐缓冲液,分别采用甲醇和乙腈作为有机相进行分离,巯基化透明质酸的保留时间约为9 min(随分子量增大保留时间稍有减小),dtt的保留时间约为37 min。结果表明,乙腈具有更好的分离度(参见图1)。因此,本实施例后续试验选择乙腈作为有机相。
31.流动相的ph较高时,dtt的峰形变宽且易出现叠加的杂峰,可能与dtt在较高ph值时易被氧化有关。结果发现,在磷酸盐缓冲液的ph在3.5~4.5时,dtt的峰形良好(参见图2)。因此,本实施例后续试验选择ph 4.0的磷酸盐缓冲液。
32.由于巯基化透明质酸衍生物不溶于有机溶剂,因此有机溶剂的体积比不能过大。结果表明,磷酸盐缓冲液和乙腈的体积比在85:15~95:5比较合适,可以实现良好的分离度,尤其是体积比为90:10时,分离度不小于14.1,能更好保障检测结果的精确性(参见图3)。因此,本实施例后续试验选择磷酸盐缓冲液和乙腈的体积比为90:10。
33.考察dtt试验液不同进样量(10~100
ꢀµ
l)。结果表明,进样量为40
ꢀµ
l时,dtt在检测波长段内出的响应高低适中,可进一步增加检测方法的准确性,因此选择40
ꢀµ
l作为gpc的检测波长。
34.考察不同的流动相流速(0.5~1.0 ml/min)。结果表明,流速为0.7 ml/min最为合适。流速过快会导致分离物质的保留时间过短,分离度不够好,过慢会导致物质峰形变宽而影响峰形。
35.在优选的检测条件下(进样量为40
ꢀµ
l,流速为0.7 ml/min,流动相为90:10体积比的磷酸盐缓冲液(ph 4.0)和乙腈,检测波长210nm时),dtt的色谱图参见图1和图3(分离度不小于14.4),满足《中华人民共和国药典(2020版四部)》通则0512《高效液相色谱法》关于分离度需≥1.5的要求,后续实施例均采用该色谱条件。
36.实施例2. 标准曲线的建立精密称取0.100g dtt标准品,用优选流动相溶解并定容至100ml,配置成浓度为1000ppm的标准储备液;移取一定体积的标准储备液,用优选流动相稀释成1、2、5、10、20ppm的标准工作液并在优选的检测条件下进行检测。
37.检测结果如下表1:表1通过标准曲线(图4)可以看出,dtt在1~20ppm范围内有良好的线性范围,其线性相关系数(r2)为0.9994,满足检测方法关于线性相关系数(r2)大于0.998的要求。
38.实施例3. 检测限和定量限用低浓度的dtt标准工作液检测出的信号与空白样品测出的信号进行比较,以信
噪比不低于3:1时相应浓度计算检测限,以信噪比不低于10:1时相应浓度计算定量限度;结果表明最低检测限可达到1 ppm(信噪比3.67),定量限可达到2.72 ppm(信噪比10)。
39.实施例4. 精密性取6份dtt标准工作液(5ppm)在优选的检测条件下进行检测,根据各供试品得到的峰面积计算相对偏差,结果如下表2:表2rsd为0.94%,具有良好的精密性,满足rsd≤2%的目标要求。
40.实施例5. 准确性采用加样回收试验法,即在前述巯基化透明质酸衍生物水溶液中加入三个不同浓度的dtt标准工作液(8ppm、10ppm和12ppm),每个浓度的标准溶液平行制备三份,用实施例2中的标准曲线,测定其中dtt的量,并计算回收率。试验结果如下表3:表3准确性是表征检测方法测定的结果与真实值接近的程度,通常用回收率(r)表征,要求在80~120%之间(rsd不大于10%),试验结果满足要求。
41.实施例6. 耐用性取5份dtt标准工作液,在其它色谱条件不变的情况下,柱温改变
±
1℃,流速改变
±
5%,分别进样检测,记录分析峰面积,代表性结果如下表4(dtt标准工作液浓度5 ppm):表4rsd为3.2%,具有良好的耐用性,满足rsd≤5%的目标要求。
42.实施例7. 方法的应用按照实施例1确定的色谱条件测定实施例1中的三种巯基化透明质酸衍生物溶液中微量dtt残留量,根据实施例2建立标准曲线,检测结果显示两种巯基化透明质酸衍生物(mw 120 kda、200 kda)溶液中dtt的残留量《检测限(1ppm),一种巯基化透明质酸衍生物(mw 500 kda)溶液中dtt的残留量为4.2 ppm。
43.根据ich《q3ar2:新原料药中杂质》关于每日最大剂量的药物中其含有的杂质界定阈值为0.1%或者1mg的规定,上述三种巯基化透明质酸衍生物溶液以有效成份(10mg/ml)计,其dtt残留量限度是10ppm,符合规定。
44.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明保护的范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1