一种基于图模型的蛋白质复合物识别方法与流程
本发明涉及一种蛋白质复合物识别方法,尤其涉及一种基于H-index图模型的蛋白质复合物识别算法HPCMiner(H-index based Protein complex Miner),将图模型H-index看作蛋白质复合物的核,通过考察扩展该图模型的一阶邻居结点识别蛋白质复合物。
背景技术:
在后基因时代,系统分析蛋白质互作网络拓扑结构,从蛋白质网络中识别蛋白质复合物,对预测蛋白质功能、解释特定的生物进程具有重要作用。同时,蛋白质复合物识别也为研究蛋白质互作网络提供了一种研究途径。蛋白质网络可以用图模型表达,其中结点是蛋白质,边代表蛋白质之间的相互作用。这样的图模型为理解复杂的生物系统提供了一个新的视角。
虽然可以对蛋白纸互作网络利用图模型的表达方法做研究,但是目前对蛋白质互作网络上的蛋白质复合物或蛋白质模块的识别,还停留在蛋白质复合物在图模型里呈现的是一个密集的蛋白质簇这样一个概念。目前对蛋白质复合物在相互作用网络中对应的子图模型还没有严格的数学表述和统一的定义。所以,很多研究者将蛋白质复合物识别问题转化为稠密子图的求解问题。
为了在蛋白质网络中发现这些重要且有生物意义的稠密子图,Sprin和Mimy(Spirin V and Mimy LA.Protein complexes and functional modules in molecular networks.Proc Natl Acad Sci USA,100(21):12123-12128,2003.)通过挖掘蛋白质中ds=1(dense subgraph,ds)的全连通图(极大团)来识别蛋白质复合物。然而,由于目前能够获得的蛋白质相互作用还不完全,仅通过挖掘全连通图来识别蛋白质复合物存在很大的局限性。最近,有很多蛋白质复合物识别方法,Bader和Hogue(Bader G.and Hogue C.An automated method for finding molecular complexes in large protein interaction networks.BMC Bioinformatics,4:2,1.)提出的MCODE(Molcular Complex Detection)算法,算法以蛋白质互作网络为输入,通过寻找稠密的蛋白质簇发现蛋白质复合物。MCODE算法可以大致分为三个阶段:第一阶段利用基于网络密度的方式计算网络结点的权重;根据已经计算完毕网络中结点的权重,从权重较大的结点开始利用贪婪搜索的方式扩展一个区域作为寻找到的蛋白质复合物;最后通过定义好的条件过滤掉不符合条件的复合物,但是该算法不能保证预测到的蛋白质复合物之间连接紧密。Enright等(Enright A.et al.An effcient algorithm for large-scale detection of protein families.Nucleic Acids Res,30:1575-1584,2002.)提出的MCL(Markov Cluster Algorithm)发现蛋白质互作网络上稠密的簇,通过模拟随机流的方式分析网络结点的分布情况,模拟随机游走的反复迭代在蛋白质互作网络上游走。网络可以看成是引导游走的路径的集合。经过足够的次数的迭代游走之后,游走者如果进入一个稠密的连接区域,就会有很小的概率走出来。该算法利用一个膨胀系数将高密度连接区域与低密度链接区域分开,但是膨胀系数会影响算法寻找到的蛋白质簇的结构,膨胀系数越大簇的数量越大。Rungarityotin等(Rungsarityotin W.et al.Identifying protein complexes directly from high-throughput tap data with markov random fields.BMC Bioinformatics,8:482.2007.)提出一个新的基于马尔科夫随机域MRF(Markov Random Feilds)的蛋白质复合物识别算法,MRF应用概率模型确定蛋白质互作网络上假阴性和假阳性的结点的质量,定义每个蛋白质的质量分数由其周围的邻居决定,根据已经确定的蛋白质的质量分数寻找蛋白质复合物。King等(King AD,Przulj N,and Jurisica I.Protein complex prediction via cost-based clustering.Bioinformatics,20(17):3013-20,2004.)提出的RNSC(the Restricted Search Clustering)尽力发现蛋白质复合物通过把网络中的定点集划分为不同的簇。RNSC算法开始随机指定一个簇类,然后不断地对这些定点集进行再划分使得划分分数达到最大值,最后根据划分的网络子集,即功能模块的最小、密度以及功能同源性,对这些功能模块进行过滤。Palla等(Palla G,Derényi I,Farkas I,and Vicsek T.Uncovering the overlapping community structure of complex networks in nature and society.Nature,435(7043):814-818,2005.)提出基于图论的计算方法CPM,是一种基于团渗透的算法,通过挖掘图中相互连通的若干k-团组成的一个k-团链。k-团是指包含k个顶点的全连通图。如果两个k-团有k-1个公共顶点,则称这两个k-团是邻接的。一系列邻接的k-团组成一个k-团链。如果两个k-团出现在一个k-团链中,则称这两个k-团是连通的。但是算法CPM的结果数据集与k值有关,k值较大获得连通的k-团集合的规模较小,且内部结点之间连接比较紧密。算法CPM在蛋白质网络中能够识别到的蛋白质复合物数量较少,特别是k取值比较大时能够识别到的蛋白质复合物就更少。对于较小的k取值算法CPM通常会发生规模比较庞大的k-团集合。这样的k-团集合包含了规模大于k的团结构和比较稀疏的k-团链。在实际应用中,更希望将这样的k-团集合分裂成多个比较稠密的团。
综上所述,现有的蛋白质复合物识别方法够识别出的具有生物意义的蛋白质复合物较少,且算法对输入参数较为敏感,识别结果不太理想。
技术实现要素:
本发明要解决的技术问题是提供一种能够识别出比较多的具有生物意义的蛋白质复合物,且算法对输入参数不敏感的蛋白质复合物识别方法。
为了解决上述技术问题,本发明的技术方案是提供一种基于图模型的蛋白质复合物识别方法,其特征在于:该方法由以下步骤组成:
步骤1:概念定义
将给定物种的蛋白质互作网络视为网络图G=(V,E),V是蛋白质结点,E是蛋白质相互作用边的集合,从所有的边的集合中去掉网络中自连接边和重复边;为从蛋白质互作网络G中发现所定义的蛋白质复合物,首先定义如下概念:
定义1 HP-vertices
给定蛋白质互作网络G=(V,E),H-index结点代表HP-vertices蛋白质集合,定义为HP=v:v V,d(v)≥h,假如此时|HP|=h,v(V\H),d(v)≤h;HP-vertices蛋白质集合包括h个蛋白,这h个蛋白的度至少为h;从HP-vertices扩展至概念HP-neighbors;
其中,v是代表蛋白质结点,d(v)是结点v的度,v(V\H)是度为H的蛋白质结点;H-index,又称为H指数或H因子,是一种评价学术成就的新方法。H代表“高引用次数”,一名科研人员的H指数是指他至多有H篇论文分别被引用了至少H次;
定义2 HP-neighbors
HP-neighbors定义为HP-vertices蛋白质集合的一阶邻居的集合;
定义3 HP-graph
蛋白质互作网络G的子图HP-graph由HP-vertices和它的HP-neighbors,除去一阶邻居HP-neighbors之间的边;
对于一个蛋白质互作网络,HP-graph从一个原始蛋白质互作网络里分离出来有可能是一个非连通子图,因此从非连通子图HP-graph中分离出所有的子图,最终得到的蛋白质互作网络的所有子图为所要识别的蛋白质复合物;
定义4 HP-complex
如果HP-graph是非连通的,HP-complex定义为HP-graph的所有子图;
所有从HP-graph中分离的子图都是想要找到的蛋白质复合物;
步骤2:获取蛋白质复合物的核蛋白HP-vertices顶点集,扩展其边缘结点一阶邻居HP-neighbors,形成HP-graph图模型;
步骤3:判别HP-graph的连通性,找到所有的稠密子图,即蛋白质复合物。
本发明提供的算法能够识别出具有重要生物意义的蛋白质复合物,另一方面本算法只需要扫描一次给定的蛋白质互作网络,不需要重复计算,且算法对单数不敏感,因此本算法是非常有效的。
本发明提供了一种新的基于H-index图模型的蛋白质复合物识别算法HPCMiner,将图模型H-index看作蛋白质复合物的核,通过考察扩展该图模型的一阶邻居结点识别蛋白质复合物。将算法应用于已知的酵母蛋白质网络,实验结果表明算法HPCMiner能够识别出比较多的具有生物意义的蛋白质复合物,且算法对输入参数不敏感。
附图说明
图1为一个给定物种的蛋白质互作网络示意图;
图2为执行算法结果得到的子图HP-graph;
图3为由算法执行结果HP-graph分离出两个蛋白质复合物;(a)为第一个蛋白质复合物,(b)为第二个蛋白质复合物;
图4为Yeast蛋白质相互作用网络利用算法HPCMiner得到的蛋白质复合物统计数据;
图5为边的密度增加算法执行时间变化图;
图6为算法HPCMiner在Yeast数据集上的执行速率展示;
图7为Rcp值为0.6、0.8、1时蛋白质复合物的质量;
图8为369个蛋白质复合物中随机选取的蛋白质复合物。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
本实施例中,给定物种的蛋白质互作网络可以视为网络图G=(V,E),V是蛋白质结点,E是蛋白质相互作用边的集合,从所有的边的集合中去掉网络中自连接边和重复边。为从蛋白质互作网络G中发现所定义的蛋白质复合物,首先定义一些概念,为从蛋白质网络中识别蛋白质复合物做准备。
一、定义
定义1 HP-vertices
给定蛋白质互作网络G=(V,E),H-index结点代表HP-vertices,定义为HP=v:v V,d(v)≥h,假如此时|HP|=h,v(V\H),d(v)≤h。HP-vertices蛋白质集合包括h个蛋白,这h个蛋白的度至少为h。从HP-vertices扩展至概念HP-neighbors。
其中,v是代表蛋白质结点,d(v)是结点v的度,v(V\H)是度为H的蛋白质结点;H-index,又称为H指数或H因子,是一种评价学术成就的新方法。H代表“高引用次数”,一名科研人员的H指数是指他至多有H篇论文分别被引用了至少H次;
定义2 HP-neighbors
HP-neighbors定义为HP-vertices蛋白质集的一阶邻居的集合。
定义3 HP-graph
蛋白质互作网络G的子图HP-graph由HP-vertices和它的HP-neighbors,除去一阶邻居HP-neighbors之间的边。
对于一个蛋白质互作网络,HP-graph从一个很大原始蛋白质互作网络里分离出来有可能是一个非连通子图。因此本实施例的方式是从非连通子图HP-graph中分离出所有的子图,最终得到的蛋白质互作网络的所有子图为所要识别的蛋白质复合物。
定义4 HP-complex
如果HP-graph是非连通的,HP-complex定义为HP-graph的所有子图。
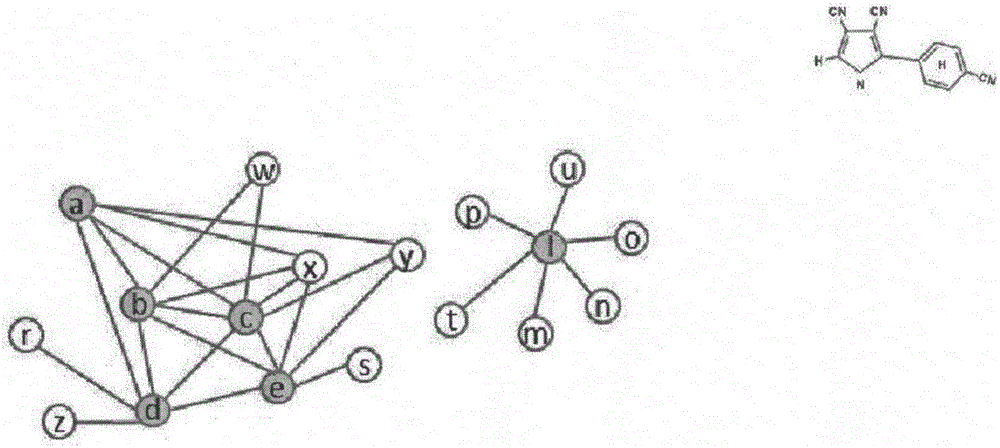
所有从HP-graph中分离的子图都是想要找到的蛋白质复合物。下面举个实例说明这些概念的具体含义。假设图1是一个给定物种的蛋白质互作网络,包括19个结点和33条边。蛋白质互作网络G中HP-vertices是H={a,b,c,d,e,l},这意味着H=6。在图1中很容易要检测出这6个蛋白质结点,即图1中带有阴影的结点,这些节点的度都为6。
从图1中可以看到,HP-vertices的HP-neighbors集合是{r,s,w,x,y,z,o,p,m,n,t,u}。H集合中的多有顶点,H={a,b,c,d,e,l,r,s,w,x,y,z,o,p,m,n,t,u},即是HP-graph包含的所有顶点。顶点q没有包含在H中,因为它不是H中顶点的一阶邻居。HP-graph包含HP-vertices与HP-neighbors点之间所有的边除去HP-neighbors本身蛋白质之间的边。
二、算法
首先获取蛋白质复合物的核蛋白HP-vertices顶点集,然后扩展其边缘结点一阶邻居HP-neighbors,形成HP-graph。值得注意的是HP-graph可能是一个非连通子图HP-index图模型的定义决定的,在之前的举例说明中也介绍过这个特点。算法最终目的是从很大的蛋白质互作用网络中能够将所有的稠密子图,分离出来作为蛋白质复合物。根据HP-graph图模型的特征,识别出的蛋白质复合物中每个蛋白质复合物中至少有一个核蛋白。算法HPCMiner主要步骤包括寻找HP-vertices顶点集、扩展至边缘顶点集合HP-neighbors、生成初始图模型HP-graph、判别HP-graph的连通性终找到所有的稠密子图即蛋白质复合物。由于纸张的篇幅大小限制,算法RWSPFinder分两个部分描述,一部分是算法的输入和输出,见算法2,另一部分是算法的核心执行代码,见算法3。
算法2识别复杂网络中蛋白质复合物算法
利用图1的蛋白质互作网络,演示算法HPCMiner在该网络上执行识别蛋白质复合物人物的结果。首先输入蛋白质互作网络G即图1,通过我们定义的图模型HP-vertices和HP-neighbors找到第一个h=6的HP-graph子图,用G1表示,其对应的子图即是图2中那些阴影的结点。明显的,子图G1包含两个子图是非连通的,算法将分解该子图为两个蛋白质复合物。
算法3识别复杂网络中蛋白质复合物算法
图3显示了算法找到的两个蛋白质复合物。
三、算法分析
HPCMiner算法第一步将蛋白质互作网络转化为无向简单图的时间复杂度为O(m),第二部迭代穷举蛋白胡互作网络中所有的HP-graph,包括更新图过程需要花费,当然,此部分还要再加上每个蛋白n次在h和度h-index的对比的时间花费。最后一步识别蛋白质复合物的时间花费。这暗含时间复杂度的上限,这里h表示每个子图HP-graph中h-index的值,n是蛋白质互作网络中积淀的数量,u是找到的所有子图HP-graph的数量。本实施例提出的算法能够识别出具有重要生物意义的蛋白质复合物,另一方面本实施例提出的算法只需要扫描一次给定的蛋白质互作网络,不需要重复计算,且算法对单数不敏感,因此本实施例提出的算法HPCMiner是非常有效的。
四、算法有效性验证
为了评估所提出算法的有效性,利用C语言实现了算法HPCMiner,在真实的数据集和模拟数据集上做了大量的实验。Graphweb是一个公共的基于图分析数据的生物网络web服务器,也是一个生物网络图数据分析工具,该生物工具可以分析包括基因、蛋白质和基因表达微阵列数据等有向生物网络、无向生物网络、加权网络、无权网络生物数据集。用户可以上传GraphWeb规定的生物数据集格式,该web平台会自动映射到集合数据的基因标识,经处理和统计得出指定的统计分析指标。在本实验中,将四个生物数据集经过处理,得到GraphWeb可以识别的规定的数据格式,利用该实验工具,分别上传生物蛋白质相互作用数据集包括Yeast,得到每个蛋白质互作网络的结点(Nodes)、边(Edges)、边密度(Edge Density,ED)和结点的平均度密度(Average node Degree,AveD)信息,来了解和认识这些生物网络的特征,详细的统计信息见表1。
表1蛋白质相互作用网络的详细信息
本实验以酵母蛋白质网络作为研究对象,因为酵母是所有物种中蛋白质相互作用数据最完备的。实验所用的蛋白质相互作用数据和用于评估的标准蛋白质复合物数据来源于MIPS数据库。在数据预处理阶段,去除了蛋白质相互作用数据中的自相互作用和冗余的相互作用,最终的相互作用网络包括1812个酵母蛋白质和6838对相互作用。首先对蛋白质复合物的核蛋白以及蛋白质复合物做基于GO注释的评估upcitepcGuang:159。GO是一个层次结构,一个蛋白质能够映射在不同的类层次或者相同的。利用GO词典映射机制评估找到的蛋白质之间的相似程度。另外,功能富集分析进一步识别蛋白质复合物的生物意义,计算每个蛋白质复合物对应的P-value。很多研究者根据超几何聚集分布的P-value来注释识别蛋白质复合物的主要功能。P-value体现了识别的蛋白质复合物对某个功能的富集程度,其计算公式为:
其中,N表示蛋白质网络的规模,C表示蛋白质复合物中蛋白质数量,k表示蛋白质复合物中含有某个功能的蛋白质数量,F表示蛋白质网络中含有该功能的蛋白质数量。如果P-value越小,越接近0,则说明蛋白质复合物能够随机出现这种功能的概率就越低,当然可能更有生物学意义。同一个蛋白质复合物内的蛋白质通常具有相同或相似的功能。一般,将P-value的最小值对应的功能作为该蛋白质复合物的主要功能。通过给每个识别的蛋白质复合物赋予其P-value最小时对应的功能,可以预测位置蛋白质的功能。这里计算P-value所用的蛋白质功能注视信息来源于FunCat。通过计算P-value,可以给每个识别的蛋白质复合物一个最佳功能注视信息。
为了清晰的表达蛋白质复合物的识别结果,将识别出的酵母蛋白质网络的复合物数量分为几组,进行统计分析。对于酵母蛋白质网络,利用HPCMiner算法识别出蛋白质复合物369个,在这些蛋白质复合物中包含蛋白质数量大于100个的有2个,大于50小于100个蛋白质的蛋白质复合物2个,大于30个蛋白质小于50个蛋白质的蛋白质复合物5个,大于2个蛋白质小于30个蛋白质的蛋白质复合物360个,利用算法HPCMiner找到的蛋白质复合物复合meso-scale要求的5至25个,这个统计结果如图4所示。
这个统计结果也证实了酵母蛋白质网络仅有几个很大的簇,约97.56%的蛋白质复合物包含的蛋白质个数都小于30。意味着通过本模型和算法得到的蛋白质复合物的大小在2到30正常的范围内。为了展示本算法HPCMiner处理大数据集的能力,利用模拟的四个数据集分别包含10000个顶点、20000个顶点、30000个顶点、40000个顶点的网络模拟数据集,边的密度设置为0.2%,然后改变边的密度直到10%。图5是HPCMiner算法在这四个数据集上随着边的密度增加算法执行时间变化图。
为了评估核蛋白在一个蛋白质复合物中的关键作用,利用BiNGO工具计算核蛋白与边缘蛋白之间的P-value值,保留那些满足P-value阈值的蛋白质对。在蛋白质复合物中,核蛋白集合为Cc,边缘蛋白为Cr。那些与核蛋白构成的蛋白质对为Cnb。当计算完Cnb中那些关键的蛋白质对的P-value值后,那些剩余的符合P-value值阈值的蛋白质对留下来为Pnb,每个蛋白质复合物都计算比例为Rcp。
对于图6通过随机抽取出酵母蛋白质网络上10%、30%、50%、80%、100%数量的边,展示了HPCMiner算法在酵母蛋白质互作网络上的执行效率。测试蛋白质复合物中核蛋白与边缘蛋白的紧密关系。定义的拓扑结构图模型HP-graph包含HP-vertices及HP-neighbors,在这个图模型里,HP-vertices蛋白之间联系是非常紧密的,但是需要确定HP-vertices蛋白与HP-neighbors蛋白的联系是否紧密,联系的紧密程度在生物学上体现的是两个物质之间的相似程度。
下面计算核蛋白与边缘蛋白之间的P-value值的方法,计算HP-vertices中每个核蛋白与边缘蛋白HP-value值的方法,计算HP-vertices中每个核蛋白与边缘蛋白HP-neighbors之间的P-value值,如果P-value小于一定的阈值就保留该对蛋白,在一个蛋白质复合物中,剩余越多的蛋白质对,说明检测到的蛋白质复合物越优越。在计算P-value值的时候,使用生物工具BiNGO,这个生物工具是做蛋白质功能的统计分析工具。为了评估核蛋白在一个蛋白质复合物中的关键作用,利用BiNGO工具计算核蛋白与边缘蛋白之间的P-value值,保留那些满足P-value阈值的蛋白质对。在蛋白质复合物中,核蛋白集合为Cc,边缘蛋白为Cr。那些与核蛋白构成的蛋白质对为Cnb。当计算完Cnb中那些关键的蛋白质对的P-value值后,那些剩余的符合P-value值阈值的蛋白质对留下来为Pnb。然后计算Cnb与Pnb的交集,如果交集越大说明检测到的蛋白质复合物越是准确,越具有生物意义的。利用找到的蛋白质复合物作为实际例子,阐述测试的实验过程。如图3所示,由算法HPCMiner检测出的蛋白质复合物,其核蛋白Cc=l,Cr=m,n,o,u,p,t,则Cnb={(l,m),(l,n),(l,o),(l,u),(l,p),(l,t)},假设P-value值为0.0001,小于该阈值的要加入到Pnb中。用如下公式计算衡量找到的蛋白质复合物的质量。
对每个蛋白质复合物都计算比例此Rcp,根据设定的不同的P-value阈值测量出的所有蛋白质复合物的Rcp,根据所统计的Rcp的平均值展示所找到的蛋白质复合物的质量如图7所示。图7展示了分别设定Rcp值为0.6,0.8,1来观察和衡量蛋白质复合物的质量。Rcp=1意味着整个蛋白质复合物所有的蛋白质都符合测定P-value阈值。
由图7可见,当设置P-value为0.01时,满足比例Rcp平均值0.8以上的蛋白质复合物达到84.8%。甚至当设定P-value阈值为0.001时,满足比例Rcp平均值0.6以上的蛋白质复合物达到78.9%。
这个实验结果表明,基于HP-graph模型的蛋白质复合物识别方法是非常有效的,能够找到具有生物意义的蛋白质复合物。为了进一步估计蛋白质复合物在GO分类词典上的生物相关性,考虑了蛋白质复合物在GO上所有的分类映射。从识别的369酵母蛋白质复合物中,随机选择一个蛋白质复合物,结构如图8所示,计算该蛋白质复合物中所有蛋白之间的蛋白质语义相似性。
在图1-8中核蛋白是YMR268C,其一阶邻居为YER112W,YBR055C,YPR178W,YBL026W,YJR022W,YNL147W,YDR378C,YER146W,YLR438C-A。语义相似性度量采用的是James Z.Wang的语义度量生物工具测得。
由表1可以看出,所有蛋白质之间的语义相似性的值都大于0.7,其中最大的语义相似性值达到完全相似为1(非对角线元素)。此结果表明通过本实施例提供的算法识别到的蛋白质复合物是具有生物意义的。
- 还没有人留言评论。精彩留言会获得点赞!