复合高分子电解质膜及使用其的膜电极复合体、固体高分子型燃料电池的制作方法
本发明涉及具有复合层(其是高分子电解质与纳米纤维无纺布复合化而成的)的复合高分子电解质膜及使用其的膜电极复合体、固体高分子型燃料电池。
背景技术:
燃料电池是通过将氢、甲醇等燃料以电化学方式氧化来获取电能的一种发电装置,近年来作为清洁能源供给源而受到关注。其中,固体高分子型燃料电池的标准工作温度低,为100℃左右,并且能量密度高,因此可期待作为较小规模的分散型发电设施、汽车或船舶等移动物体的发电装置而广泛应用。另外,作为小型移动设备、便携式设备的电源也受到关注,可期待代替镍氢电池、锂离子电池等二次电池而装载于手机、个人电脑等。
燃料电池通常以电池单元(cell)为单位而构成,所述电池单元如下形成,即,由发生承担发电的反应的阳极和阴极的电极与用作阳极和阴极间的质子传导体的高分子电解质膜构成膜电极复合体(以下有时简称为MEA),将该MEA用隔膜(separator)夹持而形成电池单元。以往,作为高分子电解质膜,广泛使用了作为全氟磺酸系聚合物的Nafion(注册商标)(Dupont公司制)制的膜。然而,Nafion(注册商标)制的高分子电解质膜虽然通过由团簇结构(cluster structure)带来的质子传导通道而在低加湿条件下显示出高的质子传导性,但另一方面,由于Nafion制的高分子电解质膜是经过多步合成而制造的,所以非常昂贵,此外,还存在因上述团簇结构而导致燃料渗透(crossover)大这样的问题。另外,还被指出使用后的废弃处理或材料的循环再利用困难之类的问题。
为了克服这样的课题,可替代Nafion(注册商标)的廉价且膜特性优异的烃系高分子电解质膜的开发近年越来越活跃(例如专利文献1)。然而,烃系高分子电解质膜存在干湿循环中的尺寸变化大的倾向,为了提高物理耐久性,要求减少尺寸变化。
因此,出于对燃料电池的干湿循环所伴随的电解质膜的尺寸变化进行抑制的目的,尝试了增强材料与高分子电解质材料的复合化。在专利文献2中提出了用含有聚四氟乙烯的多孔材料对电解质膜进行增强而成的复合高分子电解质膜。在专利文献3中提出了用含有聚苯并唑(polybenzazole)系聚合物的多孔材料对电解质膜进行增强而成的复合高分子电解质膜。在专利文献4中,提出了一种复合高分子电解质膜,其使用磺化聚酰亚胺作为高分子电解质材料,出于作为质子输送部位而发挥功能的目的,与掺杂有磷酸的聚苯并咪唑纳米纤维无纺布进行复合化,由此有效地抑制了尺寸变化。
现有技术文献
专利文献
专利文献1:国际公开第2006/087995号
专利文献2:日本特开2013-62240号公报
专利文献3:日本特开2004-217715号公报
专利文献4:日本特开2012-238590号公报
技术实现要素:
发明要解决的问题
然而,专利文献2中记载的复合高分子电解质膜因烃系电解质与含有聚四氟乙烯的多孔材料的亲和性差,得到的复合电解质膜存在大量空隙,所以在燃料透过、机械强度方面存在问题。专利文献3中记载的复合高分子电解质膜虽然通过使用含有作为烃系聚合物的聚苯并噁唑的多孔材料而可期待更高的亲和性,但由于多孔材料是利用湿式凝固法制成的缺乏均匀性的多孔材料,所以在燃料电池中使用时,在假定的干湿循环时有多孔材料的稀疏部位断裂而形成复合化电解质膜针孔的可能性。对于专利文献4中记载的复合高分子电解质膜而言,虽然使用了均匀性高的聚苯并咪唑纳米纤维无纺布作为多孔材料,但是为了提高质子传导性,用磷酸等对纳米纤维无纺布进行了被覆,因此有时在发电时磷酸溶出而降低复合高分子电解质膜的耐久性。另外,在不掺杂酸的情况下,存在质子传导性不足的问题。
本发明欲提供即便在低加湿、低温条件下也具有优异的质子传导性、而且尺寸变化率小、机械强度和化学稳定性优异、并且在制成固体高分子型燃料电池时能够实现高输出、优异的物理耐久性的复合高分子电解质膜及使用其的膜电极复合体、固体高分子型燃料电池。
用于解决问题的手段
用于解决上述问题的本发明是复合高分子电解质膜,其是具有复合层的复合高分子电解质膜,所述复合层是含有聚唑的纳米纤维无纺布(A)与含有离子性基团的高分子电解质(B)复合化而成的,所述含有聚唑的纳米纤维无纺布(A)为碱性。
发明效果
根据本发明的复合高分子电解质膜及使用其的膜电极复合体、固体高分子型燃料电池,即便在低加湿、低温条件下也具有优异的质子传导性,而且尺寸变化率小,机械强度和化学稳定性优异,并且在制成固体高分子型燃料电池时能够实现高输出、优异的物理耐久性。
附图说明
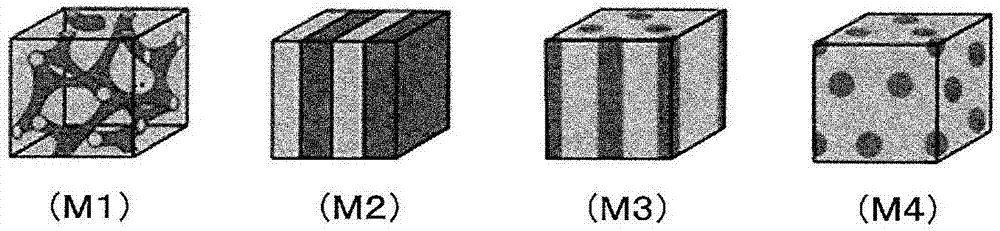
[图1](M1)~(M4)为示意性地示出含有离子性基团的高分子电解质(B)中的相分离结构的形态的说明图,(M1)示例出共连续状,(M2)示例出层状,(M3)示例出柱状(cylinder)结构,(M4)示例出海岛结构。
具体实施方式
以下,对本发明进行详细说明。以下本说明书中“~”表示包含其两端的数值的范围。
本发明针对上述问题(即,即便在低加湿、低温条件下也具有优异的质子传导性、而且尺寸变化率小、机械强度和化学稳定性优异、并且在制成固体高分子型燃料电池时能够实现高输出、优异的物理耐久性的复合高分子电解质膜)反复进行了潜心研究,结果发现电解质膜的尺寸变化率及物理耐久性很大程度上取决于多孔材料的种类和多孔材料的原料聚合物,此外,质子传导性很大程度上取决于高分子电解质材料在多孔材料中的相分离结构。换言之,探明了使用下述复合高分子电解质膜时能够解决上述问题,所述复合高分子电解质膜是具有复合层的复合高分子电解质膜,所述复合层是含有聚唑的纳米纤维无纺布(A)与含有离子性基团的高分子电解质(B)复合化而成的,上述含有聚唑的纳米纤维无纺布(A)为碱性。
<含有聚唑的纳米纤维无纺布(A)>
本发明的复合高分子电解质膜具有复合层,所述复合层是碱性的含有聚唑的纳米纤维无纺布(A)(以下有时称为“聚唑系纳米纤维无纺布”、“纳米纤维无纺布”或者简称为“无纺布”)与含有离子性基团的高分子电解质(B)(以下有时称为“高分子电解质材料”或者“高分子电解质”)复合化而成的。
碱性的聚唑系纳米纤维无纺布是指由在聚合物链中具有唑结构的聚唑系聚合物的纳米纤维形成的无纺布,通过在不利用磷酸等酸性物质进行处理的情况下使用,从而处于呈现出源自唑结构的碱性的状态。认为通过使用碱性的聚唑系聚合物,从而聚唑系聚合物的氮原子与高分子电解质材料的离子性基团发生酸碱相互作用,能够提高无纺布与高分子电解质材料的亲和性。因此,本发明中,如下所述,聚唑系纳米纤维无纺布在不进行酸性物质的掺杂的情况下以碱性的状态与芳香族烃系高分子电解质进行复合化。
聚唑系聚合物是主链具有唑结构的聚合物的统称。其中,唑结构是指包含在环内含1个以上氮原子的五元杂环结构的化合物。需要说明的是,在五元杂环中,除氮以外,还可以含有氧、硫等原子。作为本发明的聚唑,例如,可举出聚咪唑、聚苯并噁唑、聚苯并噻唑、聚苯并咪唑、聚苯并吡唑、聚苯并吡咯、聚苯并呋咱(polybenzofurazan)等,但并没有特别限定。其中,从获得性的方面考虑,优选使用聚苯并噁唑、聚苯并噻唑、聚苯并咪唑等聚苯并唑,其中,更优选使用碱性强的具有氮原子的聚苯并咪唑。
本发明中,聚苯并咪唑是指具有由下述化学式1-1或1-2表示的重复单元的聚合物。另外,聚苯并咪唑还包括具有由化学式1-1表示的重复单元及由化学式1-2表示的重复单元这两者的情况。
(化学式1-1中,Ar1为具有至少一个芳香族环的4价基团,Ar2为具有至少一个芳香族环的2价基团。化学式1-2中,Ar3为具有至少一个芳香族环的3价基团。Ar1、Ar2、Ar3分别可以被脂肪族基团、芳香族基团、卤素基团、羟基、硝基、氰基或者三氟甲基取代。)
Ar1、Ar2或Ar3所具有的芳香族环可以为苯环等单环,也可以为萘、蒽、芘等稠环。另外,可以为芳香族烃环,也可以为含有N、O、S作为芳香族环的构成原子的芳香族杂环。
化学式1-1中,Ar1优选为由下述化学式2-1或2-2表示的基团。
(化学式2-1中,Y1、Y2表示CH或N。化学式2-2中,Z表示直接键合、-O-、-S-、-SO2-、-C(CH3)2-、-C(CF3)2-、或者-CO-。)
此外,化学式1-1中,Ar2优选为从由下述化学式表示的2价基团中选择的基团。
(上述化学式中,W表示-O-、-S-、-SO2-、-C(CH3)2-、-C(CF3)2-、或者-CO-,Me表示甲基。)
另外,化学式1-2中,Ar3优选为由下述化学式表示的3价基团。
聚苯并咪唑可以为仅含有由化学式1-1或1-2表示的重复单元的均聚物,也可以为将具有多个不同结构的重复单元进行组合而成的无规共聚物、交替共聚物或者嵌段共聚物。
作为由化学式1-1或1-2表示的重复单元的具体例,可以举出由下述结构式表示的单元。
对于聚唑系聚合物而言,只要在碱性不会变得不充分的范围内,则可以含有除由化学式1-1或1-2表示的重复单元以外的追加的结构单元,可以为与追加的聚合物结构单元形成的无规共聚物、交替共聚物或者嵌段共聚物。作为这样的追加的结构单元,含有聚酰亚胺系结构单元、聚酰胺系结构单元、聚酰胺酰亚胺系结构单元、聚噁二唑(polyoxydiazole)系结构单元、聚偶氮甲碱(polyazomethine)系结构单元、聚苯并唑酰亚胺系结构单元、聚醚酮系结构单元、聚醚砜系结构单元时,能够提高力学特性,因而优选。
含有聚唑的纳米纤维无纺布(A)优选含有80重量%以上的聚唑,更优选含有90重量%以上的聚唑,进一步优选含有95重量%以上的聚唑。尤其为仅包含聚唑的纳米纤维无纺布时,机械强度高,减少尺寸变化的效果提高,因而优选。聚唑的含量低于80重量%时,有时力学强度不足,无法充分获得减少尺寸变化的效果。
使含有聚唑的纳米纤维无纺布(A)在30℃的N-甲基-2-吡咯烷酮中静置1小时时的重量变化率优选为50%以下,更优选为30%以下,特别优选为20%以下。重量变化率大于50%的情况下,有时在制膜时纳米纤维无纺布溶解,无法充分获得减少尺寸变化的效果。另外,使含有聚唑的纳米纤维无纺布(A)在80℃的N-甲基-2-吡咯烷酮中静置1小时时的重量变化率优选为50%以下,更优选为30%以下,特别优选为20%以下。重量变化率大于50%的情况下,有时在制膜时,特别是在溶剂干燥时纳米纤维无纺布溶解,无法充分获得减少尺寸变化的效果。
使含有聚唑的纳米纤维无纺布(A)在30℃的N-甲基-2-吡咯烷酮中静置1小时时的尺寸变化率优选为20%以下,更优选为10%以下,特别优选为5%以下。尺寸变化率大于20%的情况下,有时在制膜时纳米纤维无纺布收缩而产生褶皱,由此得不到均匀的膜。另外,使含有聚唑的纳米纤维无纺布(A)在80℃的N-甲基-2-吡咯烷酮中静置1小时时的尺寸变化率优选为20%以下,更优选为10%以下,特别优选为5%以下。尺寸变化率大于20%的情况下,有时在制膜时,特别是在溶剂干燥时纳米纤维无纺布收缩而产生褶皱,由此得不到均匀的膜。
作为含有聚唑的纳米纤维无纺布(A)在X射线衍射测定中的2θ,优选具有10°以上的半峰宽,更优选具有11°以上的半峰宽。为具有10°以上的半峰宽的纳米纤维无纺布时,有时能够进一步减少复合高分子电解质膜的尺寸变化。详细机理尚不明确,但认为是由于聚唑分子彼此交联而提高了机械强度。
含有聚唑的纳米纤维无纺布(A)在由反射光谱计算的Kubelka-Munk函数中,对于450nm的光而言,优选显示0.35以上的值,更优选显示0.40以上的值,特别优选显示0.45以上的值。为在Kubelka-Munk函数中具有0.35以上的值的纳米纤维无纺布时,有时能够进一步减少复合高分子电解质膜的尺寸变化。详细机理尚不明确,但认为是由于聚唑分子彼此交联而提高了机械强度。
含有聚唑的纳米纤维无纺布(A)在发射光谱测定中,以450nm的光激发的发射光谱的峰强度I450与以300nm的光激发的发射光谱的峰强度I300之比(I450/I300)优选为0.30以上,更优选为0.35以上,特别优选为0.40以上。另外,I450/I300优选为1.40以下,优选为1.20以下,特别优选为1.00以下。为I450/I300为0.30以上且1.40以下的纳米纤维无纺布时,有时能够进一步减少复合高分子电解质膜的尺寸变化。详细机理尚不明确,但认为:在I450/I300为0.30以上且1.40以下的含有聚唑的纳米纤维无纺布中,聚唑形成分子间交联结构而使得高分子链的共轭扩大,从而吸收·发射光谱向长波长位移,I450/I300显示该范围内的值,通过形成上述交联结构而提高了机械强度。I450/I300小于0.30时,有时交联结构少,吸收·发射光谱的长波长位移也小,以450nm的光激发时的峰强度变小,为该无纺布时,交联结构的形成变得不充分而导致机械强度不足。另一方面,I450/I300大于1.40时,有时交联结构变得过多,含有聚唑的纳米纤维无纺布变硬变脆,由此导致工艺性、抑制尺寸变化的能力变得不充分。
构成聚唑系纳米纤维无纺布的聚唑系纳米纤维可以为具有1nm以上且小于1μm的纳米级的纤维直径的聚唑系聚合物纤维。作为聚唑系纳米纤维的纤维直径,优选为50nm~800nm,更优选为50nm~500nm,进一步优选为50~200nm。在纤维直径比50nm细的情况下,有时力学强度不足,减少尺寸变化的效果变得不充分,在纤维直径比800nm粗的情况下,存在难以均匀地形成无纺布的厚度的倾向。本发明中,聚唑系纳米纤维无纺布可以由聚苯并唑系纳米纤维构成,其纤维直径可以为50nm~500nm。
本发明中使用的聚唑系纳米纤维无纺布的厚度没有特别限制,应该根据复合高分子电解质膜的用途来决定,但具有0.5~50μm的膜厚的聚唑系纳米纤维无纺布可在实际中使用。
与高分子电解质材料复合化之前的纳米纤维无纺布的空隙率没有特别限定,从兼顾获得的复合高分子电解质膜的质子传导性及机械强度的观点考虑,优选为50~98%,更优选为80~98%。需要说明的是,聚唑系纳米纤维无纺布的空隙率Y1(体积%)被定义为通过下述数学式求出的值。
Y1=(1-Db/Da)×100
Da:构成聚唑系纳米纤维无纺布的纤维的比重
Db:包含空隙部分的聚唑系纳米纤维无纺布整体的比重
聚唑系纳米纤维无纺布中的孔隙尺寸、即利用扫描式电子显微镜从纳米纤维无纺布的表面观察到的纤维间的距离的平均值优选为100nm以上且2000nm以下,更优选为100nm以上且1000nm以下。孔隙尺寸大于2000nm的情况下,有时减少尺寸变化的效果变得不充分,孔隙尺寸小于100nm的情况下,有时无法形成高分子电解质材料的共连续状的相分离结构,质子传导性降低。另外,如果孔隙尺寸大于下文的高分子电解质材料的平均畴(domain)间距离,则纤维对相分离结构的离子性畴所形成的质子通道进行阻碍的频率变小,因而优选。需要说明的是,孔隙尺寸按照实施例第(9)项中记载的方法测定。
另外,聚唑系纳米纤维的孔隙尺寸相对于高分子电解质材料的相分离的畴间距离而言,优选为2~200倍,更优选为10~100倍。小于2倍的情况下,有时无法形成共连续状的相分离结构,质子传导性降低,大于200倍的情况下,有时减少尺寸变化的效果变得不充分。
作为含有聚唑的纳米纤维无纺布(A)的制造方法,没有特别限定,从能够提高减少复合高分子电解质膜的尺寸变化的效果这样的观点考虑,优选按照以下的工序1~3制造。
即,
工序1:将含有聚唑的纳米纤维无纺布的原料聚合物溶解的工序;
工序2:使用工序1中得到的溶液进行静电纺纱,得到纳米纤维无纺布前体的工序;
工序3:对工序2中得到的纳米纤维无纺布前体实施不熔化处理的工序。
另外,作为工序3的不熔化处理,更优选于满足下述式(F1)的温度T(℃)实施热处理。
Tg1-50(℃)≤T≤Tg1+20(℃) (F1)
(式(F1)中Tg1表示含有聚唑的纳米纤维无纺布所含的聚唑的玻璃化转变温度(℃)。)
在上述工序3的不熔化处理中,于低于Tg1-50(℃)的温度实施热处理的情况下,有时抑制尺寸变化的效果变得不充分。另外,于超过Tg1+20(℃)的温度实施热处理时,有时无纺布变硬变脆,由此工艺性明显降低而制膜困难。
在上述工序3的不熔化处理中,更优选在将含有聚唑的纳米纤维无纺布层合于具有满足下述式(F2)的玻璃化转变温度Tg2(℃)(由不具有玻璃化转变温度的材料形成的基材的情况下,为熔点Tm(℃))的基材上的状态下实施热处理。
Tg2(Tm)>T (F2)
(式(F2)中Tg2表示构成基材的材料的玻璃化转变温度(℃),Tm(℃)表示构成基材的材料的熔点。)
上述基材只要为满足式(F2)的基材就没有特别限定,优选使用聚酰亚胺膜、玻璃、不锈钢等。
通过使用按照上述制造方法制造的纳米纤维无纺布,有时能够进一步减少复合高分子电解质膜的尺寸变化。详细的机理尚不明确,但认为是由于聚唑分子彼此交联而提高了机械强度。
<含有离子性基团的高分子电解质(B)>
接下来,对本发明中使用的含有离子性基团的高分子电解质(B)(高分子电解质材料)进行说明。
作为本发明中使用的含有离子性基团的高分子电解质(B),只要能够兼顾发电特性和化学稳定性,则全氟系聚合物和烃系聚合物均可。
此处,全氟系聚合物是指聚合物中的烷基及/或亚烷基的氢的大部分或全部被氟原子取代而成的聚合物。作为具有离子性基团的全氟系聚合物的代表例,可举出Nafion(注册商标)(Dupont公司制)、Flemion(注册商标)(旭硝子公司制)及Aciplex(注册商标)(旭化成公司制)等市售品。
这些全氟系聚合物存在非常昂贵且气体透过大这样的问题。另外,从机械强度、物理耐久性、化学稳定性等方面考虑,本发明中使用的含有离子性基团的高分子电解质(B)也优选为烃系聚合物,更优选为主链中具有芳香环的芳香族烃系聚合物。其中,优选作为工程塑料使用的那种具有充分的机械强度及物理耐久性的聚合物。此处,芳香环不仅为烃系芳香环,也可以包括杂环等。另外,一部分脂肪族系单元可以与芳香环单元一起构成聚合物。
芳香族烃系聚合物是指包含在主链中具有芳香环的烃骨架的聚合物,作为具体例,可举出在主链中具有芳香环及选自聚砜、聚醚砜、聚苯醚、聚芳醚系聚合物、聚苯硫醚、聚苯硫醚砜、聚对苯撑、聚芳撑系聚合物、聚芳撑酮、聚醚酮、聚芳撑氧化膦(polyarylene phosphine oxide)、聚醚氧化膦、聚苯并噁唑、聚苯并噻唑、聚苯并咪唑、聚酰胺、聚酰亚胺、聚醚酰亚胺、聚酰亚胺砜中的结构的聚合物。需要说明的是,此处提及的聚砜、聚醚砜、聚醚酮等是在其分子链中具有砜键、醚键、酮键的结构的统称,包括聚醚酮酮、聚醚醚酮、聚醚醚酮酮、聚醚酮醚酮酮、聚醚酮砜等。烃骨架可以具有这些结构中的多个结构。其中,作为芳香族烃系聚合物,最优选特别是具有聚醚酮骨架的聚合物,即聚醚酮系聚合物。
作为本发明的高分子电解质材料,也优选具有共连续状或层状的相分离结构的材料。这样的相分离结构可在例如包含具有离子性基团的亲水性化合物和不具有离子性基团的疏水性化合物这样的不相溶的2种以上的物质的共混成型体、或者包含含有离子性基团的链段(B1)和不含离子性基团的链段(B2)这样的不相溶的2种以上的链段的嵌段共聚物等中呈现,其结构形态大致被分为共连续状(M1)、层状(M2)、柱状结构(M3)、海岛结构(M4)这四种(参照图1)。构成本发明的复合高分子电解质膜的含有离子性基团的高分子电解质(B)优选为分别具有1个以上的含有离子性基团的链段(B1)和不含离子性基团的链段(B2)的嵌段共聚物。
在本发明的这种包含含有离子性基团的高分子电解质(B)的高分子电解质膜中,上述相分离结构大多由亲水性畴和疏水性畴形成,所述亲水性畴包含含有离子性基团的链段(B1),所述疏水性畴包含不含离子性基团的链段(B2)。在图1(M1)~(M4)中,浅色的连续相由选自亲水性畴、疏水性畴中的一方的畴形成,深色的连续相或分散相由另一方的畴形成。尤其在包含共连续状(M1)及层状(M2)的相分离结构中,亲水性畴及疏水性畴均形成连续相。
上述相分离结构例如在物理化学年度评论(Annual Review of Physical Chemistry),41,1990,p.525等中有记载。通过控制这些构成亲水性畴的化合物和构成疏水性畴的化合物的结构、组成,即便在低加湿及低温条件下也能够实现优异的质子传导性,特别是该结构为包含图1所示的(M1)、(M2)即共连续状(M1)、层状(M2)的结构时,通过形成连续的质子传导通道能够得到质子传导性优异的高分子电解质成型体,同时,利用疏水性畴的结晶性能够实现具有极佳的燃料阻挡性、耐溶剂性、机械强度、物理耐久性的高分子电解质膜。其中,特别优选为共连续状(M1)的相分离结构。
另一方面,认为即便是图1所示的(M3)、(M4)即柱状结构(M3)、海岛结构(M4)的相分离结构的情况,也能够形成连续的质子传导通道。然而,两种结构都是在构成亲水性畴的成分的比率较构成疏水性畴的成分而言相对较少的情况、或者构成疏水性畴的成分的比率较构成亲水性畴的成分而言相对较少的情况下能够构建的结构。前者的情况下,起到质子传导作用的离子性基团量绝对性地减少,特别是对于海岛结构,由于本身就未形成连续的质子传导通道,所以质子传导性差。另外,后者的情况下,虽然质子传导性优异,但是由于结晶性的疏水性畴少,所以燃料阻挡性、耐溶剂性、机械强度、物理耐久性差,均无法充分获得本发明的效果。
此处,畴是指在一个成型体中,类似的物质、链段凝聚而成的块。
另外,本发明中,也包括将多种这些聚合物进行混合而成的聚合物,统称为芳香族烃系聚合物。特别是将具有离子性基团的芳香族烃系聚合物与不具有离子性基团的芳香族烃系聚合物共混而成的聚合物作为形成共连续状的相分离结构的聚合物是有效的。
作为含有离子性基团的高分子电解质(B),可举出具有离子性基团的芳香族烃系嵌段共聚物(以下有时简称为“嵌段共聚物”)。具有离子性基团的芳香族烃系嵌段共聚物为特别优选的芳香族烃系聚合物。具有离子性基团的芳香族烃系嵌段共聚物是指包含:含有离子性基团的芳香族烃链段(B1)和不含离子性基团的芳香族烃链段(B2)的嵌段共聚物。此处,链段是指包含表示特定性质的重复单元的共聚物聚合物链中的部分结构,表示分子量为2000以上的结构。通过使用嵌段共聚物,与聚合物共混物相比较,能够呈现出具有微细的畴的共连续状的相分离结构,能够实现更优异的发电性能、物理耐久性。
以下,有时将含有离子性基团的芳香族烃链段(B1)或聚合物表述为“离子性嵌段”,将不含离子性基团的芳香族烃链段(B2)或聚合物表述为“非离子性嵌段”。不过,本说明书中的“不含离子性基团”这样的记载不排除该链段或聚合物在不阻碍共连续状的相分离结构的形成的范围含有少量的离子性基团的形态。
本发明中,优选高分子电解质材料形成了共连续状的相分离结构,即,芳香族烃系聚合物的离子性嵌段和非离子性嵌段呈现共连续状的相分离结构。利用共连续状的相分离结构中的包含离子性嵌段的畴,即便不进行利用酸性物质的掺杂也可形成良好的质子传导通道,同时,利用包含非离子性嵌段的畴能够呈现良好的机械强度、燃料阻挡性。
聚合物的相分离结构可在2种以上的不相溶的链段键合而成的高分子、例如嵌段共聚物或接枝共聚物、或者2种以上的不相溶的高分子混合而成的聚合物共混物中呈现,作为本发明的高分子电解质材料,为了在未掺杂酸性物质的条件下充分构建质子传导通道,优选使用呈现共连续状的相分离结构的高分子电解质材料。对于两种不同的嵌段一个一个连接而成的二嵌段聚合物而言,已明确了柱状结构、海岛结构的相分离结构为可在离子性嵌段的比率较非离子性嵌段而言相对较少的情况、或者离子性嵌段的比率较非离子性嵌段而言相对较多的情况下构建的结构。前者的情况下,因起到质子传导作用的离子性基团量减少,所以质子传导性降低,相反,后者的情况下,因离子性基团量增加,所以机械强度降低。层状的相分离结构的情况下,通过在与膜面方向垂直的方向上形成连续的层状结构,可构建良好的质子传导通道,但与纳米纤维无纺布复合化时,由于构成纳米纤维无纺布的纤维阻碍连续的层状结构的形成,所以得不到本发明这样的效果。
高分子电解质材料形成共连续状的相分离结构可以通过高分子电解质材料满足下述(1)及(2)来确认。
(1)相分离结构的平均畴间距离为2nm以上
平均畴间距离可以由从实施了图像处理的透射式电子显微镜(TEM)的观察图像测量的各畴间距离的平均值来求得。通过相分离结构的平均畴间距离为2nm以上,首先能够确认形成了相分离结构。本发明中,平均畴间距离优选在2nm以上且小于1μm的范围,更优选为在5nm以上且500nm以下的范围,进一步优选为在10nm以上且100nm以下的范围。平均畴间距离小于2nm时,相分离结构变得不清楚,可能无法形成良好的质子传导通道。另一方面,平均畴间距离为1μm以上时,虽形成质子传导通道,但可能因溶胀而使机械强度、物理耐久性差。另外,如上文所述,优选平均畴间距离小于聚唑系无纺布的孔隙尺寸。需要说明的是,平均畴间距离的测定按照实施例第(4)项中记载的方法进行。
(2)利用透射式电子显微镜(TEM)观察来观察共连续状的相分离结构
呈现共连续状的相分离结构可以通过下述方法来确认,即,针对通过透射式电子显微镜(TEM)断层成像观察而得到的三维图,对从纵、横、高这三个方向切下的数字切片三视图分别显示的样态进行比较。具有共连续状的相分离结构或层状的相分离结构时,在全部三视图中,包含离子性嵌段且含有离子性基团的畴(以下称为“离子性畴”)和包含非离子性嵌段且不含离子性基团的畴(以下称为“非离子性畴”)均形成连续相,特别是共连续状的相分离结构的情况下,显示各个连续相互缠绕的样态。另一方面,层状的情况下,显示连续层连接成层状的样态。柱状结构、海岛结构的情况下,上述畴中的任一个在至少一面没有形成连续相。此处,连续相从宏观上来看,是指各个畴不独立而彼此连接的相。
需要说明的是,在TEM观察、TEM断层成像中,为了明确离子性畴与非离子性畴的凝聚状态、对比度,优选使用通过将电解质膜在2wt%乙酸铅水溶液中浸渍2日由此用铅对离子性基团进行了离子交换的样品。
组成含有离子性基团的高分子电解质(B)的芳香族烃系聚合物中的离子性畴与非离子性畴的体积比优选为80/20~20/80,更优选为60/40~40/60。上述范围外的情况下,有时质子传导性不足,尺寸稳定性、机械特性不足。
作为芳香族烃系聚合物,离子性嵌段与非离子性嵌段的摩尔组成比(A1/A2)优选为0.20以上,更优选为0.33以上,进一步优选为0.50以上。另外,摩尔组成比A1/A2优选为5.00以下,更优选为3.00以下,进一步优选为2.50以下。在摩尔组成比A1/A2小于0.20或者大于5.00的情况下,有时低加湿条件下的质子传导性不足,耐热水性、物理耐久性不足。此处,摩尔组成比A1/A2表示离子性嵌段中存在的重复单元的摩尔数与非离子性嵌段中存在的重复单元的摩尔数之比。“重复单元的摩尔数”是指用离子性嵌段、非离子性嵌段的数均分子量除以各自对应的结构单元的分子量而得的值。
另外,为了得到充分的尺寸稳定性、机械强度、物理耐久性、燃料阻挡性、耐溶剂性,芳香族烃系聚合物优选为具有结晶性的聚合物。此处,“具有结晶性”是指具有可结晶化的性质(升温时可结晶化)、或者已经结晶化。
结晶性的有无的确认可通过差示扫描量热分析法(DSC)或广角X射线衍射来实施。本发明中,在制膜后通过差示扫描量热分析法测定的结晶热优选为0.1J/g以上、或者通过广角X射线衍射测定的结晶度优选为0.5%以上。即,在差示扫描量热分析法中未确认到结晶峰的情况下,认为存在已经结晶化的情况和高分子电解质为非晶性的情况,已经结晶化的情况下,通过广角X射线衍射测定的结晶度为0.5%以上。
具有结晶性的芳香族烃系聚合物有时高分子电解质膜的加工性不良。此时,可以向芳香族烃系聚合物中导入保护基,暂时抑制结晶性。具体而言,在导入了保护基的状态下与聚唑系纳米纤维无纺布复合化,其后进行脱保护,由此可以将具有结晶性的芳香族烃系聚合物在本发明中用作高分子电解质材料。
芳香族烃系聚合物所具有的离子性基团只要为具有质子交换能力的离子性基团即可。作为这样的官能团,优选使用磺酸基、磺酰亚胺基、硫酸基、膦酸基、磷酸基、羧酸基。在聚合物中可以含有2种以上的离子性基团。其中,从质子传导率高的方面考虑,聚合物更优选具有选自磺酸基、磺酰亚胺基、硫酸基中的至少一种,从原料成本的方面考虑,最优选具有磺酸基。
从质子传导性与耐水性的平衡来考虑,作为芳香族烃系聚合物整体的离子交换容量(IEC)优选为0.1meq/g以上且5.0meq/g以下。IEC更优选为1.4meq/g以上,进一步优选为2.0meq/g以上。并且IEC更优选为3.5meq/g以下,进一步优选为3.0meq/g以下。IEC小于0.1meq/g的情况下,有时质子传导性不足,IEC大于5.0meq/g的情况下,有时耐水性不足。
另外,从低加湿条件下的质子传导性的方面考虑,优选离子性嵌段的IEC高,具体而言,离子性嵌段的IEC优选为2.5meq/g以上,更优选为3.0meq/g以上,进一步优选为3.5meq/g以上。另外,作为上限,优选为6.5meq/g以下,更优选为5.0meq/g以下,进一步优选为4.5meq/g以下。离子性嵌段的IEC小于2.5meq/g的情况下,有时低加湿条件下的质子传导性不足,离子性嵌段的IEC大于6.5meq/g的情况下,有时耐热水性、物理耐久性不足。
从耐热水性、机械强度、尺寸稳定性、物理耐久性的方面考虑,优选非离子性嵌段的IEC低,具体而言,非离子性嵌段的IEC优选为1.0meq/g以下,更优选为0.5meq/g以下,进一步优选为0.1meq/g以下。非离子性嵌段的IEC大于1.0meq/g的情况下,有时耐热水性、机械强度、尺寸稳定性、物理耐久性不足。
此处,IEC是指向芳香族烃系聚合物的每单位干燥重量中导入的离子性基团的摩尔量,该值越大,表示离子性基团的导入量越多。本发明中,将IEC定义为利用中和滴定法求得的值。利用中和滴定进行的IEC的计算按照实施例第(2)项中记载的方法进行。
本发明中,作为组成含有离子性基团的高分子电解质(B)的芳香族烃系聚合物,优选使用芳香族烃系嵌段共聚物,更优选为聚醚酮系嵌段共聚物。特别是,可最优选使用含有包含如下所述的结构单元(S1)(其含有离子性基团)的链段和包含结构单元(S2)(其不含离子性基团)的链段的聚醚酮系嵌段共聚物。
(通式(S1)中,Ar1~Ar4表示任意的2价亚芳基,Ar1及/或Ar2可以含有离子性基团,Ar3及Ar4可以含有离子性基团,也可以不含离子性基团。Ar1~Ar4可以被任意地取代,可以相互独立地使用2种以上的亚芳基。*表示与通式(S1)或其他结构单元的键合部位。)
(通式(S2)中,Ar5~Ar8表示任意的2价亚芳基,可以被任意地取代,但不含离子性基团。Ar5~Ar8可以相互独立地使用2种以上的亚芳基。*表示与通式(S2)或其他结构单元的键合部位。)
此处,作为Ar1~Ar8优选的2价亚芳基,可举出亚苯基、亚萘基、亚联苯基、芴二基等烃系亚芳基;吡啶二基、喹喔啉二基、噻吩二基等亚杂芳基等,但不限于这些例子。Ar1~Ar8优选为亚苯基和含有离子性基团的亚苯基,最优选为对亚苯基和含有离子性基团的对亚苯基。另外,Ar5~Ar8可以被除离子性基团以外的基团取代,但从质子传导性、化学稳定性、物理耐久性的方面考虑,更优选为无取代。
<复合高分子电解质膜>
本发明的复合高分子电解质膜具有复合层,所述复合层是将上述含有离子性基团的高分子电解质(B)(高分子电解质材料)与上述聚唑系纳米纤维无纺布(A)复合化而成的。通过复合化,在聚唑系纳米纤维无纺布的空隙中填充高分子电解质材料。复合层中的高分子电解质材料的填充率优选为50%以上,更优选为60%以上。如果复合层的填充率下降,则失去质子的传导通道,从而发电性能降低。需要说明的是,本发明中的复合层的填充率为由IEC计算的值,具体而言按照实施例第(3)项中记载的方法进行。
复合高分子电解质膜可以由1层这样的复合层形成,也可以由2层以上的复合层层合而成。层合时,可以层合具有不同的填充率的多个复合层。另外,可以与复合层的两侧或一侧相接地具有仅由高分子电解质材料形成的层。通过具有这样的层,能够提高复合高分子电解质膜与电极的粘合性,能够抑制界面剥离。与复合层的两侧或一侧相接地形成仅由高分子电解质材料形成的层时,构成该层的电解质材料优选为芳香族烃系聚合物,更优选为与填充到复合层内的芳香族烃系聚合物相同的聚合物。
本发明的复合高分子电解质膜通过具有复合层,能够降低面方向的尺寸变化率。通过降低面方向的尺寸变化率,从而在用作燃料电池的电解质膜时,能够减少干湿循环时在电解质膜的边缘部分等产生的由溶胀收缩引起的应力,能够提高耐久性。复合高分子电解质膜的面方向的尺寸变化率λxy优选为10%以下,更优选为8%以下,进一步优选为5%以下。
另外,对于复合高分子电解质膜的面方向的尺寸变化率而言,优选MD、TD方向的各向异性小。各向异性大的情况下,有时制约燃料电池的电池单元设计;由溶胀收缩引起的应力集中在与尺寸变化大的方向正交的边缘,电解质膜的断裂从该部分开始。具体而言,复合高分子电解质膜的面方向中的、MD方向的尺寸变化率λMD与TD方向的尺寸变化率λTD之比λMD/λTD优选满足0.5<λMD/λTD<2.0。
此处,尺寸变化率是表示干燥状态下的复合高分子电解质膜的尺寸与湿润状态下的复合化电解质膜的尺寸的变化的指标,具体的测定按照实施例第(5)项中记载的方法进行。
另外,基于相同的理由,对于复合化电解质膜的弹性模量、屈服应力而言,也优选MD、TD方向的各向异性小。
本发明的复合化电解质膜中的复合层的厚度没有特别限定,优选为0.5μm以上且50μm以下,更优选为2μm以上且40μm以下。复合层厚时,有电解质膜的物理耐久性提高,但另一方面,膜电阻增大的倾向。相反,复合层薄时,有发电性能提高,但另一方面,物理耐久性出现问题,容易产生电短路、燃料透过等问题的倾向。
<复合高分子电解质膜的制造方法>
关于本发明的复合高分子电解质膜,作为一例,可以通过下述复合高分子电解质膜的制造方法来制造,该制造方法的特征在于,依次包括下述工序:使用形成共连续状的相分离结构的芳香族烃系聚合物作为含有离子性基团的高分子电解质(B)(高分子电解质材料),在高分子电解质材料所含的离子性基团与碱金属或碱土金属的阳离子形成盐的状态下,将碱性的聚唑系纳米纤维无纺布(A)与高分子电解质材料进行复合化的工序;将与离子性基团形成盐的碱金属或碱土金属的阳离子同质子进行交换的工序。以下,对该制造方法进行说明。需要说明的是,在本说明书中,以下将处于离子性基团与碱金属或碱土金属的阳离子形成盐的状态的高分子电解质材料表述为“盐型的高分子电解质材料”。
作为将碱性的聚唑系纳米纤维无纺布与盐型的高分子电解质材料进行复合化的方法,优选使聚唑系纳米纤维无纺布中含浸盐型的高分子电解质材料的溶液(以下有时称为“高分子电解质溶液”),其后将溶剂干燥而制造复合高分子电解质膜的方法。作为使纳米纤维无纺布中含浸盐型的高分子电解质材料的溶液的方法,可举出:(1)一边将浸渍在盐型的高分子电解质溶液中的纳米纤维无纺布进行提拉一边除去多余的溶液而控制膜厚的方法;(2)将盐型的高分子电解质溶液流延涂布到纳米纤维无纺布上的方法;(3)在流延涂布有盐型的高分子电解质溶液的支承基材上贴合纳米纤维无纺布而含浸的方法。
关于溶剂的干燥,在用(3)的方法进行了含浸的情况下,可以在原有的状态下进行。另外,在用(1)或(2)的方法进行含浸的情况下,从能够减少复合高分子电解质膜的褶皱、厚度不均等而提高膜品质的方面考虑,优选以在另外准备的支承基材上贴附纳米纤维无纺布的状态将高分子电解质材料的溶剂进行干燥的方法。纳米纤维无纺布的干燥时间、干燥温度根据实验而适当地决定,优选至少干燥至从基材剥离后也可成为独立完好的膜(日语为“自立膜”)的程度。干燥的方法可选择基材的加热、热风、红外线加热器等已知的方法。考虑到高分子电解质的分解,干燥温度优选为200℃以下,更优选为130℃以下。
盐型的高分子电解质溶液中使用的溶剂可以根据聚合物种类而适当地选择。作为溶剂,例如,优选使用N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、N-甲基-2-吡咯烷酮、二甲基亚砜、环丁砜、1,3-二甲基-2-咪唑啉酮、六甲基磷酰三胺等非质子性极性溶剂;γ-丁内酯、乙酸乙酯、乙酸丁酯等酯系溶剂;碳酸亚乙酯、碳酸亚丙酯等碳酸酯系溶剂;乙二醇单甲醚、乙二醇单乙醚、丙二醇单甲醚、丙二醇单乙醚等亚烷基二醇单烷基醚。另外,可以使用将这些溶剂中的二种以上混合而成的混合溶剂。
另外,为了调整粘度,也可以在溶剂中混合下述各种低沸点溶剂,所述低沸点溶剂为:甲醇、乙醇、1‐丙醇、异丙醇等醇系溶剂;丙酮、甲基乙基酮、甲基异丁基酮等酮系溶剂;乙酸乙酯、乙酸丁酯、乳酸乙酯等酯系溶剂;己烷、环己烷等烃系溶剂;苯、甲苯、二甲苯等芳香族烃系溶剂;氯仿、二氯甲烷、1,2-二氯乙烷、全氯乙烯、氯苯、二氯苯、六氟异丙醇等卤代烃系溶剂;乙醚、四氢呋喃、1,4-二氧杂环己烷等醚系溶剂;乙腈等腈系溶剂;硝基甲烷、硝基乙烷等硝基化烃系溶剂;水;等等。
使用的盐型的高分子电解质溶液的浓度优选为5~40重量%,更优选为10~25重量%。如果为该范围的浓度,则能够在纳米纤维无纺布的空隙中充分地填充聚合物,并且能够获得表面平滑性优异的复合层。如果盐型的高分子电解质溶液的浓度过低,则高分子电解质材料向纳米纤维无纺布的空隙中的填充效率下降,有时需要多次的浸渍处理。另一方面,如果高分子电解质溶液的浓度过高,则有时溶液粘度过高,无法在纳米纤维无纺布的空隙中充分填充聚合物,复合层中的填充率下降,复合化电解质膜的表面平滑性变差。
需要说明的是,盐型的高分子电解质溶液的溶液粘度优选为100~50,000mPa·s,更优选为500~10,000mPa·s。如果溶液粘度过低,则溶液的滞留性差,有时从纳米纤维无纺布流出。另一方面,溶液粘度过高的情况下,有时产生如上所述的问题。
高分子电解质材料与纳米纤维无纺布的复合化优选在将纳米纤维无纺布固定于支承基材上的状态下进行。作为支承基材,没有特别限制,可使用已知的支承基材,例如,可举出由不锈钢等金属形成的环形带或圆形物(drum),由聚对苯二甲酸乙二醇酯、聚酰亚胺、聚苯硫醚、聚砜等聚合物形成的膜,玻璃,剥离纸等。优选对金属的表面实施镜面处理而使用,对聚合物膜的涂覆面实施电晕处理、实施易剥离处理而使用。此外,在连续涂覆成卷状的情况下,也可以在涂覆面的背面实施易剥离处理,防止在卷绕后电解质膜与涂覆基材的背面侧粘合。膜的情况下,厚度没有特别限定,从处理性的观点考虑,厚度优选为50μm~600μm。
另外,作为对盐型的高分子电解质溶液进行流延涂布的方法,可采用刮刀涂布、直接辊涂布、迈耶棒涂布、凹版涂布、逆向涂布、气刀涂布、喷雾涂布、刷涂、浸涂、模涂、真空模涂、帘式涂布、流涂、旋涂、丝网印刷、喷射涂布等手法。
另外,在盐型的高分子电解质溶液的含浸时,也优选实施减压或加压,实施高分子电解质溶液的加热、基材或含浸气氛的加热等,由此更容易含浸。
在本制造方法中,在盐型的高分子电解质材料与碱性的聚唑系纳米纤维无纺布进行复合化后,包括将与离子性基团形成盐的碱金属或碱土金属的阳离子同质子进行交换的工序。
该工序优选为使纳米纤维无纺布与盐型的高分子电解质材料的复合层与酸性水溶液接触的工序。另外,该接触更优选为将复合层浸渍在酸性水溶液中的工序。在该工序中,酸性水溶液中的质子被置换为与离子性基团进行离子键合的阳离子,并且同时除去残留的水溶性的杂质、残留单体、溶剂、残留盐等。
酸性水溶液没有特别限定,优选使用硫酸、盐酸、硝酸、乙酸、三氟甲磺酸、甲磺酸、磷酸、柠檬酸等。酸性水溶液的温度、浓度等也应适当地决定,但从生产率的观点考虑,优选于0℃以上且80℃以下的温度,使用3重量%以上且30重量%以下的硫酸水溶液。
需要说明的是,使用支承基材进行复合化的情况、制造厚度30μm以下的复合层的情况下,优选在不从支承基材剥离复合层的状态下进行与酸性水溶液的接触。在无支承基材的条件下进行时,由于电解质膜薄,所以有时与水及/或酸性水溶液的接触·溶胀时的机械强度下降,容易发生膜断裂。另外,有时在最终将复合层干燥时出现褶皱,产生表面缺陷。
出于提高机械强度及提高离子性基团的热稳定性、提高耐水性、提高耐溶剂性、提高耐自由基性、提高涂布液的涂覆性、提高保存稳定性等目的,可以在不违背本发明的目的的范围内向复合层中添加交联剂、常规的高分子化合物中使用的结晶成核剂、增塑剂、稳定剂、脱模剂、抗氧化剂、自由基清除剂、无机微粒等添加剂。
本发明的复合高分子电解质膜可用于各种用途。例如,可用于人工皮肤等医疗用途、过滤用途、耐氯性反渗透膜等离子交换树脂用途、各种结构材料用途、电化学用途、加湿膜、防雾膜、防静电膜、脱氧膜、太阳能电池用膜、气体阻隔膜。其中,可更优选用于各种电化学用途。作为电化学用途,例如,可举出固体高分子型燃料电池、氧化还原液流电池、水电解装置、氯碱电解装置、电化学氢泵、水电解式氢产生装置。
在固体高分子型燃料电池、电化学氢泵、和水电解式氢产生装置中,高分子电解质膜以在两面依次层合有催化剂层、电极基材及隔膜的结构状态使用。其中,将在电解质膜的两面层合有催化剂层的电解质膜(即催化剂层/电解质膜/催化剂层这样的层构成的电解质膜)称为带催化剂层的电解质膜(CCM),将在电解质膜的两面进一步依次层合有催化剂层及气体扩散基材的电解质膜(即,气体扩散基材/催化剂层/电解质膜/催化剂层/气体扩散基材这样的层构成的电解质膜)称为膜电极复合体(MEA)。本发明的复合高分子电解质膜可适当地用作构成这样的CCM及MEA的电解质膜。
实施例
以下,通过实施例对本发明进行更详细的说明,但本发明并不限于这些。需要说明的是,各种测定条件如下。
(1)聚合物的分子量
通过GPC测定聚合物溶液的数均分子量及重均分子量。使用TOSOH公司制HLC-8022GPC作为紫外检测器和差示折光仪的一体型装置,另外使用2根TOSOH公司制TSK gel SuperHM-H(内径6.0mm、长度15cm)作为GPC柱,利用N-甲基-2-吡咯烷酮溶剂(含有10mmol/L溴化锂的N-甲基-2-吡咯烷酮溶剂),以流量0.2mL/min进行测定,通过标准聚苯乙烯换算而求出数均分子量及重均分子量。
(2)离子交换容量(IEC)
利用中和滴定法进行测定。测定实施3次,取其平均值。
1.进行质子置换,将用纯水充分清洗过的复合高分子电解质膜的膜表面的水分拭去后,于100℃真空干燥12小时以上,求出干燥重量。
2.在复合高分子电解质膜中加入50mL的5wt%硫酸钠水溶液,静置12小时而进行离子交换。
3.使用0.01mol/L氢氧化钠水溶液对生成的硫酸进行滴定。加入市售的滴定用酚酞溶液0.1w/v%作为指示剂,以变为浅红紫色的点作为终点。
4.IEC通过下述式求出。
IEC(meq/g)=〔氢氧化钠水溶液的浓度(mmol/ml)×滴加量(ml)〕/试样的干燥重量(g)
(3)复合层中的芳香族烃系高分子电解质的填充率
用光学显微镜或扫描式电子显微镜(SEM)观察复合高分子电解质膜的截面,将包含芳香族烃系高分子电解质和聚唑系纳米纤维无纺布的复合层的厚度设为T1,在复合层的外侧具有其他层时,将它们的厚度设为T2、T3。将形成复合层的高分子电解质的比重设为D1,将形成复合层的外侧的其他层的高分子电解质的比重分别设为D2、D3,将复合高分子电解质膜的比重设为D。将形成各个层的聚合物的IEC设为I1、I2、I3,将复合高分子电解质膜的IEC设为I时,复合层中的芳香族烃系高分子电解质的含有率Y2(体积%)通过下式求出。
Y2=[(T1+T2+T3)×D×I-(T2×D2×I2+T3×D3×I3)]/(T1×D1×I1)×100
(4)通过透射式电子显微镜(TEM)断层成像观察相分离结构
将复合高分子电解质膜的试样片浸渍在作为染色剂的2wt%乙酸铅水溶液中,于25℃下静置48小时而进行染色处理。取出染色处理后的试样,用环氧树脂包埋,照射30秒的可见光进行固定。使用超微切片机.在室温下切削出100nm薄片,根据以下的条件实施观察。
装置:场发射式电子显微镜(HRTEM),JEOL制JEM2100F
图像获取:Digital Micrograph
系统:标记法(marker method)
加速电压:200kV
拍摄倍率:30,000倍
倾斜角度:+61°~-62°
重建分辨率:0.71nm/pixel
三维重建处理采用标记法。作为实施三维重建时的对位标志物,使用赋予到胶棉膜上的Au胶体粒子。以标志物为基准,在+61°~-62°的范围内,将试样以1°为单位进行倾斜,基于从拍摄TEM图像的连续倾斜图像系列取得的总计124枚的TEM图像实施CT重建处理,观察三维相分离结构。
另外,图像处理使用LUZEX(注册商标)AP(Nireco公司制),针对TEM原图像,在自动模式下执行浓度不均修正、浓度变换、空间滤波器的处理。并且,对于处理过的图像,在该装置的自动模式下,以从黑色到白色的256级灰度来表示,将0~128级定义为黑色,将129~256级定义为白色,由此用颜色区分含有离子性嵌段的畴和含有非离子性嵌段的畴,测量各畴间距离,并且将其平均值作为平均畴间距离。
(5)通过热水试验测定尺寸变化率(λxy)
将复合高分子电解质膜切成约5cm×约5cm的正方形,在温度23℃±5℃、湿度50%±5%的调温调湿气氛下静置24小时后,用游标卡尺测定MD方向的长度和TD方向的长度(MD1和TD1)。将该电解质膜在80℃的热水中浸渍8小时后,再次用游标卡尺测定MD方向的长度和TD方向的长度(MD2和TD2),通过下式计算面方向中的MD方向和TD方向的尺寸变化率(λMD和λTD)及面方向的尺寸变化率(λxy)(%)。
λMD=(MD2-MD1)/MD1×100
λTD=(TD2-TD1)/TD1×100
λxy=(λMD+λTD)/2
(6)使用复合高分子电解质膜制作膜电极复合体(MEA)
准备1对将市售的电极、BASF公司制燃料电池用气体扩散电极“ELAT(注册商标)LT120ENSI”5g/m2Pt切成5cm见方而得的电极,将它们以夹持复合高分子电解质膜的方式对置重叠而成为燃料极、空气极,于150℃、5MPa进行3分钟加热加压,得到评价用MEA。
(7)质子电导率
将膜状的试样在25℃的纯水中浸渍24小时后,于80℃、相对湿度25~95%的恒温恒湿槽中在各步骤中保持30分钟,以恒电位交流阻抗法测定质子传导率。作为测定装置,使用Solartron公司制电化学测定系统(Solartron 1287Electrochemical Interface及Solartron 1255B Frequency Response Analyzer),通过双端子法进行恒电位阻抗测定,求出质子传导率。交流振幅为50mV。样品使用宽度10mm、长度50mm的膜。测定夹具由酚醛树脂制成,将测定部分放开。作为电极,使用铂板(厚度100μm,2片)。将电极以电极间距离10mm、在样品膜的表面侧和背面侧相互平行、且与样品膜的长度方向垂直的方式配置。
(8)干湿循环耐久性
将上述(6)中制成的MEA设置于英和株式会社制JARI标准电池单元“Ex-1”(电极面积25cm2),在电池单元温度80℃的状态下,重复向两电极供给2分钟的160%RH的氮、然后向两电极供给2分钟的0%RH的氮(露点为-20℃以下)的循环。每隔1000次循环实施氢渗透量的测定,将氢渗透电流超过初始电流的10倍的时间点作为干湿循环耐久性。
氢渗透量的测定如下进行:向一方的电极供给作为燃料气体的氢,向另一方的电极供给氮,在加湿条件:氢气90%RH、氮气:90%RH下进行试验。保持到开路电压变为0.2V以下为止,以1mV/sec扫描电压直至变为0.2~0.7V为止,将0.7V时的电流值作为氢渗透电流。
(9)利用扫描式电子显微镜(FE-SEM)观察纳米纤维无纺布
对纳米纤维无纺布溅射涂布金属微粒之后,按照以下的条件实施观察。
装置:Hitachi High-Technologies Corporation.制,场发射式扫描电子显微镜(FE-SEM)SU8020
加速电压:2.0kV
图像处理使用LUZEX(注册商标)AP(Nireco公司制),针对SEM原图像,在自动模式下,执行浓度不均修正、浓度变换、空间滤波器的处理。并且,对于处理过的图像,在该装置的自动模式下,以从黑色到白色的256级灰度来表示,将0~128级定义为黑色,将129~256级定义为白色,由此用颜色区分纳米纤维无纺布的纤维存在的位置和不存在的位置,求出纳米纤维无纺布的纤维直径,并且测量构成纳米纤维无纺布的纤维间的距离,将其平均值作为纤维间距离的平均值。将得到的纤维间距离的平均值作为孔隙尺寸。
(10)通过X射线衍射(XRD)分析纳米纤维无纺布晶体结构
以总厚度成为1mm的方式重叠无纺布试样并设置于Si无反射板,通过广角X射线衍射法(2θ-θ扫描法)测定。计算得到的光谱所含的峰的半峰宽并进行比较。以下示出测定条件。
广角X射线衍射法(2θ-θ扫描法)
(i)X射线产生装置
理学电机公司制:RU-200R(旋转对阴极型)
X射线源:CuKα射线(弯晶单色器)
输出:50kV,200mA
(ii)测角器
理学电机公司制2155S2型
狭缝体系:1°-1°-0.3mm-0.45mm
检测器:闪烁计数管
(iii)计数记录装置
理学电机公司制:RINT-1400型
(iv)扫描方式
2θ-θ连续扫描
(v)测定范围(2θ)
3~60°
(vi)测定步长(2θ)
0.02°
(vii)扫描速度
1°/min
(11)吸收光谱测定
将在下述测定条件下得到的纳米纤维无纺布的漫反射率光谱转换成Kubelka-Munk函数。此处,Kubelka-Munk函数在将试样的相对漫反射率设为R时,以(1-R)2/2R表示。通过用Kubelka-Munk来表示,与透过法中的吸收光谱同样地,能够实施与吸收系数、试样浓度成比例的定量处理。
测定装置:SolidSpec-3700DUV紫外可见近红外分光光度计(岛津公司制)
狭缝宽度:8mm(870nm以下),20nm(870nm以上)
狭缝程序:正常
测定速度:低速
光源:氘灯(310nm以下)
卤素灯:(310nm以上)
检测器:PMT(870nm以下)
InGaAs(870~1650nm)
附属装置:大型试样室积分球(60mmφ)Spectralon
入射角反射:8°
参照物:标准白色板(Labsphere公司制)
(12)发射光谱测定
在下述测定条件下,测定纳米纤维无纺布的发射光谱,对300nm激发时的峰强度(I300)和450nm激发时的峰强度(I450)进行比较。需要说明的是,用各波长的激发光强度将发光强度标准化,还进行了暗计数(dark count)的修正。没有特别使用滤波器。相对于激发光,观测22.5°方向的发光。
测定装置:Fluorolog3-22(HORIBA Jobin Yvon公司制)
光源:氙气灯
检测器:PMT
激发波长:300nm及450nm
观测波长:~750nm(每隔1nm)
狭缝宽度:激发侧2nm,观测侧2nm
时间常数:0.2s
测定模式:SC/RC
(13)Tg测定
在下述测定条件下,使用差示扫描量热仪(DSC)测定纳米纤维无纺布材料的玻璃化转变温度(Tg)。
测定装置:SII公司制DSC6220
测定范围:30℃~550℃
扫描速度:2℃/分钟
合成例1
(由下述通式(G1)表示的2,2-双(4-羟基苯基)-1,3-二氧杂环戊烷(K-DHBP)的合成)
在具备搅拌器、温度计及馏出管的500ml烧瓶中,投入4,4’-二羟基二苯甲酮49.5g、乙二醇134g、原甲酸三甲酯96.9g及对甲苯磺酸一水合物0.50g进行溶解。其后于78~82℃保温搅拌2小时。进一步将内温缓慢地升温至120℃,进行加热直至甲酸甲酯、甲醇、原甲酸三甲酯的馏出完全停止。将该反应液冷却至室温后,用乙酸乙酯稀释反应液,用5%碳酸钾水溶液100ml清洗有机层并分液后,馏去溶剂。在残留物中加入二氯甲烷80ml使晶体析出,过滤并干燥而得到2,2-双(4-羟基苯基)-1,3-二氧杂环戊烷52.0g。对该晶体进行GC分析,结果为99.9%的2,2-双(4-羟基苯基)-1,3-二氧杂环戊烷、和0.1%的4,4’-二羟基二苯甲酮。
合成例2
(由下述通式(G2)表示的3,3’-二磺酸二钠-4,4’-二氟二苯甲酮的合成)
使4,4’-二氟二苯甲酮109.1g(Aldrich试剂)在发烟硫酸(50%SO3)150mL(和光纯药试药)中于100℃反应10小时。其后,少量逐次地投入到大量的水中,用NaOH中和后,加入食盐(NaCl)200g使合成物沉淀。将得到的沉淀进行过滤分离,用乙醇水溶液进行重结晶,得到由上述通式(G2)表示的3,3’-二磺酸二钠-4,4’-二氟二苯甲酮。纯度为99.3%。
合成例3
(由下述通式(G3)表示的不含离子性基团的低聚物a1的合成)
在具备搅拌机、氮气导入管、迪安-斯达克榻分水器(Dean-Stark trap)的1000mL三颈瓶中,加入碳酸钾16.59g(Aldrich试剂,120mmol)、上述合成例1中得到的K-DHBP25.8g(100mmol)及4,4’-二氟二苯甲酮20.3g(Aldrich试剂,93mmol),进行氮气置换后,在N-甲基吡咯烷酮(NMP)300mL、甲苯100mL中于160℃进行脱水后,升温而除去甲苯,于180℃进行1小时聚合。在大量的甲醇中进行再沉淀纯化,得到不含离子性基团的低聚物(末端:羟基)。数均分子量为10000。
在具备搅拌机、氮气导入管、迪安-斯达克榻分水器的500mL三颈瓶中,加入碳酸钾1.1g(Aldrich试剂,8mmol)、不含离子性基团的上述低聚物a1(末端:羟基)20.0g(2mmol),进行氮气置换后,在N-甲基吡咯烷酮(NMP)100mL、甲苯30mL中于100℃进行脱水后,升温而除去甲苯,加入十氟联苯4.0g(Aldrich试剂,12mmol),于105℃进行1小时反应。用大量的异丙醇进行再沉淀而进行纯化,得到由下述式(G3)表示的不含离子性基团的低聚物a1(末端:氟基)。数均分子量为11000。
合成例4
(由下述通式(G4)表示的含有离子性基团的低聚物a2的合成)
在具备搅拌机、氮气导入管、迪安-斯达克榻分水器的1000mL三颈瓶中,加入碳酸钾27.6g(Aldrich试剂,200mmol)、上述合成例1中得到的K-DHBP12.9g(50mmol)及4,4’-联苯二酚(4,4’-biphenol)9.3g(Aldrich试剂,50mmol)、上述合成例2中得到的3,3’-二磺酸二钠-4,4’-二氟二苯甲酮39.3g(93mmol)、以及17.9g的18-冠-6(和光纯药82mmol),进行氮气置换后,在N-甲基吡咯烷酮(NMP)300mL、甲苯100mL中于170℃进行脱水后,升温而除去甲苯,于180℃进行1小时聚合。用大量的异丙醇进行再沉淀而进行纯化,得到由下述式(G4)表示的含有离子性基团的低聚物a2(末端:羟基)。数均分子量为16000。
(式(G4)中,M表示H、Na或K。)
合成例5
(由下述式(G5)表示的3-(2,5-二氯苯甲酰基)苯磺酸新戊酯的合成)
在具备搅拌机、冷却管的3L的三颈瓶中,加入氯磺酸245g(2.1mol),接下来加入2,5-二氯二苯甲酮105g(420mmol),在100℃的油浴器中反应8小时。在规定时间后,将反应液缓慢地注入到碎冰1000g中,用乙酸乙酯进行提取。用食盐水清洗有机层,并用硫酸镁干燥后,馏去乙酸乙酯,得到淡黄色的粗结晶3-(2,5-二氯苯甲酰基)苯磺酰氯。粗结晶未被纯化,直接用于下一工序。
将2,2-二甲基-1-丙醇(新戊醇)41.1g(462mmol)加入到吡啶300mL中,冷却至约10℃。经约30分钟向其中缓慢地加入上述中得到的粗结晶。添加全量后,进一步搅拌30分钟进行反应。反应后,将反应液注入到盐酸水溶液1000mL中,将析出的固体回收。使得到的固体溶解于乙酸乙酯,用碳酸氢钠水溶液、食盐水清洗后,用硫酸镁干燥后,馏去乙酸乙酯,得到粗结晶。将其用甲醇进行重结晶,得到由上述结构式表示的3-(2,5-二氯苯甲酰基)苯磺酸新戊酯的白色晶体。
合成例6
(由下述通式(G6)表示的不含离子性基团的低聚物的合成)
在安装有搅拌机、温度计、冷却管、迪安-斯达克管、导入氮气的三通阀的1L的三颈瓶中,称取2,6-二氯苄腈49.4g(0.29mol)、2,2-双(4-羟基苯基)-1,1,1,3,3,3-六氟丙烷88.4g(0.26mol)、碳酸钾47.3g(0.34mol)。进行氮气置换后,加入环丁砜346ml、甲苯173ml进行搅拌。将烧瓶置于油浴器,使其回流并加热至150℃。使由反应生成的水与甲苯共沸,一边用迪安-斯达克管将其排出到体系外一边进行反应时,约3小时几乎观察不到水的生成。边缓慢地提高反应温度边除去大部分的甲苯后,于200℃继续反应3小时。接下来,加入2,6-二氯苄腈12.3g(0.072mol),进一步反应5小时。
将得到的反应液静置冷却后,加入甲苯100ml进行稀释。将副反应生成的无机化合物的沉淀物过滤除去,将滤液投入到2L的甲醇中。将已沉淀的生成物过滤分离,回收并干燥后,溶于四氢呋喃250ml中。将其在甲醇2L中进行再沉淀,得到由下述通式(G6)表示的目标化合物107g。数均分子量为11000。
合成例7
(包含由下述式(G8)表示的链段和由下述式(G9)表示的链段的聚醚砜(PES)系嵌段共聚物前体b2’的合成)
将无水氯化镍1.62g和二甲基亚砜15mL混合,调整至70℃。向其中加入2,2’-联吡啶2.15g,于该温度搅拌10分钟,制备含有镍的溶液。
此时,在使2,5-二氯苯磺酸(2,2-二甲基丙基)酯1.49g和由下述式(G7)表示的SUMIKAEXCEL PES5200P(住友化学公司制,Mn=40,000,Mw=94,000)0.50g溶于二甲基亚砜5mL而得到的溶液中加入锌粉末1.23g,并调整至70℃。向其中注入上述含有镍的溶液,于70℃进行4小时聚合反应。在甲醇60mL中加入反应混合物,接下来,加入6mol/L盐酸60mL搅拌1小时。通过过滤而分离析出的固体,进行干燥,以99%的收率得到1.62g灰白色的包含由下述式(G8)和下述式(G9)表示的链段的嵌段共聚物前体b2’。重均分子量为23万。
[高分子电解质溶液A]包含嵌段共聚物的高分子电解质溶液,所述嵌段共聚物含有作为含有离子性基团的链段的上述(G4)表示的低聚物和作为不含离子性基团的链段的上述(G3)表示的低聚物
在具备搅拌机、氮气导入管、迪安-斯达克榻分水器的500mL三颈瓶中,加入碳酸钾0.56g(Aldrich试剂,4mmol)、合成例4中得到的含有离子性基团的低聚物a2(末端:羟基)16g(1mmol),进行氮气置换后,在N-甲基吡咯烷酮(NMP)100mL、环己烷30mL中于100℃进行脱水后,升温而除去环己烷,加入合成例3中得到的不含离子性基团的低聚物a1(末端:氟基)11g(1mmol),于105℃进行24小时反应。通过在大量的异丙醇中的再沉淀纯化,得到嵌段共聚物b1。重均分子量为34万。
使溶解有得到的嵌段共聚物的5重量%N-甲基吡咯烷酮(NMP)溶液在久保田制作所制变频·小型高速冷冻离心机(在型号6930上设置角转子RA-800,25℃,30分钟,离心力20000G)中进行聚合原液的直接离心分离。由于沉降固体物质(滤饼)和上清液(涂布液)完全地分离开,所以回收上清液。接下来,边搅拌边于80℃进行减压蒸馏,使用1μm的聚丙烯制过滤器进行加压过滤,得到高分子电解质溶液A。高分子电解质溶液A的粘度为1300mPa·s。
[高分子电解质溶液B]包含下述通式(G10)表示的聚芳撑系嵌段共聚物的高分子电解质溶液
将干燥的N,N-二甲基乙酰胺(DMAc)540ml在氮气下加入到3-(2,5-二氯苯甲酰基)苯磺酸新戊酯135.0g(0.336mol)与合成例6中合成的由式(G6)表示的不含离子性基团的低聚物40.7g(5.6mmol)、2,5-二氯-4’-(1-咪唑基)二苯甲酮6.71g(16.8mmol)、双(三苯基膦)二氯化镍6.71g(10.3mmol)、三苯基膦35.9g(0.137mol)、碘化钠1.54g(10.3mmol)、锌53.7g(0.821mol)的混合物中。
将反应体系在搅拌下加热(最终加热至79℃),反应3小时。在反应中途观察体系中的粘度上升。用DMAc730ml稀释聚合反应溶液,搅拌30分钟,过滤助剂使用硅藻土,进行过滤。
将上述滤液用蒸发器浓缩,在滤液中加入溴化锂43.8g(0.505mol),于内温110℃在氮气氛下反应7小时。反应后,冷却至室温,注入到丙酮4L中,使其凝固。过滤收集凝固物,并风干后,用搅拌机粉碎,用1N盐酸1500ml边搅拌边进行清洗。过滤后,用离子交换水清洗生成物直至清洗液的pH达到5以上,然后于80℃干燥一晚,得到目标聚芳撑系嵌段共聚物23.0g。该脱保护后的聚芳撑系嵌段共聚物的重均分子量为19万。将得到的聚芳撑系嵌段共聚物溶于N-甲基-2-吡咯烷酮/甲醇=30/70(质量%)有机溶剂中使其成为0.1g/g,得到高分子电解质溶液B。高分子电解质溶液B的粘度为1200mPa·s。
[高分子电解质溶液C]包含无规共聚物的高分子电解质溶液C
在具备搅拌机、氮气导入管、迪安-斯达克榻分水器的5L的反应容器中,加入合成例1中合成的2,2-双(4-羟基苯基)-1,3-二氧杂环戊烷129g、4,4’-联苯二酚93g(Aldrich试剂)、以及合成例2中合成的3,3’-二磺酸二钠-4,4’-二氟二苯甲酮422g(1.0mol),进行氮气置换后,加入N-甲基-2-吡咯烷酮(NMP)3000g、甲苯450g、232g的18-冠-6(和光纯药试药),在确认单体全部溶解后,加入碳酸钾304g(Aldrich试剂),边回流边于160℃进行脱水后,升温而除去甲苯,于200℃进行1小时脱盐缩聚。重均分子量为32万。
接下来,以聚合原液的粘度成为500mPa·s的方式添加NMP进行稀释,在久保田制作所制变频·小型高速冷冻离心机(在型号6930上设置角转子RA-800,25℃,30分钟,离心力20000G)中进行聚合原液的直接离心分离。由于沉降固体物质(滤饼)与上清液(涂布液)完全地分离,所以回收上清液。接下来,边搅拌边于80℃进行减压蒸馏,除去NMP直至聚合物浓度变成20重量%,进一步用5μm的聚乙烯制过滤器进行加压过滤而得到高分子电解质溶液C。高分子电解质溶液C的粘度为1000mPa·s。
[高分子电解质溶液D]包含聚醚砜系嵌段共聚物的高分子电解质溶液D
将0.23g的合成例7中得到的嵌段共聚物前体b2’加入到溴化锂一水合物0.16g与NMP8mL的混合溶液中,于120℃反应24小时。将反应混合物注入到6mol/L盐酸80mL中,搅拌1小时。通过过滤分离析出的固体。将已分离的固体干燥,得到灰白色的包含由上述式(G8)表示的链段和由下述式(G11)表示的链段的嵌段共聚物b2。得到的聚醚砜系嵌段共聚物的重均分子量为19万。将得到的聚醚砜系嵌段共聚物溶于N-甲基-2-吡咯烷酮/甲醇=30/70(质量%)有机溶剂中使其成为0.1g/g,得到高分子电解质溶液D。高分子电解质溶液D的粘度为1300mPa·s。
合成例8
(聚苯并咪唑(PBI)的合成)
在氮气氛下,聚合溶剂使用多聚磷酸(PAA),称取3,3’-二氨基联苯胺(DAB)22.7g(106mmol)、4,4’-氧代双苯甲酸(OBBA)27.3g(106mmol),将多聚磷酸(PPA)以成为3质量%溶液的方式加入,边搅拌边缓慢地提高温度,于140℃搅拌12小时,进行缩聚。反应后,冷却至室温,注入到离子交换水中使其凝固后,用氢氧化钠水溶液中和。过滤,并用离子交换水清洗后,于80℃减压干燥一夜,得到目标聚苯并咪唑。重均分子量为42万,Tg为427℃。
合成例9
(聚苯并噁唑(PBO)的合成)
在氮气氛下,聚合溶剂使用多聚磷酸(PAA),称取3,3’-二氨基-4,4’-二羟基联苯二盐酸盐31.7g(106mmol)、4,4’-氧代双苯甲酸(OBBA)27.3g(106mmol),将多聚磷酸(PPA)以成为10质量%溶液的方式加入,边搅拌边缓慢地提高温度,于140℃搅拌12小时,进行缩聚。反应后,冷却至室温,注入到离子交换水中使其凝固后,用氢氧化钠水溶液中和。过滤,并用离子交换水清洗后,于80℃减压干燥一夜,得到目标聚苯并噁唑。重均分子量为38万,Tg为416℃。
[由聚苯并咪唑纤维形成的纳米纤维无纺布A]
将合成例8中得到的聚苯并咪唑溶于二甲基亚砜(DMSO)使其成为8重量%,使用Kato Tech公司制静电纺丝设备,在电压为20kV、注射泵喷出速度为0.12mL/时、注射器与靶间的距离为100mm的条件下进行纺纱,同时制作纳米纤维无纺布。将得到的纳米纤维无纺布于80℃减压干燥1小时后,层合在厚度125μm的Kapton(注册商标)基材上,在氮气氛中于400度加热10分钟,得到纤维直径为150nm、厚度为7μm的由聚苯并咪唑纤维形成的纳米纤维无纺布A。空隙率为87%。
[由聚苯并噁唑纤维形成的纳米纤维无纺布B]
将聚苯并咪唑变更为合成例9中得到的聚苯并噁唑,除此之外,与上述由聚苯并咪唑纤维形成的纳米纤维无纺布A的制造同样地进行,得到纤维直径为160nm、厚度为8μm的由聚苯并噁唑纤维形成的纳米纤维无纺布B。空隙率为86%。
[掺杂有磷酸的由聚苯并咪唑纤维形成的纳米纤维无纺布C]
基于日本特开2015-28850号公报,将由聚苯并咪唑纤维形成的纳米纤维无纺布A(0.10g)浸渍在60重量%的磷酸水溶液中。于室温浸渍1小时后,于110℃真空干燥12小时,得到掺杂有磷酸的由聚苯并咪唑纤维形成的纳米纤维无纺布C。刚刚干燥后的纳米纤维无纺布的重量为0.13g,纤维的直径为170nm,厚度为8μm,空隙率为83%。
[由聚酰亚胺纤维形成的纳米纤维无纺布D]
将8重量%聚苯并咪唑溶液变更为市售的聚(均苯四甲酸二酐-CO-二氨基二苯醚)酰胺酸12重量%溶液(溶剂80重量%NMP/20重量%甲苯,Aldrich公司制),除此之外,与上述由聚苯并咪唑纤维形成的纳米纤维无纺布A的制造同样地进行,得到纤维直径为160nm、厚度为8μm的由聚酰亚胺纤维形成的纳米纤维无纺布D。空隙率为89%。另外,通过热处理得到的聚酰亚胺无纺布的Tg为410℃。
[热处理温度为450℃的由聚苯并咪唑纤维形成的纳米纤维无纺布E]
将制作无纺布后的氮气氛下的加热温度变更为450℃,除此之外,与上述由聚苯并咪唑纤维形成的纳米纤维无纺布A的制造同样地进行,得到纤维直径为150nm、厚度为8μm的由聚苯并咪唑纤维形成的纳米纤维无纺布E。空隙率为88%。
[热处理温度为350℃的由聚苯并咪唑纤维形成的纳米纤维无纺布F]
将制作无纺布后的氮气氛下的加热温度变更为350℃,除此之外,与上述由聚苯并咪唑纤维形成的纳米纤维无纺布A的制造同样地进行,得到纤维直径为150nm、厚度为8μm的由聚苯并咪唑纤维形成的纳米纤维无纺布F。空隙率为86%。
[热处理温度为500℃的由聚苯并咪唑纤维形成的纳米纤维无纺布G]
将制作无纺布后的氮气氛下的加热温度变更为500℃,除此之外,与上述由聚苯并咪唑纤维形成的纳米纤维无纺布A的制造同样地进行,得到纤维直径为150nm、厚度而8μm的由聚苯并咪唑纤维形成的纳米纤维无纺布G。空隙率为88%。
[实施例1]
使用刮刀涂布机,将高分子电解质溶液A在玻璃基板上流延涂布,并贴合由聚苯并咪唑纤维形成的纳米纤维无纺布A。于室温保持1小时,使纳米纤维无纺布A充分含浸高分子电解质溶液A后,于100℃干燥4小时。再次将高分子电解质溶液A流延涂布到干燥后的膜的上表面,于室温保持1小时后,于100℃干燥4小时,得到膜状的聚合物。于80℃在10重量%硫酸水溶液中浸渍24小时而进行质子置换、脱保护反应后,在大量过剩的纯水中浸渍24小时而充分清洗,得到复合高分子电解质膜(膜厚11μm)。
[实施例2]
使用由聚噁唑纤维形成的纳米纤维无纺布B代替由聚苯并咪唑纤维形成的纳米纤维无纺布A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[实施例3]
使用高分子电解质溶液B代替高分子电解质溶液A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[实施例4]
使用高分子电解质溶液C代替高分子电解质溶液A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[实施例5]
使用高分子电解质溶液D代替高分子电解质溶液A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[实施例6]
使用市售的Nafion 10重量%分散液(Aidrich公司制,Available Acid Capacity0.92meq/g品)(以下有时记载为“高分子电解质溶液E”)代替高分子电解质溶液A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[比较例1]
使用掺杂有磷酸的由聚苯并咪唑纤维形成的纳米纤维无纺布C代替由聚苯并咪唑纤维形成的纳米纤维无纺布A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[比较例2]
使用由聚酰亚胺纤维形成的纳米纤维无纺布D代替由聚苯并咪唑纤维形成的纳米纤维无纺布A,除此之外,与实施例1同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[比较例3]
使用刮刀涂布机,将高分子电解质溶液A流延涂布到玻璃基板上,在不贴合含有聚唑的纳米纤维无纺布(A)的状态下,于100℃干燥4小时,得到膜状的聚合物。于80℃在10重量%硫酸水溶液中浸渍24小时而进行质子置换、脱保护反应后,在大量过剩的纯水中浸渍24小时而充分清洗,得到复合高分子电解质膜(膜厚10μm)。
[比较例4]
使用高分子电解质溶液B代替高分子电解质溶液A,除此之外,与比较例3同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[比较例5]
使用高分子电解质溶液C代替高分子电解质溶液A,除此之外,与比较例3同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[比较例6]
使用高分子电解质溶液D代替高分子电解质溶液A,除此之外,与比较例3同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[比较例7]
使用高分子电解质溶液E代替高分子电解质溶液A,除此之外,与比较例3同样地进行,得到复合高分子电解质膜(膜厚12μm)。
[参考例1]
使用热处理温度为450℃的由聚苯并咪唑纤维形成的纳米纤维无纺布E代替由聚苯并咪唑纤维形成的纳米纤维无纺布A,除此之外,与实施例1同样地进行,尝试制作复合高分子电解质膜。但是由于无纺布过度固化而变脆,所以在与高分子电解质溶液A进行贴合时,无纺布破损,无法制作复合高分子电解质膜。
[参考例2]
使用热处理温度为350℃的由聚苯并咪唑纤维形成的纳米纤维无纺布F代替由聚苯并咪唑纤维形成的纳米纤维无纺布A,除此之外,与实施例1同样地进行,尝试制作复合高分子电解质膜。但是由于在加热干燥工序中无纺布收缩,所以无法制作复合高分子电解质膜。
[参考例3]
使用热处理温度为500℃的由聚苯并咪唑纤维形成的纳米纤维无纺布G代替由聚苯并咪唑纤维形成的纳米纤维无纺布A,除此之外,与实施例1同样地进行,尝试制作复合高分子电解质膜。但是由于无纺布过度固化而变脆,所以在与高分子电解质溶液A进行贴合时,无纺布破损,无法制作复合高分子电解质膜。
对于各实施例、比较例中制造的复合高分子电解质膜,评价离子交换容量(IEC)、复合层中的高分子电解质的填充率、尺寸变化率λxy、质子电导率、以及干湿循环耐久性。另外对于构成复合高分子电解质膜的含有离子性基团的高分子膜,评价有无相分离结构及其形态和相分离结构的平均畴间距离,对于纳米纤维无纺布,评价纤维直径、空隙率、通过X射线衍射(XRD)测得的晶体结构(半峰宽2θ)、吸收光谱(Kubelka-Munk函数)、以及发射光谱(I450/I300)。将这些评价结果示于表1。(其中,表1中“相分离结构”×是指没有显示出明确的相分离结构。另外,关于干湿循环耐久性,在即使超过30000次循环氢渗透电流也没有超过初始电流的10倍的情况下,在30000次循环时结束评价。)
- 还没有人留言评论。精彩留言会获得点赞!