一种基于机器学习选择最优特征的DNA甲基化预测方法与流程
本发明涉及dna甲基化预测领域,更具体的,涉及基于机器学习选择最优特征的dna甲基化预测方法。
背景技术:
复杂疾病是由多基因、基因与基因相互作用、基因与环境相互作用共同引起的。这些基因与基因、基因与环境的相互作用形成一个多层次的复杂生物网络,正是这些复杂网络的变异引起了疾病的发生与发展。因此,生物数据分析的一个难点就是数据之间存在复杂的关联性。在基因组中,特定cpg位点的甲基化与邻近或其他区域的cpg位点相关联。例如在预测某一个cpg位点的数据时,多数其它位点的数据对建模和预测没有帮助,属于冗余信息,而某些特殊cpg位点的数据对于建模和预测精度非常重要,这些位点属于最强相关cpg位点。
dna甲基化直接影响细胞分化和组织器官发育,与冠心病等复杂疾病密切相关,是表观遗传学研究的热点问题。获取人体器官的dna甲基化表达数据对于研究该器官的病变具有重要理论研究意义和临床实用价值。但在多数情况下,很难直接对人体病变器官采样。能否用替代组织器官(例如外周血)中甲基化表达数据来预测目标组织器官(例如心脏)中的甲基化表达数据,仍是一个急需解决的问题。
科研人员对dna甲基化的预测开展了很多研究,但多数只是粗略的估计cpg岛(cpgisland,基因组中长度为300~3000bp的富含cpg二核苷酸的一些区域,主要存在于基因的5′区域)片段的甲基化状态,分辨率较低,预测甲基化状态一般只分为甲基化(常用1表示)和非甲基化(常用0表示)。
dna甲基化数据维度很高,一般上万甚至几十万,因此,如何高效搜索最强cpg位点成为迫切需要解决的问题。预测模型中包含最强相关cpg位点会进一步提高其预测性能。
技术实现要素:
本发明的目的在于克服现有技术存在的上述缺陷,提供一种基于机器学习选择最优特征的dna甲基化预测方法。本发明首先使用不同的机器学习算法一搜索不同个数的最强相关cpg位点,构建不同的模型的最强相关cpg位点集合,然后利用不同的机器学习算法二对不同的模型的最强相关cpg位点集合进行测试,并对测试结果与真实实验的结果进行比较,根据评价指标确定最优的最强相关cpg位点个数、机器学习算法一和机器学习算法二,最后建立基于多个最强cpg位点的预测模型。
为实现上述目的,本发明的技术方案如下:
一种基于机器学习选择最优特征的dna甲基化预测方法,其特征在于,包含以下步骤:
s1:根据n个训练样本的m个cpg位点的数据和p个测试样本的m个cpg位点的数据,分别生成包含训练样本替代器官dna甲基化值的数据矩阵x(n×m),包含训练样本目标器官dna甲基化值的数据矩阵y(n×m),包含测试样本替代器官dna甲基化值的数据矩阵w(p×m),包含测试样本目标器官dna甲基化值的数据矩阵z(p×m);
其中n是训练数据集样本个数,p是测试集样本个数,m是cpg位点个数,元素xit(i=1~n,t=1~m)的含义是训练样本中替代器官里第i个样本中第t个cpg位点的dna甲基化的值,元素yit(i=1~n,t=1~m)的含义是训练样本中目标器官里第i个样本中第t个cpg位点的dna甲基化的值,元素wit(i=1~p,t=1~m)的含义是测试样本中替代器官里第i个样本中第t个cpg位点的dna甲基化的值,元素zit(i=1~p,t=1~m)的含义是测试样本中目标器官里第i个样本中第t个cpg位点的dna甲基化的值,xi是矩阵x(n×m)的第i行,x.j是矩阵x(n×m)的第j列,yi.是矩阵y(n×m)的第i行,y.j是矩阵y(n×m)的第j列,wi.是矩阵w(p×m)的第i行,w.j是矩阵w(p×m)的第j列,zi.是矩阵z(p×m)的第i行,z.j是矩阵z(p×m)的第j列;
s2:指定位点j,去掉包含训练样本替代器官dna甲基化值的数据矩阵x(n×m)的第j列,形成替代器官训练矩阵xtrain,提取包含训练样本目标器官dna甲基化值的数据矩阵y(n×m)的第j列,形成替代器官训练向量ytrain;
s3:指定最强相关cpg位点的个数l,并使用机器学习算法一建立模型xopt=f(xtrain,ytrain);
s4:从xopt筛选出l-1个最强相关cpg位点{x.k}并与x.j合并为最强相关cpg位点集合x1(n×l);
s5:使用机器学习算法二对最强相关cpg位点集合x1(n×l)进行建模;
s6:使用s5中的生成模型对包含测试样本替代器官dna甲基化值的数据矩阵w(p×m)进行预测,得到预测的包含测试样本目标器官dna甲基化值的数据矩阵z*(p×m);
s7:比较预测的包含测试样本目标器官dna甲基化值的数据矩阵z*(p×m)和包含测试样本目标器官dna甲基化值的数据矩阵z(p×m),进行评价指标计算;
s8:变更s3中的最强相关cpg位点的个数l和机器学习算法一的类型以及变更s5中机器学习算法二的类型,重复s3~s7,共生成q个模型及其评价指标;
s9:对q个模型的评价指标进行综合比较,确定机器学习算法一、机器学习算法二的类型以及最强相关cpg位点的个数l。
优选地,所述步骤s3中所述的机器学习算法一为随机森林或过滤式或包裹式或嵌入式特征选择算法。
优选地,所述步骤s5中所述的机器学习算法二为支持向量机或深度学习算法。
优选地,所述步骤s7中所述评价指标共有四个,分别是,样本间相关系数
优选地,所述步骤s8中所述的最强相关cpg位点的个数l变更的最小值为10。
从上述技术方案可以看出,本发明通过使用机器学习算法选择dna甲基化数据的最优特征,建立替代器官和目标器官dna甲基化数据的最优数学模型,完成了使用多个最强cpg位点预测目标组织中甲基化水平的方法。因此,本发明具有提高搜索最强cpg位点效率,减少计算代价,提高预测模型预测性能的显著特点。
附图说明
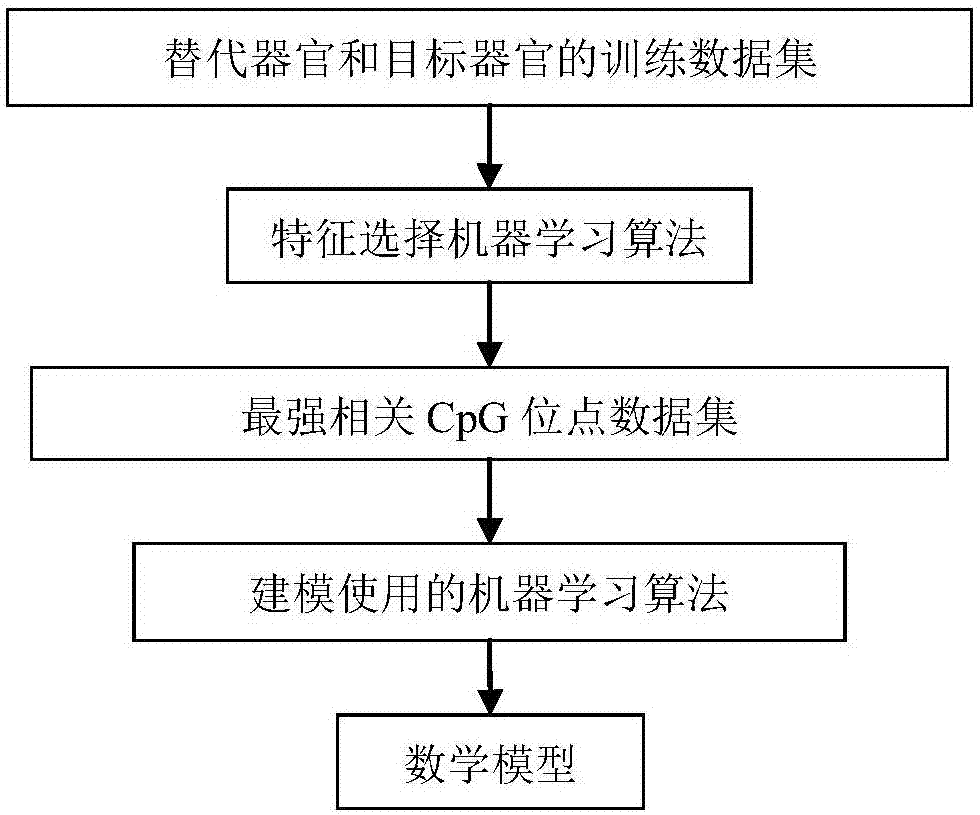
图1是本发明的流程示意图;
图2是本发明针对指定位点和最强相关cpg位点的个数建立预测模型的流程示意图;
图3是本发明使用预测模型进行测试并对预测模型进行评价的流程示意图。
具体实施方式
下面结合附图,对本发明的具体实施方式作进一步的详细说明。
请参阅图1,图1是本发明的流程示意图,并参阅图2-3。
一种基于机器学习选择最优特征的dna甲基化预测方法,其特征在于,包含以下步骤:
s1:根据n个训练样本的m个cpg位点的数据和p个测试样本的m个cpg位点的数据,分别生成包含训练样本替代器官dna甲基化值的数据矩阵x(n×m),包含训练样本目标器官dna甲基化值的数据矩阵y(n×m),包含测试样本替代器官dna甲基化值的数据矩阵w(p×m),包含测试样本目标器官dna甲基化值的数据矩阵z(p×m);其中n是训练数据集样本个数,p是测试集样本个数,m是cpg位点个数,元素xit(i=1~n,t=1~m)的含义是训练样本中替代器官里第i个样本中第t个cpg位点的dna甲基化的值,元素yit(i=1~n,t=1~m)的含义是训练样本中目标器官里第i个样本中第t个cpg位点的dna甲基化的值,元素wit(i=1~p,t=1~m)的含义是测试样本中替代器官里第i个样本中第t个cpg位点的dna甲基化的值,元素zit(i=1~p,t=1~m)的含义是测试样本中目标器官里第i个样本中第t个cpg位点的dna甲基化的值,xi.是矩阵x(n×m)的第i行,x.j是矩阵x(n×m)的第j列,yi.是矩阵y(n×m)的第i行,y.j是矩阵y(n×m)的第j列,wi.是矩阵w(p×m)的第i行,w.j是矩阵w(p×m)的第j列,zi.是矩阵z(p×m)的第i行,z.j是矩阵z(p×m)的第j列。
将训练数据集合和测试数据集均各自分成2个矩阵,分别是包含训练样本替代器官dna甲基化值的数据矩阵x(n×m),包含训练样本目标器官dna甲基化值的数据矩阵y(n×m),包含测试样本替代器官dna甲基化值的数据矩阵w(p×m),包含测试样本目标器官dna甲基化值的数据矩阵z(p×m),矩阵的行数分别为训练样本个数n和测试样本个数p,列数为cpg位点个数m。
s2:指定位点j,去掉包含训练样本替代器官dna甲基化值的数据矩阵x(n×m)的第j列,形成替代器官训练矩阵xtrain,提取包含训练样本目标器官dna甲基化值的数据矩阵y(n×m)的第j列,形成替代器官训练向量ytrain。
由于x.j与y.j对应同一个cpg位点,通常默认它们是最相关的cpg位点,所以替代器官数据矩阵x中的第j列不作为训练集参与建模过程,故而将其从包含训练样本替代器官dna甲基化值的数据矩阵x(n×m)中剔除,构建出针对指定位点j的替代器官训练矩阵xtrain。提取包含训练样本目标器官dna甲基化值的数据矩阵y(n×m)的第j列,形成替代器官训练向量ytrain。
s3:指定最强相关cpg位点的个数l,并使用机器学习算法一建立模型xopt=f(xtrain,ytrain);
s4:从xopt筛选出l-1个最强相关cpg位点{x.k}并与x.j合并为最强相关cpg位点集合x1(n×l);
指定最强相关cpg位点的个数,使用随机森林或过滤式或包裹式或嵌入式特征选择算法建立模型xopt=f(xtrain,ytrain),筛选出l-1个最强相关cpg位点{x.k}并与x.j合并为最强相关cpg位点集合x1(n×l)。
s5:使用机器学习算法二对最强相关cpg位点集合x1(n×l)进行建模。
所使用机器学习算法二为支持向量机或深度学习算法,对s4中建立的最强相关cpg位点集合x1(n×l)进行建模。
s6:使用s5中的生成模型对包含测试样本替代器官dna甲基化值的数据矩阵w(p×m)进行预测,得到预测的包含测试样本目标器官dna甲基化值的数据矩阵z*(p×m)。
s7:比较预测的包含测试样本目标器官dna甲基化值的数据矩阵z*(p×m)和包含测试样本目标器官dna甲基化值的数据矩阵z(p×m),进行评价指标计算。
评价指标共有四个,分别是,样本间相关系数
s8:变更s3中的最强相关cpg位点的个数l和机器学习算法一的类型以及变更s5中机器学习算法二的类型,重复s3~s7,共生成q个模型及其评价指标。
变更s3中的最强相关cpg位点的个数l和机器学习算法一的类型,重复s3~s7,并在s5中变更机器学习算法二的类型,共生成q个模型及其评价指标。
在变更最强相关cpg位点的个数l时,最小变更值为10个,即以10为间隔对l进行取值。
s9:对q个模型的评价指标进行综合比较,确定机器学习算法一、机器学习算法二的类型以及最强相关cpg位点的个数l。
从构建的q个模型中选取相关系数大、绝对误差小的模型,从而确定最优算法,该算法对应的机器学习算法一、机器学习算法二的类型以及最强相关cpg位点的个数l即为最优选的模型参数。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!