一种微生物的检测识别方法和系统与流程
本发明属于生物工程领域,尤其涉及一种微生物的检测识别方法和系统。
背景技术:
决定生物性状的蛋白质和RNA分子都是以DNA四种碱基的编码序列形式,将信息储存于生物细胞中。这种DNA分子包含了生物体的全套遗传信息。为了从整体角度去了解遗传信息的功能和作用,最重要的一步是将该生物的全套遗传信息测定出来,即知道该生物所有的DNA碱基排列顺序。传统的基因组测序主要采用“sanger”法测序技术,也称作“末端终止法”测序技术。这种测序方法的最大缺点是:成本高、产量低。近年来,以solexa为代表的“新一代高通量测序技术”悄然兴起。以“边合成边测序”为原理的solexa测序技术,有效地改进了传统sanger测序法的不足,具有成本低、通量高、时间短、测序准确率高、操作简便等诸多优点。
微生物在自然界中是无处不在,无处不有的,数目巨大。微生物对于地球上的生命是至关重要的,它们可以将重要的元素转换为能量,保持大气中的化学平衡,为植物和动物提供养分。微生物还可以用于实现许多商业目的,如制造抗生素、提高农业效率以及生产生物燃料。此外还有一小部分微生物对人有害,导致各种疾病的发生。从历史观点来看,微生物研究主要集中于研究个体物种。但大多数微生物是以群落的形式存在于各种环境中(生物内环境、外环境、极端环境等),而无法在实验室里单独培养。对于环境中复杂的微生物群落,传统的研究方法是针对特定的保守基因(如16S rRNA等)使用PCR技术扩增后进行测序。通过对这些保守基因的进化分类分析,从而将环境微生物进行分类。这是从物种、甚至较高的分类级别来对环境微生物进行检测的方法。这种方法可以检测出环境中未知的微生物,并且具有操作简单、技术完备、成本低廉等优点。但是随着微生物研究的不断深入、已公布的微生物基因组数目日益增多,我们发现基于保守基因测序的检测方法存在如下局限性:
1、无法识别痕量的物种。通过PCR扩增测序得到的都是丰度较高物种的基因序列。对于丰度较低的物种,需要大量的Sanger测序才能发现。
2、不能简单地由几个基因来对物种进行检测。通过对现有703种细菌基因组序列的比较分析和对真实环境样品的16S rRNA测序分析后发现:很多近缘物种的16S rRNA基因非常保守,几乎不存在差异,但是在表型上、功能上却差异显著。
3、检测只能在物种或更高的分类级别上,所得到较高级别的分类信息对以后的功能研究没有太大的作用。而即使是同一种细菌,不同菌株之间也会存在很大差异。
技术实现要素:
本发明的目的在于提供一种微生物的检测识别方法和系统,旨在解决现有的环境微生物检测方法难以识别痕量的物种的问题。
本发明是这样实现的,一种环境微生物检测方法,所述方法包括下述步骤:
采用高通量的测序技术对从环境样本中提取的DNA进行测序,得到DNA标签序列;
去除所述DNA标签序列中存在的载体污染;
将去除载体污染后得到的DNA标签序列与已知数据库中的已知序列进行比对,并根据比对结果确定所述DNA标签序列所属的分类。
作为一个实施例,该方法还包括下述步骤:
对已知数据库中的已知序列进行预处理,得到能唯一代表一个物种的DNA序列片段;
计算特有序列中每一位碱基上DNA标签序列的覆盖次数,通过泊松分布拟合得到特有序列的平均测序深度;
计算特有序列中有多少位碱基被DNA标签序列覆盖,从而得到特有序列的覆盖度;
计算整条序列中有多少位碱基被DNA标签序列覆盖,从而得到整条序列的覆盖度;
根据所述特有区域的平均测序深度、特有序列的覆盖度以及整条序列的覆盖度判断出所述特有序列代表的物种被发现的可信度。
本发明的另一目的在于提供一种环境微生物检测系统,所述系统包括:
DNA测序单元,用于采用高通量的测序技术对从环境样本中提取的DNA进行测序,得到DNA标签序列;
载体污染去除单元,用于去除所述DNA标签序列中存在的载体污染;
所属分类确定单元,用于将去除载体污染后得到的DNA标签序列与已知数据库中的已知序列进行比对,并根据比对结果确定所述DNA标签序列所属的分类。
作为一个实施例,该系统还包括:
已知序列预处理单元,用于对已知数据库中的已知序列进行预处理,得到能唯一代表一个物种的DNA序列片段;
测序深度计算单元,用于计算特有序列中每一位碱基上DNA标签序列的覆盖次数,通过泊松分布拟合得到特有序列的平均测序深度;
覆盖度计算单元,用于计算特有序列中有多少位碱基被DNA标签序列覆盖,从而得到特有序列的覆盖度,并计算整条序列中有多少位碱基被DNA标签序列覆盖,从而得到整条序列的覆盖度;
可信度判断单元,用于根据所述特有区域的平均测序深度、特有序列的覆盖度以及整条序列的覆盖度判断出所述特有序列代表的物种被发现的可信度的高低。
本发明提供的环境微生物检测方法和系统,在对环境样本中提取的DNA进行测序过程中引入了高通量的测序技术,并在序列比对时,首先去除载体污染,再将该DNA标签序列与已知数据库中的已知序列进行全面比对,可以对环境采样中更多的DNA测序,甚至能够实现对全部DNA进行测序,并更加全面地对DNA序列进行比对,从而能够有效地识别痕量的物种。可以检测到环境样本中可能存在哪些微生物物种或哪一类微生物物种。进一步通过在已知数据库中对更多的,甚至所有特有序列进行处理得到平均测序深度、覆盖度以及整条序列的覆盖度来确定特有序列代表的物种被发现的可信度的高低,从而将检测精度细致到可以区分近缘物种、甚至不同菌株。
附图说明
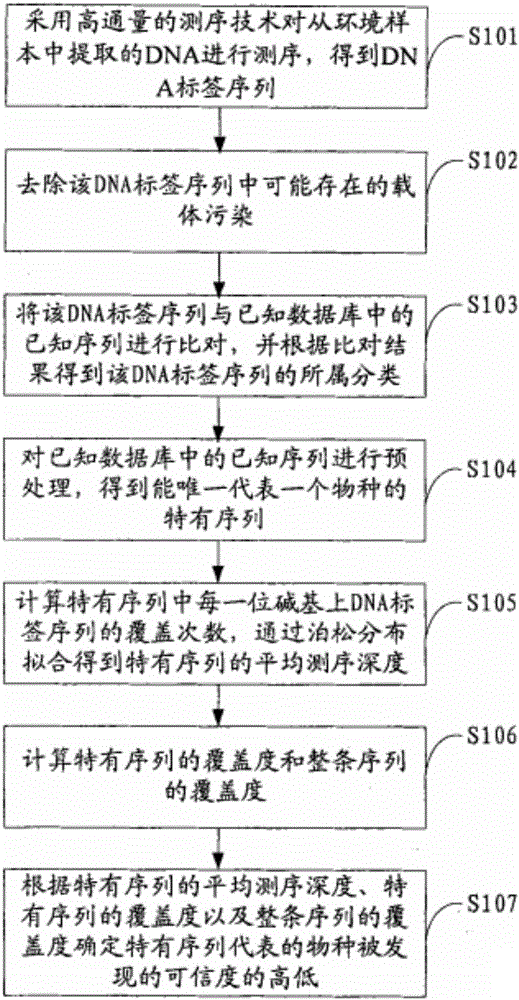
图1是本发明实施例提供的环境微生物检测方法的实现流程图;
图2是本发明实施例提供的将DNA标签序列与已知序列进行比对,确定DNA标签序列的所述分类的示意图;
图3是本发明实施例提供的连续的映射到唯一位置的模拟标签序列确定特有序列的示意图;
图4是本发明实施例提供的环境微生物检测系统的结构框图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
在本发明实施例中,采用高通量的测序技术对从环境样本中提取的DNA进行测序,得到DNA标签序列,去除该DNA标签序列中可能存在的载体污染后,将该DNA标签序列与已知数据库中的已知序列进行比对,从而得到该DNA标签序列的所属分类。
图1示出了本发明实施例提供的环境微生物检测方法的实现流程,详述如下:
在步骤S101中,采用高通量的测序技术对从环境样本中提取的DNA进行测序,得到DNA标签序列。
其中高通量的测序技术为以Solexa、Solid等为代表的第二代测序技术。由于采高通量的测序技术对DNA进行测序的具体过程是现有技术,因此,在本发明实施例中,仅简述采用高通量的测序技术对从环境样本中提取的DNA进行测序的过程:
A.从环境样本中提取DNA样品。在提取DNA样品时,需要保证样品中DNA的高质量和微生物的多样性。
B、对上述DNA样品进行文库制备。在本发明实施例中,如果需要构建双向测序文库,则为了有效的解决高含量物种的测序难题,在文库制备过程中,插入片段的长度一般小于200恥较为合适。
C、进行高通量的DNA测序反应,得到大量的DNA标签序列。
本步骤中,为提高检测的精确性,优选可以对从环境样本中提取的全部DNA进行测序。
在步骤S102中,去除步骤S101得到的该DNA标签序列中可能存在的载体污染。
由于在测序反应中所使用的载体序列是特定的,因此,由测序反应得到的DNA标签序列中可能包含这些特定的载体序列或者特定的载体序列的一部分。通过在DNA标签序列中搜索特定的载体序列字串,即可判断该DNA标签序列是否被特定的载体序列污染,进而去除该DNA标签序列中存在的载体污染。
在步骤S103中,将经去除污染后的该DNA标签序列与已知数据库中的已知序列进行比对,并根据比对结果得到该DNA标签序列的所属分类。
其中已知数据库包括但不限于细菌基因组数据库、真菌基因组数据库、病毒Genbank数据库、核糖体数据库(RDP数据库)、环境微生物的非冗余核酸序列数据库、非冗余核酸序列数据库。本发明实施例中,可以根据环境微生物的检测需求,从上述多个已知数据库中选择一个或者多个已知数据库中的已知序列与该DNA标签序列进行比对。而当环境样本较复杂时,则可以选择将所有的已知数据库中的已知序列与DNA标签序列进行比对。
在本发明实施例中,采用短串序列的映射方法将DNA标签序列与已知数据库中的已知序列进行比对,将DNA标签序列与已知序列之间的最佳匹配序列所属的分类确定为该DNA标签序列的所属分类。
其中DNA标签序列与已知序列之间的最佳匹配序列是指DNA标签序列比对到已知序列上具有最少碱基错配的序列。当采用短串序列的映射方法将DNA标签序列与已知数据库中的已知序列进行比对时,可能得到的多个最佳匹配序列,即DNA标签序列可以同时以最佳的匹配形式比对上多条已知序列,此时,将该DNA标签序列比对上的多条已知序列的最近的共同所属分类作为该DNA的所属分类。
由于微生物基因组的突变率较高,所以在将DNA标签序列与已知数据库中的已知序列进行比对时,允许预设个数的错配以及小的插入缺失序列。其中预设个数的错配可以根据经验设置。
通过上述步骤,可以得到环境样品在不同分类水平上的多样性信息。
通过上述微生物检测方法可以检测到环境样本中可能存在哪些微生物物种或哪一类微生物物种,但难以检测到物种存在的可信度,以及在物种存在的可信度高时,该物种在环境中所占的比例。
因此为了合理地解决上述两个问题,在本发明另一实施例中,可以进一步包括如下步骤S104-S107。其中,步骤S104-S107在步骤S103将DNA标签序列与已知数据库中的已知序列进行比对之前执行,也可以与步骤S103同步或在步骤S103之后进行。
在步骤S104中,对已知数据库中的已知序列进行预处理,得到能唯一代表一个物种的特有序列。其具体步骤如下:
A、根据已知数据库中的已知序列产生模拟标签序列。其具体过程如下:
从已知序列的第一位碱基开始,取预设长度的DNA序列作为第一个模拟标签序列,接着从已知序列的第二位碱基开始,取同样长度的DNA序列作为第二个模拟标签序列,依此类推,从已知序列的每一位碱基开始,取同样长度的DNA序列作为模拟标签序列。
B、将得到的各模拟标签序列映射到已知序列上,并记录映射到唯一位置的模拟标签序列。
在本发明实施例中,可以采用任意一种序列映射方法,例如SOAP比对方法,将模拟标签序列映射到已知序列上,因此,在此不再赘述。将模拟标签序列映射到已知序列上时,由于经测序得到的测序片段总会有一定的错误率存在,为了避免在实际操作中因为该测序错误而将真实DNA标签序列映射到另一位置,在本发明实施例中,在允许测序错误的前提下,将模拟标签序列映射到已知序列上。
C、查找连续的映射到唯一位置的模拟标签序列,得到能唯一代表一个物种的特有序列。其中特有序列是指能唯一代表一个物种的DNA序列片段。一般,特有序列的个数会有多个,为提闻检测的精确性,本实施例中优选找出所有的特有序列。所述特有序列的测序深度代表该物种在样品中的含量。其具体过程如下:
查找连续的映射到唯一位置的模拟标签序列,得到唯一映射的模拟标签序列的连续区域。将该连续区域的头尾两部分各去掉(模拟标签序列长度-1)个位点后的连续区域内的序列作为特有序列。因为该连续区域的头尾两部分中只被部分的模拟标签序列唯一映射,而理想的情况是每一个位点都被模拟标签序列的长度个序列唯一映射的连续区域才能唯一的代表一个物种。因此,需要将上述连续区域的头尾两部分各去掉(模拟标签序列长度-1)个位点后的连续区域作为特有序列。最后,将已知序列上全部特有序列的连接起来,做为能唯一代表这个物种DNA序列片段的“特有序列”。在本发明实施例中,当需要了解所有从环境样本中检测到的微生物物种的存在的可信度和在环境中所占的比例时,则需要对已知数据库中的所有已知序列进行上述预处理,得到能唯一代表一个物种的特有区域,由于已知数据库中可能包括多个物种,因此经预处理后,得到能唯一代表一个物种的特有区域有多个,分别唯一代表不同的物种。
请参阅图3,当查找到的连续的映射到唯一位置的模拟标签序列为短序列1至短序列II,将查找到的连续的唯一比对上的区域的头尾两部分各去掉(模拟标签序列长度个位点后的连续区域作为特有序列)。
在步骤S105中,计算特有序列中每一位碱基上DNA标签序列的覆盖次数,通过泊松分布拟合得到特有序列的平均测序深度(记为d)。其中,本步骤所述DNA标签序列对应于步骤S102经去除污染后的该DNA标签序列。根据试验结果,特有序列所代表的物种在样品中的含量是随着特有序列的平均测序深度的增加而增加的,因此,当需要了解从环境样本中检测到的物种的相对含量比时,在计算特有序列的平均测序深度时,计算唯一代表每种物种的特有序列的平均测序深度,此时,该方法还包括下述步骤:
根据计算得到的唯一代表每种物种的特有序列的平均测序深度比,得到每种特有序列代表的物种的相对含量比。由于特有序列所代表的物种在样品中的含量是随着特有序列的平均测序深度的增加而增加的,因此,计算得到的唯一代表每种物种的特有序列的平均测序深度比即为每种特有序列代表的物种的相对含量比。
如假设计算得到的唯一代表物种C的特有序列的平均测序深度为20,唯一代表物种8的特有序列的平均深度为100,唯一代表物种C的特有序列的平均深度为30时,则根据上述计算结果,可以得到物种A、物种B和物种C之间的相对含量比为20:100:30。
在步骤S106中,计算特有序列中有多少位碱基被DNA标签序列覆盖,将被覆盖的碱基位数除以特有序列中总的碱基位数,从而得到特有序列的覆盖度(记为C)。并计算整条序列中〈包括特有序列和DNA标签序列非唯一比对上的序列〉有多少位碱基被DNA标签序列覆盖,将被覆盖的碱基位数除以整条序列中的碱基位数,从而得到整条序列的覆盖度,记为(c)。比如:某一序列中有100位碱基〈即长度为100bp〉,其中80位碱基被覆盖,则计算得到该序列的覆盖度是0.8。
在步骤S107中,根据DNA标签序列的平均测序深度么特有序列的覆盖度。以及整条序列的覆盖度C’,计算特有序列代表的物种序列被发现的可信度,例如可采用如下算法计算可信度:可信度
(当P接近1时,可信度最高;当P接近0时,可信度最低),其中θ表示测序的校正因子,不同的测序方法,θ的值可能不同。通常情况下,式c<c’成立;如果实际数据中c>c’,则表明该物种序列有异常情况。
图4示出了本发明实施例提供的环境微生物检测系统的结构,为了便于说明,仅不出了与本发明实施例相关的部分。其中:
DNA测序单元41采用高通量的测序技术对从环境样本中提取的DNA进行测序,得到DNA标签序列。其中高通量的测序技术为以Solexan、Solid等为代表的第二代测序技术。该DNA测序单元41包括DNA样品提取模块411、文库制备模块412和测序模块413。其中DNA样品提取模块411从环境样本中提取DNA样品。在提取DNA样品时,需要保证样品中DNA的高质量和微生物的多样性。文库制备模块412对上述DNA样品进行文库制备。测序模块413进行高通量的DNA测序反应,得到大量的DNA标签序列。由于测序模块413的具体测序过程属于现有技术,因此,此处不再赘述。
载体污染去除单元42去除DNA测序单元41得到的DNA标签序列中可能存在的载体污染。在本发明实施例中,由于在测序反应中所使用的载体序列是特定的,因此,由测序反应得到的DNA标签序列中可能包含这些特定的载体序列或者特定的载体序列的一部分。通过在DNA标签序列中搜索特定的载体序列字串,即可判断该DNA标签序列是否被特定的载体序列污染,进而去除该DNA标签序列中存在的载体污染。
所属分类确定单元43将载体污染去除单元42处理后的DNA标签序列与已知数据库中的已知序列进行比对,并根据比对结果得到该DNA标签序列所属的分类。其中已知数据库为细菌基因组数据库、真菌基因组数据库、病毒数据库Genbank数据库、RDP社数据库、nt数据库中一种或者多种组合。
在本发明实施例中,采用短串序列的映射方法将DNA标签序列与已知数据库中的已知序列进行比对,得到DNA标签序列与已知序列之间的最佳匹配形式。其中DNA标签序列与已知序列之间的最佳匹配形式是指DNA标签序列比对到已知序列上具有最少碱基错配的位置。根据得到的DNA标签序列与已知序列中之间的最佳匹配形式即可得到该DNA标签序列所属的分类。当采用短串序列的映射方法将DNA标签序列与已知数据库中的已知序列进行比对时,可能得到的多个最佳匹配形式,即DNA标签序列可以同时以最佳的匹配形式比对上多条已知序列,此时,将该DNA标签序列比对上的多条已知序列的最近的共同所属分类作为该DNA的所属分类。
通过上述微生物检测方法可以检测到环境样本中可能存在哪些微生物物种或哪一类微生物物种,但难以检测到物种存在的可信度,以及在物种存在的可信度高时,该物种在环境中所占的比例。因此为了合理地解决上述两个问题,在本发明另一实施例中,该系统还包括已知序列预处理单元44、测序深度计算单元45、覆盖度计算单元46和可信度判断单元47。
其中已知序列预处理单元44对已知数据库中的已知序列进行预处理,得到能唯一代表一个物种的DNA序列片段。其包括模拟标签序列产生模块441、模拟标签序列映射模块442、特有序列获取模块443。
其中模拟标签序列产生模块441从已知序列的每一位碱基开始,取同样长度的DNA序列作为模拟标签序列。
模拟标签序列映射模块442将得到的各模拟标签序列映射到已知序列上,并记录映射到唯一位置的模拟标签序列。
特有序列获取模块443查找连续的映射到唯一位置的模拟标签序列区域,并将该区域的头尾两部分各去掉(模拟标签序列长度-1〉个位点后的连续区域内的序列作为特有序列。最后,将已知序列上全部特有序列连接起来,做为能唯一代表这个物种DNA序列片段的“特有序列”。由于该区域的头尾两部分中只被部分的模拟标签序列唯一映射,而理想的情况是每一个位点都被模拟标签序列的长度个序列唯一映射的连续区域才能唯一的代表一个物种。因此,需要将上述连续区域的头尾两部分各去掉(模拟标签序列长度-1)个位点后的连续区域作为特有序列,以使该特有序列的DNA序列片段能唯一代表一个物种。
测序深度计算单元45计算特有序列中每一位碱基上DNA标签序列的覆盖次数,通过泊松分布拟合得到特有序列的平均测序深度(记为d)。该特有序列的平均测序深度即为比对到该特有序列的DNA标签序列代表的物种在样品中的含量。
覆盖度计算单元46计算特有序列和整条序列的覆盖度。其包括特有序列覆盖度计算模块461和整条序列覆盖度计算模块462。特有序列覆盖度计算模块461计算特有序列中有多少位碱基被DNA标签序列覆盖,从而得到特有序列的覆盖度(记为C)。整条序覆盖度计算模块462计算整条序列中(包括特有序列和DNA标签序列非唯一比对上的序列)有多少位碱基被DNA标签序列覆盖,从而得到整条序列的覆盖度(记为C’)。
可信度判断单元47根据特有序列的平均测序深度么特有序列的覆盖度c以及整条序列的覆盖度C’判断出该特有序列所代表的物种序列被发现的可信度。在本发明实施
例中,当c近似等于且c≤c'且时,则认为该物种序列被发现的可信度高,其中θ表示测序的校正因子,不同的测序方法,θ的值可能不同。否则认为该物种序列被发现的可信度低。
当需要了解从环境样本中检测到的物种的相对含量比时,在本发明另一实施例中,该系统还包括含量比计算单元48。该含量比计算单元48根据计算得到的唯一代表每种物种的特有序列的平均测序深度比,得到每种特有序列代表的物种的相对含量比。由于特有序列所代表的物种在样品中的含量是随着特有序列的平均测序深度的增加而增加的,因此唯一代表每种物种的特有序列的平均测序深度比即为每种特有序列代表的物种的相对含量比。
在本发明实施例中,通过采用高通量的测序技术对提取的DNA样品进行测序,得到DNA标签序列,再将测序序列与已知数据库中的已知序列进行比对,根据比对结果得到DNA标签序列的所属分类,从而可以检测到环境样本中可能存在哪些微生物物种或哪一类微生物物种。通过对已知数据库中的已知序列进行预处理,得到能唯一代表一个物种的特有序列,再通过计算特有序列中每一位碱基上DNA标签序列的覆盖次数,采用泊松分布拟合得到特有序列的平均测序深度,从而检测出该特有序列代表的物种在样品中的含量。同时通过计算特有区域的覆盖度和整条序列的覆盖度,从而根据特有序列的平均测序深度、特有区域的覆盖度和整条序列的覆盖度可以判断特有序列所代表的物种被发现的可信度。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!