预测气态含硫化合物常温下在活性炭上的吸附速率常数的方法与流程
本发明涉及一种通过建立定量构效关系模型(QSAR)预测气态含硫化合物常温下在活性炭上的吸附速率常数的方法,属于生态风险评价的定量结构与活性关系技术领域。
背景技术:
有机化合物结构-活性定量相关的研究,最初作为定量药物设计的一个研究分支,是为了适应合理设计生物活性分支的需要而发展起来的。定量构效关系研究是应用化学计量学方法研究有机物的分子结构与理化性质或活性之间的定量相关关系,通过选取分子的理化参数或结构参数,用化学计量学和数理统计法研究有机化合物结构与其理化性质或生物活性之间的定量关系,建立定量构效模型方程来预测有机化合物分子理化性质或生物活性。它对于设计和筛选生物活性显著的药物,以及阐明药物的作用机理等均具有指导作用。特别是近二三十年来,由于计算机技术的发展和应用,使QSAR不仅已成为定量药物设计的一种重要方法,而且在环境化学、环境毒理学等领域中也得到了广泛的应用。许多环境科学研究者通过各种污染物结构-毒性定理关系的研究,建立了多种具有毒性预测能力的环境模型,如大连理工大学发明的专利“通过定量构效关系模型预测大气中有机物与羟基反应速率常数的方法”(中国专利申请号201310307098.0)和“一种通过定量构效关系模型预测有机物液相蒸汽压的方法”(中国专利申请号201110410088.0)及山东大学发明的专利“一种通过定量构效关系模型预测有机磷农药对水生生物急性毒性的方法”(中国专利申请号201410053184.8)。这对已进入环境的污染物及尚未投放市场的新化合物的生物活性、毒性乃至环境行为进行了成功的预测、评价和筛选,这些都说明QSAR在环境领域中已显示出极其广阔的应用前景。
硫的有机化合物广泛存在于大气环境中,来源可分为自然源和人为源。气态含硫有机化合物的人为源主要来自工业废气的排放,气态含硫有机化合物排放到大气环境中,可以进行一些物理和化学过程,从而导致它们在大气中消除或在大气中进一步转化,会对环境和生物造成非常严重的污染和危害,例如COS和CS2扩散到大气圈的平流层时,会通过光解-氧化作用生产SO2气体,这是酸雨的主要来源之一,与此同时有可能转化为硫酸盐的气溶胶,引起大气层中臭氧的损耗,加剧全球气候变化;而且当大气环境中的气态含硫有机化合物含量达到一定浓度时,可以侵袭人类的神经系统,会带来巨大的危害,它通过呼吸道、消化道和皮肤进入人体,作用于人体的各个器官,产生致畸、神经衰弱、神经性麻痹、胚胎发育障碍和子代先天性缺陷等症状,危及人体健康。有效去除气态含硫有机物迫在眉睫,特别是持久性气态有机硫。目前去除方法包括吸附、还原、氧化等方法,利用活性炭吸附去除的较为常见。因此,利用定量构效关系模型预测气态含硫化合物在常温下在活性炭上的吸附速率常数能够有效、及时的帮助人们了解活性炭对某种有机硫的吸附特性,以便提出适当的解决方式。但经检索,利用建立定量构效关系模型预测气态含硫化合物在常温下在活性炭上的吸附速率常数的方法还未见报道。
技术实现要素:
针对现有技术上的不足,本发明的目的在于提供一种预测气态含硫化合物常温下在活性炭上的吸附速率常数的方法。
本发明方法具体步骤如下:
(1)通过查阅相关文献及书籍获得待测气态含硫化合物的分子结构信息,利用量子化学软件对待测气态含硫化合物进行几何结构优化,获得最优构型,从而得到相对分子质量M、轨道能量差、分子平衡电负性三个量子化学参数;其中,为最低空轨道能量,为最高占据轨道能量;
(2)运用多元线性回归分析建立QSAR模型,获得如下回归方程,通过回归方程计算气态含硫化合物在常温下在活性炭上的吸附速率常数,
,其中,N为分子中总原子数,为原子电负性,为分子中某个原子的原子数;
拟合能力:R2=0.9312。
本发明技术方案的原理是利用已知气态含硫有机化合物分子,运用量子化学软件对其进行几何全优化,得出与吸附相关的一些分子结构参数,并查得其他无法直接计算的参数。然后结合吸附速率实验值,利用多元线性回归分析方法,建立各种分子描述符与吸附速率常数之间的定量关系拟合方程,并对方程的拟合能力,预测能力进行验证。最后对模型的适合应用范围进行规范。由此,可以快捷、有效的气态含硫化合物在常温下在活性炭上的吸附速率常数。
本发明预测气态含硫化合物在常温下在活性炭上的吸附速率常数的方法通过如下步骤构建:
(1)通过进行低温水解实验或通过查阅相关数据库和文献,获得气态含硫有机化合物的吸附速率常数()数据(本工作共收集了14个气态含硫有机化合物的相关数据);
(2)利用量子化学软件对所要研究的气态含硫有机化合物进行几何结构优化,获得相对分子质量(M)、轨道能量差(,为最高占据轨道能量)、分子平衡电负性()三个量子化学参数作为分子描述符;
(3)将步骤(1)得到的吸附速率数据中抽取1/4作为验证集数据,其余为训练集数据,训练集用来构建预测模型,验证集用来验证模型的预测能力;
(4)以步骤(2)获得的分子描述符为自变量,气态含硫化合物在常温下在活性炭上的吸附速率常数为因变量,运用多元线性回归分析建立QSAR模型,最后获得如下回归方程:
拟合能力:R2=0.9312;
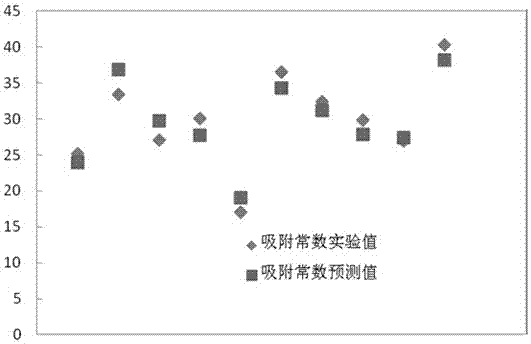
如图1所示,对于训练集来说,实验值与预测值误差较小,说明此模型具有较好的预测性及准确性,因此,此模型可以用于预测气态含硫化合物在活性炭上的吸附速率常数。
(5)将验证集数据带入获得的回归方程(图2),得到待测气态含硫有机化合物常温下在活性炭上的吸附速率常数预测值,然后根据外部预测能力评价系数的值判定外部预测能力的好坏:当大于0.7时,表示建立的模型具有良好的外部预测能力,越大,外部预测能力越好。综合实验测定,最终得其外部预测能力=0.831,说明模型具有良好的外部预测能力。其中,上述外部预测能力评价系数的计算公式如下:
(为验证集实验值,为验证集预测值,为训练集实验值均值,n为验证集个数,i表示第i个验证集)。
6)本预测模型适用于所有已知分子结构的气态含硫有机化合物。
本发明方法的优点和技术效果:
在已知气态含硫化合物分子结构的基础上,通过计算具有结构特征的分子描述符,并通过多元线性回归方法,构建了常温下活性炭对气态含硫化合物的吸附速率的QSAR模型,可快速、高效地预测气态含硫化合物在常温下在活性炭上的吸附速率常数;该方法简单、快捷、成本低,且能节省实验测试所需的人力、物力和财力;本预测模型简明、易于程序化;具有明确的应用领域、良好的拟合能力、稳健性和预测能力。
附图说明
图1 为训练集气态含硫化合物在常温下在活性炭上的吸附速率常数的实验值与预测值的拟合图;
图2 为验证集气态含硫化合物在常温下在活性炭上的吸附速率常数的实验值与预测值的拟合图。
具体实施方式
下面结合具体实施例进一步详细描述本发明,但本发明保护范围并不限于如下所述内容。
实施例1:预测二甲基三硫在常温下在活性炭上的吸附速率常数方法
首先查得二甲基三硫的分子结构信息(H3C-S-S-S-CH3),然后利用量子化学软件Gaussian 09 对分子结构进行优化,获得二甲基三硫结构的最优构型(对称构型,C-S的键长为1.89748 Å,S-S的键长为2.25417 Å,∠C-S-S= 101.4815°,∠S-S-S= 108.22126°),然后获得模型所需的描述符:相对分子质量(M为126)和轨道能量差(为4.18636eV,,其中为最低空轨道能量,为最高占据轨道能量)及分子平衡电负性(为2.3533)三个量子化学参数作为分子描述符;最后得到的吸附常数预测值为20.38647 gm/g,而查得的实验值为21 gm/g,误差仅为0.61353,与实验值非常相符。
实施例2:预测甲硫乙酸脂在常温下在活性炭上的吸附速率常数方法
首先查得甲硫乙酸脂的分子结构信息(S-C(H2)-C(H2)-O-C(O)-CH3),然后利用量子化学软件Gaussian 09 对分子结构进行优化,获得甲硫乙酸脂结构的最优构型(S-C的键长为1.88277 Å,∠S-C-C= 113.42298°),然后获得模型所需的描述符:相对分子质量(M为120)和轨道能量差(为2.5976eV,,其中为最低空轨道能量,为最高占据轨道能量)及分子平衡电负性(为2.4295)三个量子化学参数作为分子描述符。最后得到的吸附常数预测值为6.265791 gm/g,而查得的实验值为5.8 gm/g,误差仅为0.465791,与实验值非常相符。
实施例3:预测乙硫醇在常温下在活性炭上的吸附速率常数方法
首先查得乙硫醇的分子结构信息(H3C-C(H2)-SH),然后利用量子化学软件Gaussian 09 对分子结构进行优化,获得乙硫醇结构的最优构型(C-C的键长为1.52244 Å,C-S的键长为1.91543 Å,∠C-C-S= 113.40251°),然后获得模型所需的描述符:相对分子质量(M为62)和轨道能量差(为6.82584eV,,其中为最低空轨道能量,为最高占据轨道能量)及分子平衡电负性(为2.3669)三个量子化学参数作为分子描述符。最后得到的吸附常数预测值为30.74464 gm/g,而查得的实验值为30 gm/g,误差仅为0.744641,与实验值非常相符。
实施例4:预测3-甲基-2-丁烯-1-硫醇在常温下在活性炭上的吸附速率常数方法
首先查得3-甲基-2-丁烯-1-硫醇的分子结构信息(HS-C(H2)-C(H)=C(CH3)-CH3),然后利用量子化学软件Gaussian 09 对分子结构进行优化,获得3-甲基-2-丁烯-1-硫醇结构的最优构型(S-C的键长为1.94020 Å,∠S-C-C= 112.77367°),然后获得模型所需的描述符:相对分子质量(M为102)和轨道能量差(为6.02181eV,,其中为最低空轨道能量,为最高占据轨道能量)及分子平衡电负性(为2.3209)三个量子化学参数作为分子描述符。最后得到的吸附常数预测值为31.1919 gm/g,而查得的实验值为31.2 gm/g,误差仅为0.0081,与实验值非常相符。
- 还没有人留言评论。精彩留言会获得点赞!