基于粒子群算法的金属与合金势能力场开发方法与流程
本发明涉及一种金属与合金势能力场开发方法,特别涉及一种基于粒子群算法的金属与合金势能力场开发方法,属于材料的性能预测和计算领域。
背景技术
在材料的理论计算领域,分子动力学作为微观尺度的模拟手段,能够预测材料的热力学性能以及单个缺陷在材料内部的演化行为,特别是在研究核能材料的辐照损伤方面有着广泛的应用。而势能力场是分子动力学计算的前提条件,一个准确的势能力场才能保证计算结果的准确性。对于金属材料而言,嵌入原子式(eam)势能力场是最广泛使用的势能力场。传统的eam势能力场开发方法主要使用基于梯度的爬山法,其计算过程较为复杂,收敛速度慢,且开发出的各种金属势能力场往往格式与数量级不统一,难以完成多元合金势能力场的开发。
技术实现要素:
本发明的主要目的在于旨在提出一种基于粒子群算法的金属与合金势能力场开发方法,用于快速便捷的开发各类金属与合金的分子动力学势能力场,以克服现有技术的不足。
为实现前述发明目的,本发明提供的基于粒子群算法的金属与合金势能力场开发方法,包括以下步骤:
s10.基于三阶样条插值公式建立金属与合金的eam势能力场模型;
s20.使用所述eam势能力场模型计算金属和/或合金的性能参数,建立评价函数对势能力场的准确性进行评判;
s30.基于粒子群算法对所述eam势能力场模型中的参数进行优化,获取误差最小的eam势能力场。
优选地,在步骤s10中,所述eam势能力场的原子对势函数以及电子密度函数为:
其中
优选地,步骤s10中,所述eam势能力场的嵌入电子式函数为:
其中,ρ为原子周围的电子总密度,b1,b2为修正因子,数量级分别为10-4与10-8。
优选地,在步骤s20中,所述评价函数为:
fitness=∑wp(1-pcalc/ptar)2
其中,wp为各个性能参数的权重因子,pcalc为通过eam势能力场得到的计算值,ptar为目标值;所述目标值为对所述性能参数进行第一性原理计算或实验测量得到的参数值,所述性能参数包括聚合能、晶格常数、弹性常数、相转换能、点缺陷形成能;当评价函数值越小时,势能力场的误差越小,力场越准确。
优选地,采用函数公式值替代样条插值中的三阶参数,通过下式计算函数值与三阶参数的转换关系:
其中,vk为样条插值函数在rk点的计算值。
优选地,基于粒子群算法对eam势能力场模型进行优化的参数包括:原子对势函数与电子密度函数的截断距离rk,样条插值函数在rk点的计算值vk,嵌入电子式函数中的修正因子b。
进一步地,步骤s30中所述基于粒子群算法对eam势能力场模型中的参数进行优化,包括以下步骤:
s30.1.初始化粒子的速度与位置,或载入粒子群运动状态文件;
s30.2.根据粒子的位置生成势能力场文件,通过分子动力学计算得到性能参数;
s30.3.通过评价函数判断力场的准确性,选取每个粒子在计算历史中的最小评价函数值为该粒子的历史最优值;
s30.4.从所有粒子的历史最优值中选取全局最优值,根据所述历史最优值与全局最优值对粒子的速度与位置进行更新;
s30.5.判断迭代次数是否达到单次计算上限,若未达到,则返回步骤s30.2;若达到,储存粒子群运动状态并输出最优力场的评价函数值;
s30.6.判断评价函数值是否收敛,即判断两次输出的评价函数值之差是否小于收敛阈值,若小于,表示评价函数值收敛,停止运算,优化结束;否则,将粒子群运动状态文件带入步骤s30.1继续计算。
优选地,在步骤s30.4中,采用轮盘赌方法选择全局最优值,其公式为:
其中p为选择的概率,fhop为每个粒子的评价函数历史最优值。
优选地,在步骤s30.4中,所述对粒子的速度与位置进行更新采用以下公式:
vi(t+1)=w*vi(t)+c1rand1(hopi-pi(t))+c2rand2(gopi(t)-pi(t))
pi(t+1)=pi(t)+vi(t+1)
其中,vi(t)是第t次迭代中i粒子的速度,pi(t)是第t次迭代中i粒子的位置,hop、gop分别为粒子的历史最优值与全局最优值,w是速度权重系数,c1和c2是加速因子,rand1和rand2分别是范围在{0,1}的随机数。
优选地,在步骤s30.4中,采用自适应函数计算粒子的速度权重值,其公式为:
其中,wmin,wmax分别表示权重的最大值和最小值,f表示当前粒子的评价函数值,favg和fmin分别表示当前所有粒子的适应性函数平均值和最小值。
优选地,在步骤s30.5中,所述储存粒子群运动状态包括对当前粒子的位置与速度,及其每个粒子的历史最优值与全局最优值进行存储。
与现有技术相比,本发明的优点包括:
(1)利用本发明提供的技术方案,通过三阶样条插值公式,提供了普适于各种金属元素的eam势能力场公式模型,同时提供了多元合金的势能力场公式方案;通过粒子群算法对公式中的插值进行寻优,提高了搜索效率与收敛速度。
(2)通过函数公式值替代样条插值中的三阶参数的方法,使粒子群各参数间相互独立,优化了搜索效率。
(3)通过轮盘赌方法选择全局最优值,可避免搜索过程陷入局部最优的情况;并且,通过自适应速度权重参数,提高了粒子群算法的收敛速度。
(4)在粒子群计算过程中,通过对粒子群运动状态进行存储与重载,解决了内存储存数据过多引起的计算变慢的问题,从而提升运算效率,同时提高了运算的系统稳定性。
附图说明
图1是本发明一典型实施案例中一种基于粒子群算法的金属与合金势能力场开发方法流程图;
图2是本发明一典型实施案例中一种粒子群算法流程图;
图3a~图3c分别是本发明典型实施案例中fe、al、fe3al的晶体结构图;
图4是本发明一典型实施案例中优化的fe、al、fe-al原子对势函数的三阶样条插值曲线;
图5是本发明一典型实施案例中优化的fe、al、fe-al、al-fe电子密度函数的三阶样条插值曲线;
图6是本发明一典型实施案例中优化的fe、al带修正项的嵌入电子式函数的曲线。
具体实施方式
鉴于现有技术中的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案。如下将对该技术方案、其实施过程及原理等作进一步的解释说明。
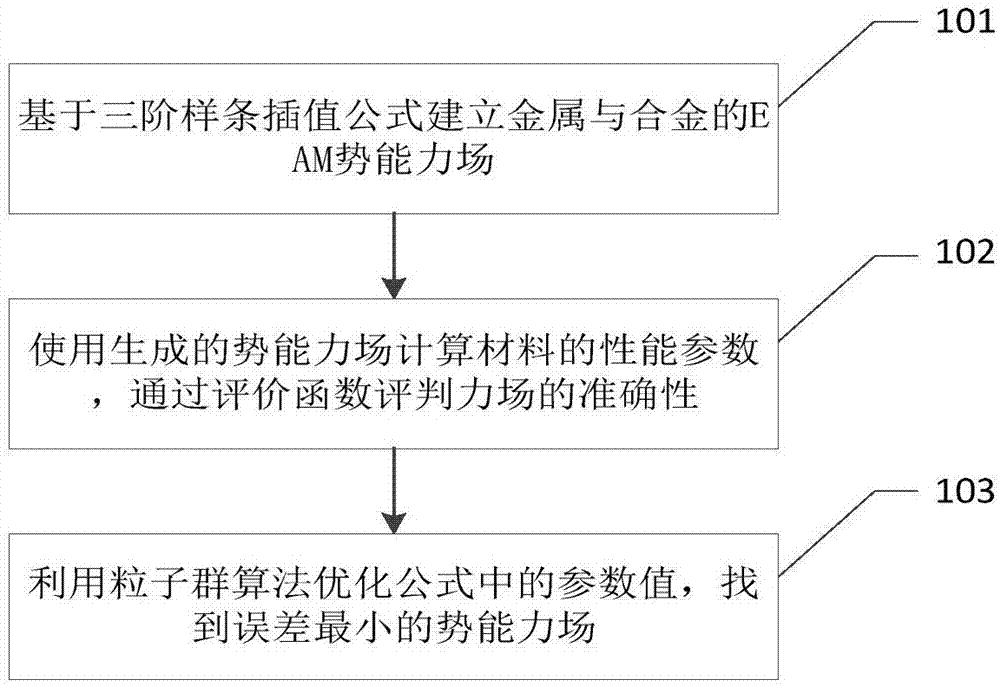
请参阅图1,本发明实施例提供了一种基于粒子群算法的金属与合金势能力场开发方法,其可以包括以下步骤:
步骤101,基于三阶样条插值公式建立金属与合金的eam势能力场;
步骤102,使用生成的势能力场计算材料的性能参数,通过评价函数评判力场的准确性;
步骤103,利用粒子群算法优化公式中的参数值,找到误差最小的势能力场。
在步骤101中,采用三阶样条插值公式描述eam力场的原子对势函数以及电子密度函数,其公式为:
其中
为保证公式中各参数的相互独立性,用函数公式值替代样条插值中的三阶参数,通过下式计算函数值与三阶参数的转换关系:
其中,vk为样条插值函数在rk点的计算值。
采用带修正的公式描述eam力场的嵌入电子式函数,其公式为:
其中ρ为原子周围的电子总密度,b1,b2为修正因子,数量级分别为10-4与10-8。
在步骤102中,采用下式对势能力场的准确性进行评判:
fitness=∑wp(1-pcalc/ptar)2
其中wp为各个性能参数的权重因子,pcalc是通过势能力场计算得到的值,ptar为目标值,包括通过第一性原理计算与实验测量得到聚合能、晶格常数、弹性常数、相转换能、点缺陷形成能等信息。
请参阅图2,一些较为具体的实施方案中,通过粒子群算法对公式中的参数进行寻优,找到误差最小的势能力场,可包含以下步骤:
步骤201,初始化粒子的速度与位置,或载入粒子群运动状态文件;
步骤202,根据粒子的位置生成势能力场文件,通过分子动力学计算得到性能参数;
步骤203,选取每个粒子的历史最优值;
计算每个粒子的评价函数值,选取每个粒子在计算历史中的最小评价函数值为该粒子的历史最优值;
步骤204,选取粒子的全局最优值,更新粒子的位置和速度;
从历史最优值中选取全局最优值,根据历史最优值与全局最优值对粒子的速度与位置进行更新。
步骤205,判断迭代次数是否达到单次计算上限,若未达到,则返回步骤202;若达到,储存粒子群运动状态并输出最优力场的评价函数值;
储存粒子群运动状态具体可包括对当前粒子的位置与速度,及其每个粒子的历史最优值与全局最优值等信息进行文件存储。
步骤206,判断评价函数值是否收敛,若收敛,停止运算,优化结束;否则将粒子群运动状态文件载入步骤201继续计算。
判断评价函数值是否收敛,即判断两次输出的评价函数值之差是否小于收敛阈值,若小于表示评价函数值收敛。
使用所述粒子群算法进行优化的参数包括原子对势函数与电子密度函数的截断距离rk,函数计算值vk,嵌入电子式函数中的修正因子b。
作为一种较佳的实施方式,可采用轮盘赌方法选择全局最优值,其公式为:
其中p为选择的概率,fhop为每个粒子的评价函数历史最优值。
进一步地,对粒子的速度与位置进行更新可采用以下公式:
vi(t+1)=w*vi(t)+c1rand1(hopi-pi(t))+c2rand2(gopi(t)-pi(t))
pi(t+1)=pi(t)+vi(t+1)
其中,vi(t)是第t次迭代中i粒子的速度,pi(t)是第t次迭代中i粒子的位置,hop、gop分别为粒子的历史最优值与全局最优值,w是速度权重系数,c1和c2是加速因子,rand1和rand2分别是范围在{0,1}的随机数。
进一步地,在速度的更新过程中,还可采用自适应函数计算粒子的速度权重值,其公式为:
其中,wmin,wmax分别表示速度权重的最大值和最小值,f表示当前粒子的评价函数值,favg和fmin分别表示当前所有粒子的评价函数的平均值和最小值。
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及具体实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
实施例1
本实施例中,对铁(fe)元素的势能力场进行开发。在常温下,铁主要以铁素体形式存在,其晶体结构为bcc,如图3a所示。分别使用5段的三阶样条插值公式描述原子对势公式,4段的三阶样条插值公式描述电子密度函数,嵌入电子式函数带两个修正项,所以计算时粒子的参数数量为20个。
在评价函数计算时,选择的比对参数包括:第一性原理计算所得的晶格常数、聚合能;bcc与fcc相的相互转化能;实验测得的单晶弹性常数c11、c12、c44;第一性原理计算的点缺陷形成能,包括空位、100哑铃对、110哑铃对、111哑铃对;聚合能随晶格常数的变化关系;第一性原理分子动力学计算所得的原子力匹配。
在粒子群寻优过程中,使用4个cpu进行并行计算,每个cpu计算20个粒子,共计80个粒子。通过储存重载的方式,将计算分为3个阶段进行,每个阶段迭代40步,共迭代120步。整个粒子群寻优过程耗时20个小时左右,优化后的势能力场函数曲线(原子对势函数、电子密度函数、嵌入电子式函数)如图4~6所示。势能力场计算结果与目标值的比对如表1所示:
表1铁(fe)势能力场计算结果与目标值
由以上比对结果可知,各参数误差值很小,可满足使用要求。使用本实施例开发的力场计算的材料的热容与热膨胀系数等热力学性能,其结果与实验结论十分接近,充分说明了本实施例开发出的力场的可靠性。
实施例2
本实施例中,进行铝(al)元素的势能力场开发。在常温下,铝的晶体结构为fcc,如图3b所示,其原子半径大于铁元素。分别使用5段的三阶样条插值公式描述原子对势公式,3段的三阶样条插值公式描述电子密度函数,嵌入电子式函数带两个修正项,所以计算时粒子的参数数量为18个。
在评价函数计算时,选择的比对参数包括:第一性原理计算所得的fcc相的晶格常数与聚合能;bcc与fcc相的相互转化能;实验测得的单晶弹性常数c11、c12、c44;第一性原理计算的点缺陷形成能,包括空位、100哑铃对、110哑铃对、111哑铃对;聚合能随晶格常数的变化关系;第一性原理分子动力学计算所得的原子力匹配。
在粒子群寻优过程中,使用4个cpu进行并行计算,每个cpu计算20个粒子,共计80个粒子。通过储存重载的方式,将计算分为3个阶段进行,每个阶段迭代40步,共迭代120步。整个粒子群寻优过程耗时28个小时左右,优化后的势能力场函数曲线(原子对势函数、电子密度函数、嵌入电子式函数)如图4~6所示,势能力场计算结果与目标值的比对如表2所示,
表2铝(al)势能力场计算结果与目标值
由以上比对结果可知,各参数误差值很小,可满足使用要求。使用本实施例开发的力场计算的材料的热容与热膨胀系数等热力学性能,其结果与实验结论十分接近,充分说明了本实施例开发出的力场的可靠性。
实施例3
本实施例中,对铁-铝(fe-al)两元体系的势能力场进行开发,由于该力场主要应用于掺杂少量al元素的fe基合金计算,所以在开发时更加侧重于al原子在bcc相fe中的缺陷行为。势能力场的中fe、al单元力场使用的是实施例1、2中得到的力场,仍需要计算的是fe原子与al原子间的原子对势函数,以及fe对于al、al对于fe的电子密度函数。分别使用5段的三阶样条插值公式描述原子对势公式,3段的三阶样条插值公式描述电子密度函数,两元体系没有引入新的嵌入电子式函数,所以计算时粒子的参数数量为22个。
在评价函数计算时,选择的比对参数包括:第一性原理分子动力学计算所得的原子力匹配;单个al原子在铁素体fe中的缺陷形成能,包括al原子替代、与fe原子形成的100、110、111哑铃对;亚稳相fe3al的晶格常数、聚合能、弹性常数以及聚合能随晶格常数变化曲线,fe3al的晶格结构如图3c所示,其中白色填充的球形图示表示fe原子,黑色填充的球形图示表示al原子。
在粒子群寻优过程中,使用4个cpu进行并行计算,每个cpu计算20个粒子,共计80个粒子。通过储存重载的方式,将计算分为3个阶段进行,每个阶段迭代40步,共迭代120步。整个粒子群寻优过程耗时6个小时左右,优化后的势能力场函数曲线(原子对势函数、电子密度函数)如图4~6所示,势能力场计算结果与目标值的比对如表3所示,
表3铁-铝(fe-al)势能力场计算结果与目标值
由以上比对结果可知,各参数误差值很小,可满足使用要求,本实施例开发出的力场具有可靠性。
采用本发明提供的技术方案,通过三阶样条插值公式,提供了普适于各种金属元素的eam势能力场公式模型,同时提供了多元合金的势能力场公式方案;通过粒子群算法对公式中的插值进行寻优,提高了寻优过程的搜索效率与收敛速度。
另外,通过函数公式值替代样条插值中的三阶参数的方法,使粒子群各参数间相互独立,优化了搜索效率;通过轮盘赌方法选择全局最优值,可避免搜索过程陷入局部最优的情况;并且,通过自适应速度权重参数,提高了粒子群算法的收敛速度;在粒子群计算过程中,通过对粒子群运动状态进行存储与重载,解决了内存储存数据过多引起的计算变慢的问题,从而提升运算效率,同时提高了运算的系统稳定性。
应当理解,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!