利用高效液相色谱-离子淌度差分质谱检测类固醇激素的方法
accucore c18等。
10.3. 根据项1或2所述的方法,其中在所述高效液相色谱中使用由流动相a和流动相b组成的混合流动相进行梯度洗脱,所述流动相a为甲酸铵或乙酸铵的水溶液,所述流动相b为甲醇水溶液。
11.4. 根据项3所述的方法,其中所述流动相a中甲酸铵的浓度为2mm至10mm,例如3mm、4mm、5mm、6mm、7mm、8mm、9mm等。在一些实施方案中,所述甲酸铵的浓度为5mm。在一些实施方案中,所述流动相a进一步包含用于调节ph值的酸。在一些实施方案中,所述酸为甲酸或乙酸。在一些实施方案中,所述酸为甲酸或乙酸,且其含量为0.05至0.2v%,例如0.1vt%、0.15v%。
12.5. 根据项4所述的方法,其中所述流动相a还进一步包含抑菌剂。在一些实施方案中,所述抑菌剂为乙腈,且所述乙腈的含量为约3v%至10v%,例如5v%。
13.6. 根据项3-5中任一项所述的方法,其中所述流动相b中水的含量为3v%-10v%。在一些实施方案中,所述流动相b中水的含量为5v%。在一些实施方案中,所述流动相b进一步包含用于调节ph值的酸。在一些实施方案中,所述酸为甲酸或乙酸。在一些实施方案中,所述酸为甲酸或乙酸,且其含量为0.05至0.2v%,例如0.1v%、0.15v%。
14.7. 根据项1-6中任一项的方法,其中梯度洗脱步骤为流动相b在所述混合流动相中的体积占比先逐渐升高再逐渐降低。
15.8. 根据项1-7中任一项所述的方法,其中流动相b在所述混合流动相中的体积占比为6:94至98:2之间,包括6:94及98:2。
16.9. 根据项7所述的方法,其中流动相b在所述混合流动相中的体积占比从6:94逐渐升高至98:2,再逐渐减少至6:94。
17.10. 根据项7的方法,其中梯度洗脱过程为:第一阶段,流动相b和流动相a的体积比保持6:94;第二阶段,流动相b和流动相a的体积比逐渐变化至为50:50并维持一段时间;第三阶段,流动相b和流动相a的体积比逐渐变化至98:2;第四阶段,流动相b和流动相a的体积比逐渐变化至6:94。
18.在一些实施方案中,在第四阶段结束后,还有第五阶段,其流动相b和流动相a的体积比将保持6:94一段时间。
19.在一些实施方案中,所述梯度洗脱是一个循环过程,在第四阶段结束后,即重新进入第一阶段。
20.在一些实施方案中,所述梯度洗脱过程为:第一阶段,在第0-0.5分钟,流动相b和流动相a的体积比为6:94;第二阶段,在第0.5-3分钟,流动相b和流动相a的体积比逐渐变化至45:55;第三阶段,在第3-5.3分钟,流动相b和流动相a的体积比逐渐变化至为50:50;第四阶段,保持流动相b和流动相a的体积比为50:50直至第6分钟;第五阶段,在第6至7.5分钟,流动相b和流动相a的体积比为98:2;第六阶段,在第7.5-7.8分钟,流动相b和流动相a的体积比逐渐减少至6:94;第七阶段,保持流动相b和流动相a的体积比为6:94至第10分钟。
21.11. 根据项1-10中任一项所述的方法,其中所述原位衍生化使用的试剂为肼类化
合物或羟胺类化合物。
22.12. 根据项11所述的方法,在一些实施方案中,所述肼类化合物是吡啶鎓肼;在一些实施方案中,所述羟胺类化合物为盐酸羟胺。
23.13. 根据项12所述的方法,其中所述衍生试剂选自:n, n, n-三乙基-2-肼-2-氧乙基氯化铵(thob)、n, n-二丁基-n-(2-肼-2-氧乙基)-丁基-1-氯化铵(dhob)、吉拉德试剂p(gp)、吉拉德试剂t(gt)、2-(2-肼基-2-氧乙基)-异喹啉-2-肼溴化物(iqhb)和盐酸羟胺(hahc)。在一些实施方案中,所述衍生剂为gp。
24.14. 根据项1-13中任一项所述的方法,其中所述衍生剂与样品的体积比为40/200至160/200,例如80/200至160/200,50/200、60/200、70/200、80/200、90/200、100/200,100/200、120/200等。在一些实施方案中,所述样品体积为200μl,所述衍生试剂的体积为100μl。在一些实施方案中,所述衍生化的温度是30-60℃,40-50℃,35℃,45℃或55℃。在一些实施方案中,所述衍生化的时间为3-6小时,例如3.5-5.5h,4-5h等。
25.15. 根据项1-14中任一项所述的方法,其中所述类固醇激素选自:t、dhea、a4、11oha4、11oht、11ka4、17ohp、11kt、dht和11kdht。在一些实施方案中,所述样品包含一种、两种或更多种下述类固醇激素:t、dhea、a4、11oha4、11oht、11ka4、17ohp、11kt、dht和11kdht。在一些实施方案中,所述样品包含下述全部类固醇激素:睾酮、dhea、雄烯二酮、11oha4、11oht、11ka4、17ohp、11kt、dht和11kdht。在一些实施方案中,所述样品包含下述全部类固醇激素:t、dhea、a4、11oha4、11oht、11ka4、17ohp和11kt。
26.16. 根据项1-15中任一项所述的方法,其中所述样品是包含蛋白质的生物样品,所述方法还进一步包含在原位衍生化之前使所述类固醇激素与蛋白的结合解离。
27.在一些实施方案中,在衍生化前或使所述类固醇激素与蛋白质解离后,还进一步包含使用分散磁固相萃取技术吸附所述类固醇激素。
28.17. 根据项16所述的方法,其使用氯化铵缓冲液使类固醇激素与蛋白质解离。在一些实施方案中,所述氯化铵缓冲液为饱和氯化铵缓冲液。
29.18. 根据项17所述的方法,其中所述饱和氯化铵缓冲液与所述血清的体积比为1:1。例如,在一些实施方案中,所述血清为200μl,所述饱和氯化铵缓冲液为200μl。
30.19. 根据项16所述的方法,其使用磁性吸附剂进行分散磁固相萃取。在一些实施方案中,所述磁性吸附剂为mgo、ni@mwcnt或fe3o4@sio2。
31.20. 根据项19所述的方法,其中所述吸附剂为mgo,且在一些实施方案中,所述吸附时间大于20分钟,或为20至80分钟,大于30分钟,大于35分钟,大于40分钟,大于45分钟,大于50分钟,大于55分钟,大于60分钟,大于65分钟,大于70分钟,或大于75分钟。在一些实施方案中,所述吸附剂为mgo,且每200μl样品中加入mgo的量为0.5至2.5mg,例如1mg、1.5mg、2mg等。
32.21. 根据项16-20中任一项所述的方法,其中所述生物样品是血清。在一些实施方案中,所述样品是人血清。
附图说明
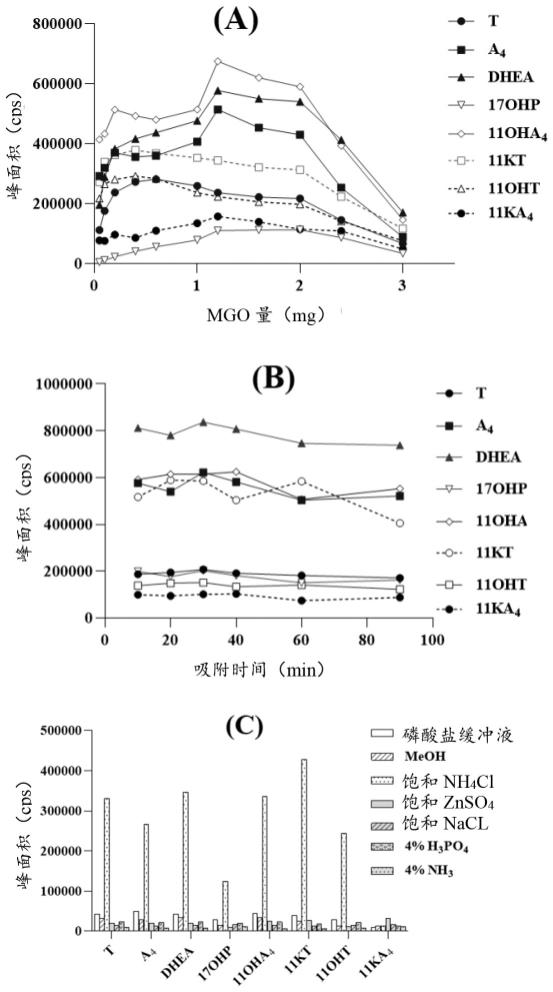
33.图1. dmspe条件对目标分析物质谱响应的影响。(a) mgo量;(b) 吸附时间;(c) 对于患者样品中类固醇-蛋白质的结合,不同溶剂的解离效果比较。
34.图2. 原位衍生化条件的优化。(a)不同衍生试剂的比较;(b)衍生试剂用量的影响;(c)衍生温度的影响;(d)衍生时间的优化。
35.图3. 空白血清和lloq的典型色谱图。(a)空白对照中的分析物;(b)空白对照中的is;(c)加标lloq中的分析物;(d)加标lloq中的is。
36.图4. t和a4采用hplc-dms/ms/ms方法和cl法检测结果之间的相关性分析。
37.图5. 质谱法检测类固醇结果与其他生化指标的相关性分析。
38.图6. 显示采用多反应监测模式(mrm)进行质谱的定量分析所监测的mrm离子对,及其中ms/ms和dms所使用的部分参数。
39.图7. 显示在lloq和4个qc水平下,用5天分别对6个平行样进行批内/批间精密度和准确度的评估结果。
40.图8. 显示用6个不同批次的活性碳处理的血清对三种qc水平的基质效应的评估结果,以及对四个qc水平和is的分析物回收率的评估结果。
41.图9. 显示在三个qc水平上对稳定性的评估结果。
42.图10. 显示对样品中的8种类固醇激素的检测结果和对应的临床指标相关性。
43.图11. 显示经本技术方案检测出的各检测物之间相关系数(r)的p值。
具体实施方式
44.本技术建立了一种快速、高选择性和高灵敏度的高效液相色谱-离子淌度差分质谱检测法,可用于在包含多种类固醇激素的复杂基质,例如血清中,分离多种类固醇激素并对其进行定量。本技术的方案,无需使用有毒的有机试剂,即可实现复杂生物样品中极微量物质的精准检测,易于在医学实验室广泛应用。
45.具体的,本技术在hplc-ms/ms系统中引入了离子淌度分离技术(dms,differential mobility spectrometry)以提高该系统的分辨率。并且,根据难以分离的类固醇激素特性和临床样品性质,为更好地分离各类固醇激素,该系统中还可进一步引入分散磁性固相萃取(dmspe,dispersive magnetic solid phase extraction)、原位衍生化、和/或类固醇蛋白质解离技术,并针对性地对各步骤进行了优化。最终实现了对至少四种11-氧化雄激素和四种经典雄激素或前体的彼此分离和定量。所述四种11-氧化雄激素和四种经典雄激素或前体为:睾酮(t)、脱氢表雄酮(dhea)、雄烯二酮(a4)、11β-羟雄甾烯二酮(11oha4)、11-羟睾酮(11oht)、11-酮基雄烯二酮(11ka4)、17-α-羟孕酮(17ohp)和11-酮基睾酮(11kt)。此外,本领域技术人员应当知晓,其他与前述类固醇分子具有一定质量差异、结构差异、和/或衍生后离子带电量差异的类固醇分子或其异构体也可以通过本技术的方案进行分离和定量,所述其他类固醇分子包含例如5α-二氢睾酮(dht)和11-酮基-5α-二氢睾酮(11kdht)。
46.本技术涉及一种利用高效液相色谱-离子淌度差分质谱检测或分离类固醇激素的方法,其包含:对待测样品进行衍生化;将经原位衍生化处理的样品注入到高效液相色谱-离子淌度差分质谱串联装置中,获得质量色谱图。
47.在一些实施方案中,所述样品是包含蛋白质的生物样品,例如血清,则所述方法还
可以进一步包含:在原位衍生化之前使所述类固醇激素与蛋白质解离;和使用dmspe法净化富集所述类固醇激素。
48.其中,在一些实施方案中,所述衍生化为原位衍生化。
49.高效液相色谱-离子淌度差分质谱高效液相色谱-离子淌度差分质谱是一种色谱、离子淌度谱与质谱联用的检测技术,即在质谱前对样品进行高效液相色谱分离及离子淌度分离。
50.在一些实施方案中,所述质谱是单级色谱。在一些实施方案中,所述色谱是多级质谱,例如三级、四级、五级或更多及质谱。在一些实施方案中,所述质谱是二级质谱,例如hplc-dms/ms/ms,即在hplc-ms/ms之间增加一步dms。本领域技术人员应当知晓,任选的hplc系统及质谱均能用于本技术的方案。例如,在本技术的后续实施例中,申请人采用了由20a hplc系统(shimadzu, chiyoda-ku, tokyo, japan)、selexion dms离子淌度分离设备、及串联质谱仪qtrap 5500(ab sciex, framingham, ma, usa)组成的系统。
51.在一些实施方案中,hplc使用的色谱柱为具有碳十八固定相的液相色谱柱(c18液相色谱柱)。在一些实施方案中,所述色谱柱为十八烷基硅烷键合多孔硅胶柱。本领域技术人员应当知晓,任何品牌的十八烷基硅烷键合多孔硅胶柱均可用于本技术的方案,仅因不同品牌色谱柱存在差异,其在进行高效液相色谱分离时,洗脱过程会有所差异。所述色谱柱包括但不限于例如agilent、waters、thermo等各厂家的市售核壳型十八烷基硅烷键合柱。在本技术中,我们仅就其中一种色谱柱的分离条件进行了优化,但本领域技术人员应当理解,使用其他各厂家色谱柱的方案也落入本技术的保护范围。
52.在一些实施方案中,所述色谱柱为任意的多孔硅胶柱。在一些实施方案中,所述色谱柱为十八烷基硅烷键合多孔硅胶柱。在一些实施方案中,所述色谱柱为任意封端的十八烷基硅烷键合多孔硅胶柱。在一些实施方案中,所述色谱柱选自:agilent poroshell 120 ec-c18、waters cortecs c18、thermo accucore c18等。
53.本领域技术人员应当知晓,任何可使待测类固醇激素达到充分分离和良好峰形的流动相均可用于本技术的方案。在一些实施方案中,使用agilent poroshell ec c18柱(50
×
2.1 mm, 2.7 μm)进行色谱分离,其进行梯度洗脱时的混合流动相由流动相a和流动相b组成。在一些实施方案中,所述流动相a为甲酸铵或乙酸铵的水溶液,所述流动相b为甲醇水溶液。在一些实施方案中,所述流动相a中甲酸铵的浓度为2mm至10mm,例如3mm、4mm、5mm、6mm、7mm、8mm、9mm等。在一些优选的实施方案中,所述甲酸铵的浓度为5mm。在一些实施方案中,所述流动相a进一步包含用于调节ph值的酸。在一些实施方案中,所述酸为弱酸。在一些实施方案中,所述酸为甲酸或乙酸。在一些实施方案中,所述酸为甲酸,且其含量为约0.1v%。
54.在一些实施方案中,所述流动相a还进一步包含抑菌剂。本领域技术人员应当知晓,任何不影响流动相a对分析成分的溶解的抑菌剂均可用于本技术的方案,例如乙腈。在一些实施方案中,所述流动相a的乙腈含量为约3v%至10v%,例如4v%、5v%、6v%、7v%、8v%、9v%。
55.在一些实施方案中,所述流动相b中水的含量为3v%-10v%,例如4v%、5v%、6v%、7v%、8v%、9v%。在一些实施方案中,所述流动相b进一步包含用于调节ph值的酸。在一些实施方案
中,所述酸为弱酸。在一些实施方案中,所述酸为甲酸或乙酸。在一些实施方案中,所述酸为甲酸,且其含量为0.1v%。
56.在一些实施方案中,使用由流动相a和b组成的混合流动相的梯度洗脱过程为流动相b在所述混合流动相中的占比先逐渐升高再逐渐降低。在一些实施方案中,流动相b在所述混合流动相中的占比在6:94至98:2之间,包括6:94及98:2。在一些实施方案中,流动相b在所述混合流动相中的占比从6:94逐渐升高至98:2,再逐渐减少至6:94。在一些实施方案中,使用由流动相a和b组成的混合流动相的梯度洗脱过程为流动相b与流动相a的比例在6:94逐渐升高至98:2及从98:2逐渐降低至6:94之间反复循环。本领域技术人员应当知晓,梯度洗脱过程中,流动相中a与b比例的变化是一个连续的过程,其可以产生6:94及98:2之间的任一比例,并且根据梯度洗脱的具体实施情况,本领域技术人员可以根据当时的操作情况在其中任一比例做一定时间的停留,或调整其在任意两个比例之间变化的速度。
57.在一些实施方案中,所述梯度洗脱过程包含:第一阶段,流动相b和流动相a的体积比保持6:94;第二阶段,流动相b和流动相a的体积比逐渐变化至为50:50并维持一段时间;第三阶段,流动相b和流动相a的体积比逐渐变化至98:2;第四阶段,流动相b和流动相a的体积比逐渐变化至6:94;其中任一阶段均可作为洗脱的初始阶段,然后依次向后经历后续阶段,例如,从第一阶段至第四阶段;例如以第四阶段为起始阶段,则流动相b和流动相a的体积比从98:2逐渐变化至6:94,并进入下一阶段,即第一阶段,流动相b和流动相a的体积比保持6:94,然后继续进行第二阶段等后续阶段。
58.在一些实施方案中,所述梯度洗脱是一个循环过程,在第四阶段结束后,即重新进入第一阶段,并反复循环以上四个阶段。
59.在一些实施方案中,所述梯度洗脱过程为:第一阶段,在第0-0.5分钟,流动相b和流动相a的体积比为6:94;第二阶段,在第0.5-3分钟,流动相b和流动相a的体积比逐渐变化至45:55;第三阶段,在第3-5.3分钟,流动相b和流动相a的体积比逐渐变化至为50:50;第四阶段,保持流动相b和流动相a的体积比为50:50直至第6分钟;第五阶段,在第6至7.5分钟,流动相b和流动相a的体积比为98:2;第六阶段,在第7.5-7.8分钟,流动相b和流动相a的体积比逐渐减少至6:94;第七阶段,保持流动相b和流动相a的体积比为6:94至第10分钟。
60.在一些实施方案中,上述实施方案在各阶段的时间及a相和b相的体积比均可依据实际情况进行调整。
61.在一些实施方案中,所述流动相的流速为0.3 ml/min。在一些实施方案中,所述进样量为5
ꢀµ
l。本领域技术人员应当知晓,上述流动相的流速及进样量仅是一个可操作的实例,实际上流动相的流速及进样量,以及上述洗脱过程的循环次数均可以根据样品量及其本身待测物含量进行调整,并且所述调整的方法,是本领域技术人员容易掌握的。
62.在一些实施方案中,在ms步骤中,所述所有的目标分析物都在正电离模式下检测。在一些实施方案中,ms使用的源温度为650℃。在一些实施方案中,所述雾化气为65 psi。在一些实施方案中,所述加热气体为65 psi。在一些实施方案中,离子源喷雾电压为5500v。上
述ms的操作条件仅是一种示例,本领域技术人员完全可以根据实际样品量及设备等情况做适应性的进一步调整。
63.在本技术的一些实施方案中,采用了多反应监测模式(mrm)进行质谱的定量分析,所监测的mrm离子对如图6所示。此外,图6也列出了一些实施方案中,ms/ms和dms所使用的部分参数。
64.衍生化本技术通过引入衍生化步骤,可使得原本因结构相似而难以分离的类固醇分子,例如包括11-羟甲睾酮(11oht)、11-酮睾酮(11kt)和11-酮-5α-双氢睾酮(11kdht)在内的11-氧化雄激素得到分离。
65.在一些实施方案中,使用经前述dmspe处理后的样品进行原位衍生化。
66.在一些实施方案中,所述原位衍生化使用的衍生剂为羰基衍生试剂。在一些实施方案中,所述衍生剂是季铵肼。在一些实施方案中,所述衍生试剂是吡啶鎓肼。吡啶鎓肼的代表包括例如吉拉德试剂p(gp)。后文实施例中以gp为例对本技术的方案进行了验证,证明了吡啶鎓肼作为衍生剂在本技术方案中的可行性。在一些实施方案中,所述衍生试剂选自:n, n, n-三乙基-2-肼-2-氧乙基氯化铵(thob)、n, n-二丁基-n-(2-肼-2-氧乙基)-丁基-1-氯化铵(dhob)、吉拉德试剂p(gp)、吉拉德试剂t(gt)、2-(2-肼基-2-氧乙基)-异喹啉-2-肼溴化物(iqhb)和盐酸羟胺(hahc)。
67.在一些实施方案中,所述衍生试剂与样品的体积比为40/200至160/200,例如80/200至160/200,50/200、60/200、70/200、80/200、90/200、100/200,100/200、120/200等。
68.使用本技术的方案,发明人成功实现了对低至200μl的临床样品中至少8种类固醇激素的分离及定量,但这并不代表本技术的方案仅能用于200μl以上的样品。以样品体积为200μl,所述衍生剂为gp为例,在一些实施方案中,所述衍生试剂包含0.2mol/l的gp甲醇溶液和2%的乙酸;在一些实施方案中衍生剂的使用体积为100μl;在一些实施方案中,所述衍生化的温度是30-60℃、40-50℃、35℃、45℃或55℃;在一些实施方案中,所述衍生化的温度是50℃;在一些实施方案中,所述衍生化的时间为3-6小时,例如3.5-5.5h,4-5h等。当样品中待测物质含量和/或样品量发生变化时,本领域技术人员完全可以适应性地调整衍生试剂的体积及衍生化时间,以及选择更加适应该实际情况的衍生化温度,以使得全部待测类固醇激素充分衍生化。
69.待衍生化完成之后,例如经过4小时的衍生化之后,衍生物可被转移至容器中进行高效液相色谱-离子淌度差分质谱操作。
70.dmspe对于包含待检的包含类固醇物质的样品,现有技术中多采用有机溶剂来进行液-液萃取(lle)、固-液相萃取(sle)、固相萃取(spe)技术来提取类固醇类物质。lle、sle和spe的程序很复杂,而且耗费较长时间,而其大量使用包括氯丁烷、甲基叔丁基醚、乙酸乙酯和二氯甲烷在内的有毒有机溶剂会损害操作者的健康、污染环境。而本技术的方案中,使用分散磁固相萃取技术(dmspe,dispersive magnetic solid phase extraction)来制备样品,可以使分析物可以在不到1分钟内用磁铁轻松地与水层分离,并且具有较高的回收率。在整个过程中,没有使用有毒的有机试剂。这种绿色程序是医学实验室的理想选择。
71.在本技术中,发明人特别优化了用于dmspe的吸附剂。在一些实施方案中,dmpse使
用磁性纳米颗粒作为吸附剂。在一些实施方案中,dmpse使用的是核壳结构的磁性纳米颗粒作为吸附剂。在一些实施方案中,所述磁性纳米颗粒包含:mgo(磁性氧化石墨烯)、ni@mwcnt(多壁碳纳米管包埋镍)或fe3o4@sio2(二氧化硅包裹的四氧化三铁磁性纳米颗粒)。本领域技术人员应当知晓,其他具有巨大比表面的磁性纳米颗粒也可实现本技术dmspe步骤吸附剂的功能。
72.在一些实施方案中,所述磁性纳米颗粒包含fe3o4@go。在一些实施方案中,所述磁性纳米颗粒为mgo纳米复合材料,且在一些实施方案中,所述吸附时间为20至80分钟。在一些实施方案中,所述吸附时间大于20分钟,或大于30分钟,大于35分钟,大于40分钟,大于45分钟,大于50分钟,大于55分钟,大于60分钟,大于65分钟,大于70分钟,或大于75分钟。在一些实施方案中,所述吸附时间为30分钟。在一些实施方案中,所述吸附剂为fe3o4@go,且每200μl样品中加入fe3o4@go的量为0.5至2.5mg,例如1mg、1.5mg、2mg等。
73.在本技术中,前述衍生化步骤可以直接在所述磁性吸附剂上进行。通过本领域技术人员所知晓的任何技术将所述衍生化步骤产生的衍生物与吸附剂分离后,对衍生物进行检测。
74.对含蛋白质样品的检测方法本技术尤其提供了在含蛋白质样品,尤其是在含有性激素结合蛋白和/或白蛋白的生物样品中检测所述类固醇激素的方法,例如,在血清中检测所述类固醇激素的方法。本技术的方法对所述含蛋白质样品的量要求低,例如可使用至少低至200μl的血清进行上述多种类固醇的净化、富集和定量。
75.对于含蛋白质的样品,可首先使类固醇激素从与蛋白质的结合态中解离。例如可以使用氯化铵缓冲液使类固醇激素与白蛋白和性激素结合蛋白解离。在一些实施方案中,所述氯化铵缓冲液为饱和氯化铵缓冲液。
76.以血清样品为例,在一些实施方案中,所述饱和氯化铵缓冲液与所述血清的体积比为1:1。在一些实施方案中,所述血清为200μl,所述饱和氯化铵缓冲液为200μl。
77.以上详细描述了本技术的优选实施方式,但是,本技术并不限于此。在本技术的技术构思范围内,可以对本技术的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本技术所公开的内容,均属于本技术的保护范围。
78.下面结合附图对本技术的示例性实施例进行描述,其中包括了本技术实施例的各种细节以助于理解,其结果则体现了本技术涵盖的实施方式普遍能够达到的技术效果。
79.下述实施例结果证实了,本技术的方案无需使用有毒有机试剂,且仅使用200μl的血清进行样品制备,便能同时实现对至少8种目标类固醇激素的灵敏且准确的检测。特别地,下述实施例4还根据clsi指南对上述方案进行了选择性、线性、低限值、提取回收率、批内和批间精度和准确性的评估。结果显示,所有分析物的批内和批间精密度均小于16.7%和12.9%(含定量下限)。批内和批间的相对误差(re%)分别为-14.7 ~ 13.3%和-9.3 ~ 11.0%。不同分析物的提取回收率在54.0%至92.7%之间。此外,实施例5还显示了该方案被应用于432个多囊卵巢综合症(pcos)临床样品的检测结果,通过与现有技术公知的检测结果相比较,显示了本技术的方案在精确度和选择度方面均优于现有技术。
80.实施例
81.实施例中使用的材料来源睾酮(t)购自european pharmacopoeia,雄烯二酮(a4)购自macklin biochemical有限公司(中国,上海)。11-酮基睾酮(11kt)和11-酮睾酮-d3(11kt-d3)购自cfwlabs公司(walnut, ca, usa)。11β-羟基睾酮(11oht)、11-羰基雄烯二酮(11ka4)和11β-羟基雄酮(11oha4)来自steraloids公司(newport, ri, usa)。11β-羟雄甾烯二酮(11oha
4-d4)和睾酮-d3(t-d3购自cambridge isotope laboratories公司(andover, ma, usa)。17-α-羟基孕酮及其内标物(is),17-α-羟基孕酮-d8,分别来自dr. ehrenstorfer gmbh和trc。
82.hplc级别的乙腈、甲醇和甲酸购自fisher scientific(fair lawn, nj, usa)。甲酸铵购自sigma-aldrich公司(st louis, mo, usa)。上述实施例使用的mgo为fe3o4@氧化石墨烯(fe3o4@go)纳米复合材料悬浮液(10 mg/ml,直径:fe3o4,~10nm;氧化石墨烯片的直径,《5μm)和多壁碳纳米管包埋镍(ni@mwcnts;mwcnt直径8-15nm;mwcnt长度50 μm;ni含量60%)购自 江苏先丰纳米材料科技有限公司(江苏省南京市,中国)。fe3o4@sio2纳米复合材料是由中国科学院杨海涛教授的实验室提供的。fe3o4@sio2~nh2(nh2的直径,0.1-1 μm)来自飞默生物科技有限公司。超纯水是使用颇尔纯化水系统(颇尔公司,北京)制作的。
83.六种衍生剂,包括n,n,n-三乙基-2-肼基-2-氧乙基氯化铵(thob),n,n-二丁基-n-(2-肼基-2-氧乙基)-1-丁基氯化铵(dhob)。吉拉德试剂t(gt)、2-(2-肼基-2-氧乙基)-异喹啉-2-肼溴化物(iqhb)、盐酸羟胺(hahc)和gp,均购自bidepharm co. ltd. (shanghai, p. r. china)。hplc级乙腈、甲醇和甲酸来自fisher scientific co. 甲酸铵购自sigma-aldrich fluka公司(st louis, mo, usa)。
84.实施例中使用的样品来源下述实施例中的临床样品来自北京大学第三医院的生殖医学中心。入组的女性患者(n=432)年龄在20至44岁(30.2
±
4.0),具有不孕症的临床特征。在月经的第二至第五天用抗凝管采血。用cl法检测生化指标,包括t、a4、抗穆勒氏激素(amh)、空腹血糖和空腹胰岛素水平。在分析之前,将0.5毫升的剩余血清在-80℃下冻存。
85.对照样品及标准品由于所有的目标分析物都是人血清中的内源性化合物,所以不能用血清作为空白基质来制备校准物或质量控制(qc)样品。在本技术的实施例中,用血清/活性碳以20/1(v/w)的比例去除内源性类固醇。血清和活性碳的混合物涡旋3分钟,在4℃下储存48小时。然后以4574g离心10分钟,用0.2μm的膜过滤,得到无类固醇的血清。用活性碳处理的血清被用作空白基质。
86.在甲醇中制备分析物的单个储备液,以获得适当浓度的储备液(其中一些是购买的溶液)。通过甲醇稀释储备液来制备混合次级储备液。用混合次级储备液通过甲醇连续稀释制备8种校准工作溶液、定量下限(lloq)和4种质控工作溶液。内标物(iss)的储备液和工作液也以同样的方法制备。所有的标准溶液在使用前都储存在-80℃。
87.每种分析物的校准范围是参照实际临床使用情况设定的,尽管可以得到更低的定量下限。将10μl工作溶液加入190μl空白基质中,制备8个非零校准物、lloq样品和4个qc标
准。校准范围、lloqs和qc的浓度列于图6。
88.实施例1 dmspe条件优化1.1 类固醇-蛋白质解离剂优化由于类固醇与性激素结合球蛋白(shbg)和白蛋白的蛋白结合率很高,必须加入解离试剂以获得分析物的总浓度。本实施例用相同的临床样品比较了七种不同的解离试剂,最后在提取前使用了等体积的饱和nh4cl(如图1中c所示)。
89.1.2 吸附剂及其使用条件的优化本实施例比较了三种不同类型磁性吸附剂的吸附能力和分离速度,包括mgo、ni@mwcnt和fe3o4@sio2。ni@mwcnt可以获得最高的吸附能力。然而,由于磁力较弱,它很难与水层分离。mgo和fe3o4@sio2的分离速度相当,但前者的吸附能力要比后者高很多。因此,采用mgo作为磁性吸附剂。吸附剂的量和吸附时间也进行了优化(如图1中的a和b所示)。考虑到所有的分析物特性,最终选择使用1.2毫克的mgo进行萃取,吸附时间为30分钟,达到了如图1所示的更优的吸附效果。
90.实施例2 原位衍生化条件的优化本实施例研究了影响原位衍生化的因素。测试了六种不同的羰基衍生试剂,包括n,n,n-三乙基-2-肼基-2-氧乙基氯化铵(thob),n,n-二丁基-n-(2-肼基-2-氧乙基)-1-丁基氯化铵(dhob),吉拉德试剂p(gp),吉拉德试剂t(gt),2-(2-肼基-2-氧乙基)-异喹啉-2-肼溴化物(iqhb)、盐酸羟胺(hahc)。图2的a中对每种衍生物的信号进行了比较。从结果可以看出,gp是对t、dhea、a4、11oha4、11oht和11ka4最敏感的衍生剂。gt和iqhb分别对17ohp和11kt是最好的。考虑到所有这些分析物,我们在后续测试中选择了属于吡啶鎓肼的gp。通过进一步优化gp量、衍生化温度和衍生化时间,最后确定在测试的条件范围内,使用100μl的0.2m gp(以甲醇为溶剂,并含有2%的乙酸作为催化剂)可以取得更优的衍生化效果(如图2的b、c、d所示),其大大提高了质谱分析的灵敏度。一些异构体(例如11oha4和11kt在衍生化后质量区别显著增加(m/z 285.2
→
80.0和m/z 436.2
→
357.3)),可以通过,例如本实施例优化的串联质谱条件进行分辨。
91.实施例3 hplc-dms/ms/ms如图6所述,本实施例使用了正离子模式的电喷雾电离源(esi)进行串联质谱(ms/ms)检测,并优化了多反应监测(mrm)条件。
92.当分析样品中含有t和dhea等具有相同二级质谱碎片的检测物,则必须首先通过色谱分离,再进行基于质谱的检测。此外,一些非异构体化合物可能产生相同的子离子,并会发生串扰。例如,a
4-2gp、17ohp-2gp和11oha
4-2gp会产生m/z 80的片段,这将导致相互间的串扰,必须在ms/ms前分离。发明人首先参照之前的论文(zhang x, etc., a sensitive hplc-dms/ms/ms method for multiplex analysis of androgens in human serum without derivatization and its application to pcos patients. j pharmaceut biomed 2021; 192:113680)进行hplc-dms/ms/ms,使用poroshell sb柱(150
ꢀ×ꢀ
2.1 mm, 2.7
ꢀµ
m)进行hplc分析。然而,由于衍生化的副产品对11-氧化雄激素有干扰。于是,发明人在本实施例中重新优化了色谱条件,最终选择了poroshell ec柱(50
ꢀ×ꢀ
2.1 mm, 2.7
ꢀµ
m)进行hplc分析。梯度流动相由5mm甲酸铵缓冲液(含5v%乙腈,a相)和甲醇(含5v%水,b相)组成,两相均含有0.1v%的甲酸。为了获得足够的分辨率,本实施例测试了不同的梯度,并采用
了如下的多步梯度:第一步在最初的0.5分钟内,流动相b的初始百分比为v6%,然后在3分钟内增加到45v%;第二步,流动相b在2.3分钟内慢慢变为50v%。第三步,在第6至7.5分钟(从开始色谱检测起算),使用高比例的有机相(98v%的流动相b)来清洗柱子。然后在0.3分钟内回到初始比例并平衡2.2分钟。在这些条件下,两种异构体t-gp和dhea-gp实现了基线分离。会产生串扰的非异构体(a
4-2gp,17ohp-2gp,11oha
4-2gp和11ka4)也分别被充分分离。然而,这四个分析物的基线相当高,这大大影响灵敏度和稳健性。因此,在hplc-ms/ms系统中引入了dms,基线有所下降。优化后的分离电压(sv)和补偿电压(cov)列于图6。
93.实施例4. 方案校验根据clsi指南,本实施例用六个不同批次的活性碳处理的血清评估了经前述实施例优化的方案的选择性、线性、低限值、提取回收率、批内和批间精度和准确性。根据clsi指南对所开发的方法进行了选择性、线性、定量下限、提取回收率、批内和批间精密度和准确度的评估。使用了6个不同批次的活性碳处理的血清来评估内源性干扰和基质效应。用双空白(活性炭处理后的空白血清)、单空白(活性炭处理后血清中加入内标)和定量下限样品证明了分析物和iss的潜在干扰。对每种提取物中目标保留时间的干扰情况进行了验证,以评估其选择性。内标归一化基质效应因子(ismf)被用来评估基质效应。在存在或不存在基质的情况下,以三种qc水平对8种分析物和内标进行了测试。ismf的计算方法是:ismf=(有基质时分析物与is的峰面积比)/(无基质时分析物与is的峰面积比)。参照临床要求设定校准范围,在整个范围内用8个非零的加标校准物为每种分析物生成标准曲线。用1/x2的权重系数进行线性回归拟合。
94.采用五个分析批(不同天)对四个qc浓度水平和lloq进行精密度和准确度评估,每个浓度平行做六份。批内/批间准确度计算为re%=(均值-标示值)/(标示值)
×
100。提取回收率在四个qc水平上进行评估,其计算方法为:回收率%=(萃取后的分析物峰面积
⁄
无萃取的is峰面积)/(无萃取的分析物峰面积
⁄
无萃取的is峰面积)
×
100。质控工作液在提取前添加,以获得提取时的分析物峰面积,在提取后添加,以获得未提取时的分析物峰面积。所有的is工作溶液都在提取后加标,以校正仪器的变化。在下一步进行衍生化处理。
95.血清样品及处理后样品在不同条件下的稳定性也在三个qc水平上进行了评估。血清样品在室温下及4℃条件下保存96小时后,采用新的标准曲线对质控样品进行分析,以评估室温放置稳定性和冷藏稳定性。对于冻融稳定性,考察了三个冻融周期。在每个冻融循环中,质控标准品在-80℃下冷冻至少12小时,然后在室温下解冻2小时。处理后样品也考察了在4℃或室温下放置28小时的稳定性。
96.结果显示,所有分析物的内源性干扰小于17.5%,所有iss的内源性干扰小于4.1%,这表明使用本方案的内源性干扰是可忽略的。图3显示了活性碳处理后空白基质的色谱图和lloq的色谱图(空白基质中待测物(a)及内标(b),lloq样本中待测物(c)及内标(d))具体地,参照临床要求设置校准范围,lloqs范围为0.01至1纳克/毫升。应该指出的是,本实施例可达到的定量限(s/n》10)或检测限(s/n》3)均远低于现有技术中参照临床需要设置的lloqs(图6)。可以看出,lloq的s/n值从29到1052不等,这表明有可能扩大校准范围,而且该方法的灵敏度也很高。八个非零校准物被用来构建校准曲线,加权系数为1/x2。本技术的方案对所有的分析物都是线性的,系数超过0.9900。在所有接受的分析批中,校准物的准确度在85%到115%之间。
97.在lloq和4个qc水平下,用5天分别对6个平行样进行了批内/批间精密度和准确度的评估。结果显示在图7中。lloq的批内/批间精密度(以变异系数表示,cv%)不超过16.7%,所有分析物的相对误差(re%)在-12.4至10.7%。对于qc品,所有分析物的批内/批间精度不超过12.9%,相对偏差在-14.7至13.3%之间。
98.用6个不同批次的活性碳处理的血清评估了三种qc水平的基质效应,结果见图8。可见,六批血清中三个水平的ismf的cv%分别小于13.9%、10.0%和9.2%。因此,基质效应是可以忽略不计的。对四个qc水平和is的分析物回收率进行了评估,结果见图8。所有目标雄激素在整个范围内的总回收率为54.0-92.7%,cv%小于12.7%。五种is的回收率接近于相应的分析物。
99.在三个qc水平上对稳定性进行的评估,结果总结在图9中。所有分析物在血清中至少稳定96小时,或提取后在室温或4℃下稳定存放28小时。所有的雄激素都能经受三次冷冻/解冻循环,在-80℃下储存至少46天均稳定。
100.实施例5. 与临床常规方法的检测效果对比发明人收集了432名患者的空腹血清,他们在申请人单位的生殖中心例行进行口服葡萄糖耐量试验。这些人通常是肥胖者或代谢性疾病的高危人群。从该生殖中心的数据库可获得检索这些患者的其他临床及基本指标。使用本技术的方案对这些样品中的8种类固醇激素进行了检测。其检测结果和对应的临床指标相关性见图10。
101.发明人比较了临床常规使用的基于免疫测定的化学发光平台和本技术的方案所测得的t和a4。图4中显示了r2。对于t和a4,两种平台之间中的r2分别为0.334和0.285。这表明对于t和a4,化学发光平台和基于ms的平台之间的相关性很差。临床上使用的基于化学发光的t只能解释33.4%的基于ms的t,而基于化学发光的a4只能解释28.5%的基于ms的a4。显然本技术的方案具有更优的检出率。图5显示了pcos的其他临床或生物学特征与雄激素之间的相关系数(r)。图11中显示了经本技术方案检测出的各检测物之间相关系数(r)的p值。例如,基于ms的t和amh之间的r为0.57,p值小于0.0001。
102.amh是被认为是pcos的驱动力之一,本实施例的结果也再次证实了amh与t和a4有很好的相关性。此外,由于本方案的改进,申请人打破了之前检测水平的限制,可以更加精确地检测复杂生物样品中的多种过去难以区分和定量的类固醇激素,因此更加有利于后续对雄激素等类固醇激素的定量分析检测。本技术的方案,可用于对pcos等疾病的进一步诊断及研究,指导临床治疗。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1