一种有机高分子交联的有机膦酸高温质子交换膜及其制备方法与流程
本发明属于燃料电池技术领域,具体涉及一种有机高分子交联的有机膦酸高温质子交换膜及其制备方法。
背景技术
质子交换膜燃料电池发电作为新一代高效、环保型发电技术,其广阔的应用前景可与计算机技术相媲美。其以固体的质子交换膜为电解质,不存在电解质腐蚀问题,同时具有能量转换效率高、污染小、启动快、寿命长和功率密度高等特点,在固定电站、分散式电站、军用特种电源、可移动电源、备用电源等方面都有广阔的应用前景,尤其是电动车的最佳驱动电源。经过多年的基础研究与应用开发,质子交换膜燃料电池用作汽车动力的研究已取得实质性进展,微型质子交换膜燃料电池便携电源和小型质子交换膜燃料电池移动电源已达到产品化程度,中、大功率质子交换膜燃料电池发电系统的研究也取得了一定成果。
传统的pemfc(质子交换膜燃料电池)一般工作在60~90℃,较低的工作温度为pemfc带来种种弊端,例如催化剂中毒以及水热管理复杂等,这些都严重限制了传统质子交换膜燃料电池的发展。因此,为了解决传统的pemfc的质子交换膜燃料电池的上述问题,高温质子交换膜燃料电池(hightemperatureprotonexchangemembranefuelcell,ht-pemfc)的发展成为必然。高温质子交换膜燃料电池具有明显的优势:(1)提高了催化剂的co耐受性;(2)提高了反应和扩散速率;(3)简化了电池系统的水管理;(4)简化了电池系统的热管理;(5)可以降低催化剂的pt载量以及开发非铂系催化剂,以降低整个pemfc的成本。而高温质子交换膜(ht-pem)是高温质子交换膜燃料电池(ht-pemfc)实现高温工作的核心所在。
目前,广泛使用的高温质子交换膜为pbi(聚苯并咪唑)类型的质子交换膜,该体系最大的缺点就是无机磷酸的流失问题。因此,为了解决无机磷酸流失的问题,有机膦酸高温质子交换膜已经成为一种必然的发展方向。
目前制备有机膦酸高温质子交换膜的方法比较多,但是还是存在一些问题。例如,有机膦酸聚合物制备困难,例如将乙烯基膦酸连接到ppo以及psu上的方法需要低温无水无氧操作,并且ppo以及psu的溶解性差,不适合大规生产;此外,现有的有机膦酸高温质子交换膜的制膜工艺复杂,如basf公司推出的产品,工艺就非常的复杂,并且产品的批量稳定性差;最后,膦酸含量无法准确控制,电导率调控困难,例如pbi膜浸泡在含有乙烯基磷酸的溶液中然后聚合,该体系中膦酸含量无法准确控制。
综上所述,本领域尚缺乏一种有机膦酸聚合物制备简单、制膜工艺简单、有机膦酸含量和质子电导率准确可控的高温质子交换膜及其制备方法。
技术实现要素:
本发明的目的是提供一种有机膦酸聚合物制备简单、制膜工艺简单、有机膦酸含量和质子电导率准确可控的高温质子交换膜及其制备方法。
本发明的第一方面,提供了一种有机高分子交联的有机膦酸高温质子交换膜,所述的有机高分子交联的有机膦酸高温质子交换膜是由(a)聚苯并咪唑类型化合物a、(b)“酸-碱”型有机膦酸聚合物b以及(c)有机高分子交联剂c作为原料复合而成的,其中,所述的a与b的摩尔比na:nb=1:0.01-99.99;
所述的“酸-碱”型有机膦酸聚合物b为碱性烯烃单体d与有机膦酸单体e发生共聚形成的共聚物,其中碱性烯烃单体d选自下组:
其中有机膦酸单体e选择下组:
r2选自下组:h、取代或未取代的c1-c9的烷基、苯基(ph)、tms(me3si,三甲基硅基)、tes(et3si,三乙基硅基)、tbs(叔丁基二甲基硅基)、tips(三异丙基硅基)、tbdps(叔丁基二苯基硅基)。
在另一优选例中,所述的聚苯并咪唑类聚合物a选自下组:
其中,n=2-10000;
r选自下组:无、o、s、nh、c(o)、s(o)2、未取代或卤代的c1-c6亚烷基、未取代或卤代的c2-c6亚烯基;
r1选自下组:
在另一优选例中,所述的聚苯并咪唑类型化合物a选自下组:
其中r1的定义如上文中所述。
在另一优选例中,单体d与单体e共聚的摩尔比为nd:ne=1:0.01-99.99;和/或
所述的a与c的摩尔比na:nc=1:0.01-10.00。
在另一优选例中,所述的“酸-碱”型有机膦酸聚合物b是通过以下方法制备的:在去离子水中,用单体d与有机膦酸单体e进行聚合反应,得到所述的聚合物b。
在另一优选例中,所述的反应在氮气保护下进行。
在另一优选例中,所述的反应在引发剂存在下进行;较佳地,所述的引发剂为自由基链引发剂;更佳地,所述的引发剂的摩尔量为单体d与有机膦酸单体e总摩尔量的0.1%-5%。
在另一优选例中,所述的含有羟基官能团或者羟基被保护基团保护的“酸-碱”型有机膦酸聚合物b是通过如下方法制备的:
(1)在氮气保护下,向三口反应瓶中依次加入单体d以及有机膦酸单体e,以去离子水作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn),其中aibn的摩尔量为单体d与有机膦酸单体e总摩尔量的0.1%-5%;
(2)搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,干燥,得“酸-碱”型有机膦酸聚合物b。
在另一优选例中,所述的有机高分子交联剂c选自下组:
其中m=2-10000。
在另一优选例中,所述的a与b的摩尔比na:nb=1:0.1-40,a与c的摩尔比na:nc=1:0.02-10.00。
本发明的第二方面,提供了一种如本发明第一方面所述的有机高分子交联的有机膦酸高温质子交换膜制备方法,所述的有机高分子交联的有机膦酸高温质子交换膜的制备方法如下:
(i)提供聚苯并咪唑类型化合物a与“酸-碱”型有机膦酸聚合物b;
(ii)在惰性气体的保护下,将两者的混合物溶于有机溶剂中,配成混合溶液;
(iii)混合溶液冷却至室温,加入有机高分子交联剂c,搅拌两个小时,搅拌均匀;
(iv)过滤除去不溶物,得到混合滤液;
(v)对所述的混合滤液进行脱气处理;
(vi)使所述进行脱气处理的混合滤液成膜,得到有机高分子交联的有机膦酸高温质子交换膜。
在另一优选例中,聚苯并咪唑类型化合物a与“酸-碱”型有机膦酸聚合物b的摩尔比例为1:0.1~40,聚苯并咪唑类型化合物a与有机高分子交联剂c的摩尔比例为1:0.02-10.00。
在另一优选例中,所述的有机溶剂为强极性有机溶剂,更佳地选自下组:dmso(二甲基亚砜)、dmf(n,n-二甲基甲酰胺)、dmac(n,n-二甲基乙酰胺),或nmp(n-甲基吡咯烷酮),或其组合。
在另一优选例中,所述的步骤(ii)中,所配溶液的固含量为1-30wt%。
在另一优选例中,所述的成膜包括:在玻璃板或塑料薄膜上涂膜并干燥,形成有机高分子交联的有机膦酸高温质子交换膜。
在另一优选例中,所述的涂膜方法为流延法。
在另一优选例中,所述的干燥包括:在70-90℃下进行初步干燥后,升温到100-160℃下进行第二步干燥,然后升温到160-300℃进行第三步干燥。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
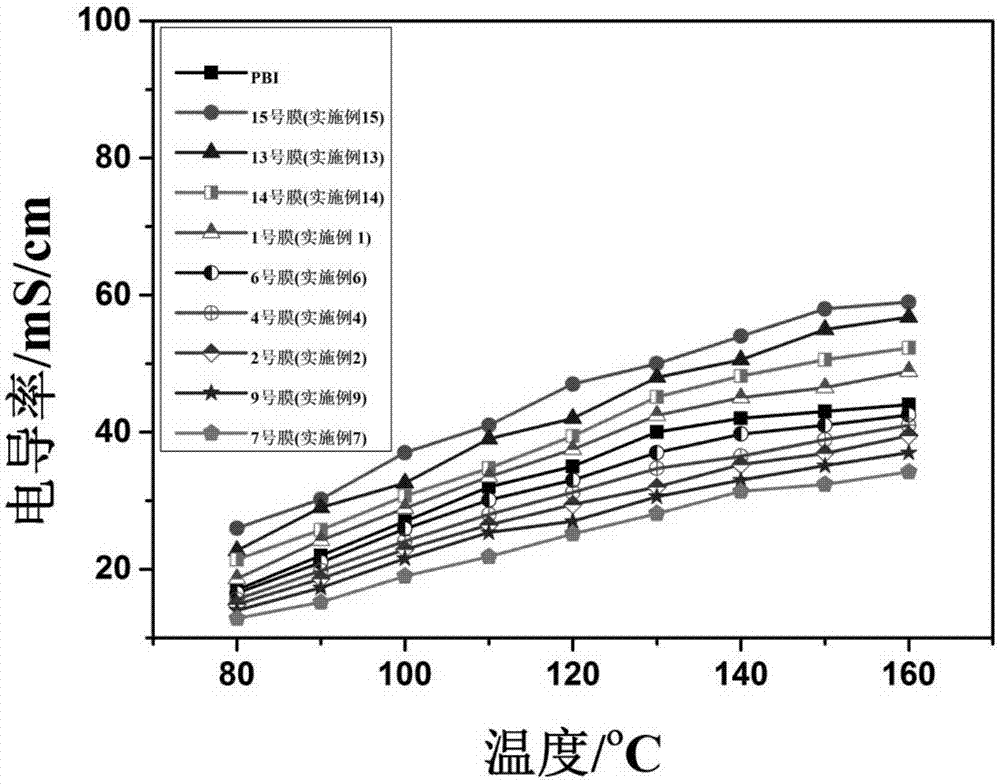
图1为实施例2、4、6、7、9、13、14和15中有机膦酸高温质子交换膜与pbi膜的质子电导率随温度变化的曲线图。
图2为实施例2、6、7和15中所制备的高温质子交换膜磷酸(膦酸)流失速率测试结果对比图。
图3为实施例1、2和14中所制备的高温质子交换膜的单电池放电曲线图。
图4为实施例1、4和15中所制备的高温质子交换膜的单电池工作稳定性测试图。
具体实施方式
本发明人经过长期而深入的研究发现,采用聚苯并咪唑类型化合物a与另外一种“酸-碱”型有机膦酸聚合物b在配成溶液以后,再加入有机高分子交联剂c,混合均匀之后再浇注成膜,经过干燥之后,可以得到具有高质子传导率、高机械强度、高热稳定性、高的抗氧化稳定性以及低溶胀率的高温质子交换膜,因此非常适合用作为高温质子导电膜燃料电池的质子交换膜。基于上述发现,发明人完成了本发明。
本发明的有机高分子交联的有机膦酸高温质子交换膜与传统的聚苯并咪唑(pbi)类型的高温质子交换膜以相比,具有以下优点:
1)不使用无机膦酸,不存在无机膦酸流失的问题;
2)不存在膜的机械强度与膜的质子电导率之间的矛盾;
3)不存在无机磷酸渗出对催化剂的毒化作用;
与报道过的有机膦酸高温质子交换膜以相比,具有以下优点:
1)有机膦酸聚合物制备方法简单,适合大规模制备;
2)制膜工艺简单,适合大批量制备,并且重复性好;
3)有机膦酸含量准确可控;
4)质子电导率可随膦酸含量的变化进行调节;
5)质子交换膜膜材料的溶胀率低;
6)膜材料的热稳定性和抗氧化稳定性显著增强;
7)聚合物膜的玻璃化转变温度提高,耐高温工作性能更好;
8)膜材料的机械强度高;
因此,采用本发明的有机高温质子交换膜将具有更高的机械强度、更高的质子传导率、更高的尺寸稳定性、更高工作稳定性和耐久性,有利于推动高温燃料电池的商业化发展。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
实施例1
m-pbi与[n-乙烯基咪唑—乙烯基膦酸二(三甲基硅)酯]共聚物b1以及有机高分子交联剂c1复合膜的制备
(1)n-乙烯基咪唑—乙烯基膦酸二(三甲基硅)酯共聚物b1(摩尔比1:4)的制备:
在氮气保护下,向三口反应瓶中依次加入n-乙烯基咪唑(282.3mg,3mmol)以及乙烯基膦酸二(三甲基硅)酯(3028.7mg,12mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,24.63mg,0.15mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b1。
(2)m-pbi与共聚物b1以及有机高分子交联剂c1(摩尔比1:2:0.1)复合膜的制备
按照1:2:0.1的摩尔比,称量干燥的m-pbi(154.2mg,0.5mmol)与共聚物b1(1103.7mg,1.0mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmso(二甲基亚砜,23.900g)中,加热配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c1(26.2mg,0.05mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为24μm(微米),dsc测试显示其玻璃化转变温度tg=321℃,力学性能测试显示其拉伸强度为109mpa。
实施例2
py-pbi与[2-乙烯基咪唑—烯丙基膦酸二(三乙基硅)酯]共聚物b2以及有机高分子交联剂c2复合膜的制备
(1)2-乙烯基咪唑—烯丙基膦酸二(三乙基硅)酯共聚物b2(摩尔比1:3)的制备
在氮气保护下,向三口反应瓶中依次加入2-乙烯基咪唑(282.3mg,3mmol)以及烯丙基膦酸二(三乙基硅)酯(3155.2mg,9mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,19.71mg,0.12mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b2。
(2)py-pbi与共聚物b2以及有机高分子交联剂c2(摩尔比1:1:0.2)复合膜的制备
按照1:1:0.2的摩尔比,称量干燥的py-pbi(154.5mg,0.5mmol)与共聚物b2(572.3mg,0.5mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmf(n,n-二甲基甲酰胺,17.443g)中,加热搅拌配成含固量为4%的溶液,冷却至室温,加入有机高分子交联剂c2(67.9mg,0.1mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到230℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为22μm(微米),dsc测试显示其玻璃化转变温度tg=320℃,力学性能测试显示其拉伸强度为107mpa。
实施例3
o-pbi与[1-乙烯基吡唑—1,1-乙烯基二膦酸四甲酯]共聚物b3以及有机高分子交联剂c3复合膜的制备
(1)1-乙烯基吡唑—1,1-乙烯基二膦酸四甲酯共聚物b3(摩尔比1:2)的制备
在氮气保护下,向三口反应瓶中依次加入1-乙烯基吡唑(282.3mg,3mmol)以及1,1-乙烯基二膦酸四甲酯(1464.7mg,6mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,14.78mg,0.09mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b3
(2)o-pbi与共聚物b3以及有机高分子交联剂c3(摩尔比1:3:0.15)复合膜的制备
按照1:3:0.15的摩尔比,称量干燥的o-pbi(200.2mg,0.5mmol)与共聚物b3(873.4mg,1.5mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,13.420g)中,加热搅拌配成含固量为8%的溶液,冷却至室温,加入有机高分子交联剂c3(90.0mg,0.15mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到250℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为26μm(微米),dsc测试显示其玻璃化转变温度tg=314℃,力学性能测试显示其拉伸强度为102mpa。
实施例4
oso2-pbi与[n-乙烯基咪唑—2-丙烯基(2-亚甲基膦酸二乙酯)膦酸二乙酯]共聚物b4以及有机高分子交联剂c4复合膜的制备
(1)n-乙烯基咪唑—2-丙烯基(2-亚甲基膦酸二乙酯)膦酸二乙酯共聚物b4(摩尔比1:4)的制备
在氮气保护下,向三口反应瓶中依次加入n-乙烯基咪唑(94.1mg,1mmol)以及2-丙烯基(2-亚甲基膦酸二乙酯)膦酸二乙酯(1464.7mg,4mmol),以去离子水(180ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,8.2mg,0.05mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b4。
(2)oso2-pbi与共聚物b4以及有机高分子交联剂c4(摩尔比1:0.5:0.05)复合膜的制备
按照1:0.5:0.05的摩尔比,称量干燥的oso2-pbi(464.1mg,1.0mmol)与共聚物b4(703.5mg,0.5mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,22.185g)中,加热搅拌配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c4(36.7mg,0.05mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到220℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为28μm(微米),dsc测试显示其玻璃化转变温度tg=322℃,力学性能测试显示其拉伸强度为109mpa。
实施例5
f6-pbi与[1-乙烯基三氮唑—乙烯基膦酸二甲酯]共聚物b5以及有机高分子交联剂c5复合膜的制备
(1)1-乙烯基三氮唑—乙烯基膦酸二甲酯共聚物b5(摩尔比1:5)的制备:
在氮气保护下,向三口反应瓶中依次加入1-乙烯基三氮唑(190.2mg,2mmol)以及乙烯基膦酸二甲酯(1360.9mg,10mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,9.9mg,0.06mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b5。
(2)f6-pbi与共聚物b5以及有机高分子交联剂c5(摩尔比1:5:0.3)复合膜的制备
按照1:5:0.3的摩尔比,称量干燥的f6-pbi(267.3mg,0.5mmol)与共聚物b5(1937.9mg,2.5mmol)。在氮气的保护下,将两者的混合物溶于干燥的nmp(n-甲基吡咯烷酮19.847g)中,加热搅拌配成含固量为10%的溶液,冷却至室温,加入有机高分子交联剂c5(99.6mg,0.15mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为29μm(微米),dsc测试显示其玻璃化转变温度tg=285℃,力学性能测试显示其拉伸强度为101mpa。
实施例6
oo-pbi与[1-乙烯基-1h-1,2,3-三氮唑—烯丙基膦酸二(三异丙基硅)酯]共聚物b6以及有机高分子交联剂c6复合膜的制备
(1)1-乙烯基-1h-1,2,3-三氮唑—烯丙基膦酸二(三异丙基硅)酯共聚物b6(摩尔比1:6)的制备
在氮气保护下,向三口反应瓶中依次加入1-乙烯基-1h-1,2,3-三氮唑(95.1mg,1mmol)以及烯丙基膦酸二(三异丙基硅)酯(2692.6mg,6mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,11.5mg,0.07mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b6。
(2)oo-pbi与共聚物b6以及有机高分子交联剂c6(摩尔比1:1:0.4)复合膜的制备
按照1:1:0.4的摩尔比,称量干燥的oo-pbi(246.8mg,0.5mmol)与共聚物b6(1393.9mg,0.5mmol)。在氮气的保护下,将两者的混合物溶于干燥的nmp(n-甲基吡咯烷酮25.704g)中,加热搅拌配成含固量为6%的溶液,冷却至室温,加入有机高分子交联剂c6(159.6mg,0.2mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为27μm(微米),dsc测试显示其玻璃化转变温度tg=316℃,力学性能测试显示其拉伸强度为105mpa。
实施例7
py-o-pbi与[n-乙烯基-1,3,4-三氮唑—1,1-乙烯基二膦酸四苯酯]共聚物b7以及有机高分子交联剂c7复合膜的制备
(1)n-乙烯基-1,3,4-三氮唑—1,1-乙烯基二膦酸四苯酯共聚物b7(摩尔比1:5)的制备
在氮气保护下,向三口反应瓶中依次加入n-乙烯基-1,3,4-三氮唑(95.1mg,1mmol)以及1,1-乙烯基二膦酸四苯酯(2460.4mg,5mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,9.9mg,0.06mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b7。
(2)py-o-pbi与共聚物b7以及有机高分子交联剂c7(摩尔比1:0.2:0.5)复合膜的制备
按照1:0.2:0.5的摩尔比,称量干燥的py-o-pbi(355.4mg,1mmol)与共聚物b7(511.1mg,0.2mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,42.460g)中,加热搅拌配成含固量为2%的溶液,冷却至室温,加入有机高分子交联剂c7(94.0mg,0.5mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为30μm(微米),dsc测试显示其玻璃化转变温度tg=330℃,力学性能测试显示其拉伸强度为109mpa。
实施例8
p-pbi与[1-乙烯基三氮唑—2-丙烯基[2-亚甲基膦酸二(三甲基硅)酯]膦酸二(三甲基硅)酯]共聚物b8以及有机高分子交联剂c8复合膜的制备
(1)1-乙烯基三氮唑—2-丙烯基[2-亚甲基膦酸二(三甲基硅)酯]膦酸二(三甲基硅)酯共聚物b8(摩尔比1:5)的制备
在氮气保护下,向三口反应瓶中依次加入1-乙烯基三氮唑(95.1mg,1mmol)以及2-丙烯基[2-亚甲基膦酸二(三甲基硅)酯]膦酸二(三甲基硅)酯(2520.8mg,5mmol),以去离子水(200ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,9.9mg,0.06mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b8。
(2)p-pbi与共聚物b8以及有机高分子交联剂c8复合膜(摩尔比1:0.1:0.6)的制备
按照1:0.1:0.6的摩尔比,称量干燥的p-pbi(616.7mg,2mmol)与共聚物b8(523.2mg,0.2mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,27.357g)中,加热搅拌配成含固量为4%的溶液,冷却至室温,加入有机高分子交联剂c8(333.6mg,1.2mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为32μm(微米),dsc测试显示其玻璃化转变温度tg=329℃,力学性能测试显示其拉伸强度为120mpa。
实施例9
so2-pbi与[n-乙烯基苯并咪唑—乙烯基膦酸二(三甲基硅)酯]共聚物b9以及有机高分子交联剂c9复合膜的制备
(1)n-乙烯基苯并咪唑—乙烯基膦酸二(三甲基硅)酯共聚物b9(摩尔比1:8)的制备
在氮气保护下,向三口反应瓶中依次加入n-乙烯基苯并咪唑(144.2mg,1mmol)以及乙烯基膦酸二(三甲基硅)酯(2016.6mg,8mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,29.6mg,0.18mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b9。
(2)so2-pbi与共聚物b9以及有机高分子交联剂c9(摩尔比1:0.3:0.8)复合膜的制备
按照1:0.3:0.8的摩尔比,称量干燥的so2-pbi(494.6mg,1mmol)与共聚物b9(648.2mg,0.3mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,21.713g)中,加热搅拌配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c9(121.6mg,0.8mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为30μm(微米),dsc测试显示其玻璃化转变温度tg=350℃,力学性能测试显示其拉伸强度为116mpa。
实施例10
s-pbi与[2-乙烯基苯并咪唑—烯丙基膦酸二(三乙基硅)酯]共聚物b10以及有机高分子交联剂c10复合膜的制备
(1)2-乙烯基苯并咪唑—烯丙基膦酸二(三乙基硅)酯共聚物b10(摩尔比1:10)的制备
在氮气保护下,向三口反应瓶中依次加入2-乙烯基苯并咪唑(144.2mg,1mmol)以及乙烯基膦酸二(三甲基硅)酯(3501.9mg,10mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,9.1mg,0.055mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b10。
(2)s-pbi与共聚物b10以及有机高分子交联剂c10(摩尔比1:10:2)复合膜的制备
按照1:10:2的摩尔比,称量干燥的s-pbi(41.6mg,0.1mmol)与共聚物b10(3645.9mg,1mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,72.009g)中,加热搅拌配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c10(30.4mg,0.2mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到240℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为36μm(微米),dsc测试显示其玻璃化转变温度tg=369℃,力学性能测试显示其拉伸强度为128mpa。
实施例11
abpbi与[5-乙烯基三氮唑—1,1-乙烯基二膦酸四甲酯]共聚物b11以及有机高分子交联剂c9复合膜的制备
(1)5-乙烯基三氮唑—1,1-乙烯基二膦酸四甲酯]共聚物b11(摩尔比1:12)的制备
在氮气保护下,向三口反应瓶中依次加入5-乙烯基三氮唑(95.1mg,1mmol)以及1,1-乙烯基二膦酸四甲酯(2929.4mg,12mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,10.7mg,0.065mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b11。
(2)abpbi与共聚物b11以及有机高分子交联剂c9(摩尔比1:10:3)复合膜的制备
按照1:10:3的摩尔比,称量干燥的abpbi(11.6mg,0.1mmol)与共聚物b11(3023.4mg,1mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,27.315g)中,加热搅拌配成含固量为10%的溶液,冷却至室温,加入加入有机高分子交联剂c9(45.6mg,0.3mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到230℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为31μm(微米),dsc测试显示其玻璃化转变温度tg=290℃,力学性能测试显示其拉伸强度为103mpa。
实施例12
聚[2,6-(对亚苯基)-苯并二咪唑]与[5-乙烯基嘧啶—2-丙烯基[2-亚甲基膦酸二(三甲基硅)酯]膦酸二(三甲基硅)酯]]共聚物b12以及有机高分子交联剂c7复合膜的制备
(1)5-乙烯基嘧啶—2-丙烯基[2-亚甲基膦酸二(三甲基硅)酯]膦酸二(三甲基硅)酯]共聚物b12(摩尔比1:16)的制备
在氮气保护下,向三口反应瓶中依次加入5-乙烯基嘧啶(159.2mg,1.0mmol)以及2-丙烯基[2-亚甲基膦酸二(三甲基硅)酯]膦酸二(三甲基硅)酯(8066.5mg,16mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,14.0mg,0.085mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b12。
(2)聚[2,6-(对亚苯基)-苯并二咪唑]与共聚物b12以及有机高分子交联剂c7(摩尔比1:16:10)复合膜的制备
按照1:16:10的摩尔比,称量干燥的聚[2,6-(对亚苯基)-苯并二咪唑](11.6mg,0.05mmol)与共聚物b12(6538.0mg,0.8mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmac(n,n-二甲基乙酰胺,26.298g)中,加热搅拌配成含固量为20%的溶液,冷却至室温,加入有机高分子交联剂c7(94.0mg,0.5mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到230℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为36μm(微米),dsc测试显示其玻璃化转变温度tg=275℃,力学性能测试显示其拉伸强度为95mpa。
实施例13
聚[2,6-[4′,4″-亚(二苯基砜基)]-苯并二咪唑]与(4-乙烯基吡啶—乙烯基膦酸)共聚物b13以及有机高分子交联剂c1复合膜的制备
(1)4-乙烯基吡啶—乙烯基膦酸共聚物b13(摩尔比1:4)的制备
在氮气保护下,向三口反应瓶中依次加入4-乙烯基吡啶(210.0mg,2.0mmol)以及乙烯基膦酸(864.0mg,8mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,14.8mg,0.09mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b13。
(2)聚[2,6-[4′,4″-亚(二苯基砜基)]-苯并二咪唑]与共聚物b13以及有机高分子交联剂c1(摩尔比1:4:1)复合膜的制备
按照1:4:1的摩尔比,称量干燥的聚[2,6-(对亚苯基)-苯并二咪唑](74.4mg,0.2mmol)与共聚物b13(429.6mg,0.8mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmso(二甲基亚砜,9.576g)中,搅拌搅拌配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c1(104.8mg,0.2mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到160℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为24μm(微米),dsc测试显示其玻璃化转变温度tg=288℃,力学性能测试显示其拉伸强度为104mpa。
实施例14
聚[2,6-[4′,4″-亚(二苯基醚)]-苯并二咪唑]与(n-乙烯基咪唑—乙烯基膦酸)共聚物b14以及有机高分子交联剂c3复合膜的制备
(1)n-乙烯基咪唑—乙烯基膦酸共聚物b14(摩尔比1:5)的制备
在氮气保护下,向三口反应瓶中依次加入n-乙烯基咪唑(188.0mg,2.0mmol)以及乙烯基膦酸(1080.0mg,10mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,19.7mg,0.12mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b14。
(2)聚[2,6-[4′,4″-亚(二苯基醚)]-苯并二咪唑]与共聚物b14以及有机高分子交联剂c3(摩尔比1:2:0.1)复合膜的制备
按照1:2:0.1的摩尔比,称量干燥的聚[2,6-(对亚苯基)-苯并二咪唑](162.2mg,0.5mmol)与共聚物b14(634.0mg,1.0mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmso(二甲基亚砜,15.128g)中,配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c3(30.0mg,0.05mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到160℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为27μm(微米),dsc测试显示其玻璃化转变温度tg=279℃,力学性能测试显示其拉伸强度为106mpa。
实施例15
聚[2,2′-(2″,6″-亚吡啶基)-5,5′-二(苯并咪唑基)砜]与[1-乙烯基三氮唑—烯丙基膦酸共聚物b15以及有机高分子交联剂c5复合膜的制备
(1)1-乙烯基三氮唑—烯丙基膦酸共聚物b15(摩尔比1:5)的制备
在氮气保护下,向三口反应瓶中依次加入n-乙烯基咪唑(190.1mg,2.0mmol)以及烯丙基膦酸(1220.0mg,10mmol),以去离子水(250ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,19.7mg,0.12mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b15。
(2)聚[2,2′-(2″,6″-亚吡啶基)-5,5′-二(苯并咪唑基)砜]与共聚物b15(摩尔比1:4:0.4)复合膜的制备
按照1:4:0.4的摩尔比,称量干燥的聚[2,2′-(2″,6″-亚吡啶基)-5,5′-二(苯并咪唑基)砜](93.3mg,0.25mmol)与共聚物b15(705.1mg,1.0mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmso(二甲基亚砜,15.170g)中,配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c5(66.4mg,0.1mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到160℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为29μm(微米),dsc测试显示其玻璃化转变温度tg=288℃,力学性能测试显示其拉伸强度为107mpa。
实施例16
聚[2,6-[4′,4″-亚(二苯基甲烷)]-苯并二咪唑]与[5-乙烯基三氮唑—1,1-乙烯基二膦酸]共聚物b16以及有机高分子交联剂c10复合膜的制备
(1)5-乙烯基三氮唑—1,1-乙烯基二膦酸共聚物b16(摩尔比1:4)的制备
在氮气保护下,向三口反应瓶中依次加入5-乙烯基三氮唑(95.1mg,1mmol)以及1,1-乙烯基二膦酸(751.9mg,4mmol),以去离子水(180ml)作为溶剂,然后加入自由基链引发剂偶氮二异丁腈(aibn,8.2mg,0.05mmol)。搅拌(机械搅拌),加热到80℃反应。反应24h后,溶液变粘稠,停止反应,冷却致室温。将反应液倒入乙酸乙酯中,并不断搅拌,有固体析出,过滤,真空干燥,得共聚物b16。
(2)聚[2,6-[4′,4″-亚(二苯基甲烷)]-苯并二咪唑]与共聚物b16以及有机高分子交联剂c10(摩尔比1:3:2)复合膜的制备
按照1:3:2的摩尔比,称量干燥的聚[2,6-[4′,4″-亚(二苯基甲烷)]-苯并二咪唑](80.6mg,0.25mmol)与共聚物b16(635.3mg,0.75mmol)。在氮气的保护下,将两者的混合物溶于干燥的dmso(二甲基亚砜,13.602g)中,配成含固量为5%的溶液,冷却至室温,加入有机高分子交联剂c10(76.0mg,0.5mmol),搅拌2小时,搅拌均匀后过滤,滤去不溶物,滤液经过脱气处理后流延到10cm×10cm的玻璃板上,然后放到鼓风烘箱中在80℃下干燥两小时,接着进一步升温到120℃下干燥一个小时,最后升温到160℃干燥一个小时,即得到有机膦酸高温质子交换膜。该膜的厚度为25μm(微米),dsc测试显示其玻璃化转变温度tg=297℃,力学性能测试显示其拉伸强度为102mpa。
测试实施例
质子电导率测试:
用membranetestsystem740装置,采用两电极法测试pbi膜以及本专利中2号膜(实施例2)、4号膜(实施例4)、6号膜(实施例6)、7号膜(实施例7)、9号膜(实施例9)、13号膜(实施例13)、14号膜(实施例14)、15号膜(实施例15)的质子电导率测试。
测试方法:
(1)测试前处理:
在60℃条件下,将pbi膜浸泡在85%的磷酸中,浸泡48小时后取出干燥,待测。
将要测试的有机膦酸膜浸泡在0.5mol/l的稀磷酸中,浸泡48小时后取出,然后用去离子浸泡,水洗直中性(洗出液为中性),干燥,待测。
(2)正式测试(测试设备membranetestsystem740):
1.将待测样品膜剪裁1cm×3cm形状;
2.在两电极夹具金属片上用导电胶粘带有pt/c催化剂的gde,上下各一片,并将剪好待测膜装在gde中间,夹紧夹具;
3.接通mts740的电源,将装有待测膜的夹具放入到本装置的测试腔室内。
4.打开本仪器测试程序,接通气瓶与仪器相连接的管路。调节n2管路压力至0.5mpa,h2管路压力至0.4mpa,检查程序中个指示灯指示正常,并检查化学工作站与740操作腔体的连接。
5.膜样品开始测试时,应当先通入n2吹扫10min以排除腔室内的空气,吹扫速度为500sccm/min,仪器运行过程中,当温度与湿度(相对湿度为2%,测试有机膦酸膜时相对湿度设定为100%)达到设定的值并稳定后,即可开始测试。
6.使用mts740程序自动获得阻抗谱图,由谱图获得膜电阻值r。
7.数据处理:电导率由ρ=l/(rs×a)计算得到。
l:膜厚度;
rs:阻抗谱读取的膜电阻
a:被测面积
8.测试结束时,必须用n2吹扫15min以排除仪器中的水汽,最后关闭程序及仪器的电源。
测试结果如图1中所示。结果显示,本申请的燃料电池用复合型有机膦酸高温质子交换膜的电导率与pbi膜/无机磷酸体系达到同一个数量级,并且有的配比膜的,其电导率明显高于pbi膜。目前,大量报道的有机膦酸膜存在的最大的问题就是质子电导率偏低,比pbi膜/无机磷酸体系低1-2个数量级,而本发明中所制备的有机膦酸膜与pbi膜/无机磷酸体系相比,可以达到同一个数量级,具有极其优异的效果。
磷酸(膦酸)流失速率测试:
我们使用pbi膜/无机磷酸体系,以及有机膦酸膜在浸泡去离子水后电导率的变化来反应磷酸(膦酸)流失速率。我们选择pbi膜/无机磷酸,以及有机膦酸2号膜(实施例2)、6号膜(实施例6)、7号膜(实施例7)、15号膜(实施例15)进行测试对比:
测试方法:
(1)测试测试样品膜的准备:
在60℃条件下,将pbi膜浸泡在85%的磷酸中,浸泡48小时后取出干燥。
将要测试的有机膦酸膜浸泡在0.5mol/l的稀磷酸中,浸泡48小时后取出,然后用去离子浸泡,水洗直中性(洗出液为中性),干燥。
(2)正式测试
1.将准备好的样品膜在去离子水中浸泡5分钟后取出,干燥。
2.按照前面电导率的测试方法测试干燥好的样品膜在160℃条件下的质子电导率(测试条件:pbi膜相对湿度2%;有机膦酸膜相对湿度100%)
3.循环上述操作1以及2测试九次。
测试结果如图2所示,pbi膜/无机磷酸体系的质子电导率下降非常的快,而有机膦酸膜的质子电导率几乎没有任何下降。这个结果说明pbi膜/无机磷酸体系中的无机磷酸流失非常的严重,而本申请的有机膦酸膜的有机膦酸几乎不存在磷酸流失的问题。实际使用过程中,燃料电池在运行的时候会产生水,质子电导膜处于不断有水分流失的状态,而本申请的有机膦酸膜则不存在膦酸随水分流失被带走的问题,因此在实际使用状态下可以有效稳定膦酸含量,进而可以保持良好的导电性能。
单电池放电性能测试:
用fuelcelltestsystem850e装置,测试本专利中1号膜(实施例1)、2号膜(实施例2)、14号膜(实施例14)组装的单电池的放电性能。
(1)测试测试样品膜的准备:
将要测试的有机膦酸膜浸泡在0.5mol/l的稀磷酸中,浸泡48小时后取出,然后用去离子浸泡,水洗直中性(洗出液为中性),干燥。
(2)单电池组装:
a)将被测膜与电极制备得到膜电极,并将其组装在单电池模型中,待测用;
b)单电池与氢燃料电池测试系统fuelcelltestsystem850e连接。氢气进气端接电池负极,空气进气端接电池正极,氢气与空气进出气端分别在石墨双极板中同侧,保证在一侧双极板中一种气体一进一出;
(3)单电池测试:
a)测试系统参数设置:
测试温度:160℃
氢气进气量:500cc/min,计量比为1.6
空气进气量:1500cc/min,计量比为1.3
b)测试程序:
采用扫描电位测试方法
电压范围:0.25—1v,每0.05v收集一个数据点
c)收集电压、电流密度、能量密度等参数的数据,并设置好储存路径;
测试结果如图3所示,说明有机膦酸高温质子交换膜具有优异的放电性能。
单电池工作稳定性测试:
我们选择pbi膜与1号膜(实施例1)、4号膜(实施例4)以及15号膜(实施例15)分别组装单电池,进行工作稳定性测试。
1.测试前膜处理:
a)在60℃条件下,将pbi膜浸泡在85%的磷酸中,浸泡48小时后取出干燥。将有机膦酸膜浸泡在0.5mol/l的稀磷酸中,浸泡48小时后取出,然后用去离子浸泡,水洗直中性(洗出液为中性),干燥。
b)将待测膜裁减成6.5cm×6.5cm的尺寸;
2.mea(膜电极)制备;
a)使用sigracet公司型号为29bc的gdl,裁剪成5×5cm尺寸,为催化剂喷涂做准备;
b)在gdl气孔层进行催化剂喷涂,阴极催化剂担载量为2mg/cm2,阳极催化剂担载量为3mg/cm2,制备gde;
c)将待测膜放在中间,其上下分别放置gde的阴、阳极,放成三明治结构,然后进行热压(热压条件为135℃、10mpa、2min),得到mea;
3.电池组装:
将制备得到的mea组装在单电池模型中。
4.单电池测试:
测试设备:fuelcelltestsystem850e
将单电池与测试设备连接好,按照设定的参数:阳极:氢气500毫升/分钟;阴极:空气1500毫升/分钟;测试温度:160℃;pbi膜相对湿度:2%,有机膦酸膜相对湿度100%,恒定扫描电压0.7v.在测试条件下仪器自动记录电流密度的数据。测试结果如图4所示。
从测试结果可以看出,pbi膜组装的单电池在测试1000多小时后就可以看到明显的性能下降,而1号膜(实施例1)、4号膜(实施例4)以及15号膜(实施例15)组装的单电池的性能一直很稳定,没有明显的下降,说明本申请的有机膦酸质子交换膜制备的单电池取得了更好的稳定性。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!