一种Pt/金属碳化物/碳纳米材料催化剂及其制备方法与流程
本发明涉及纳米复合催化剂制备技术领域,尤其涉及一种应用于直接甲醇燃料电池(dmfc)阳极的pt/金属碳化物/碳纳米材料催化剂及其制备方法。
背景技术:
燃料电池是近年来兴起的清洁能源技术装置,是继水力、火力和核能发电之后的新一代发电技术。它是一种不经燃烧直接以电化学反应方式将燃料和氧化剂的化学能转变为电能的高效连续发电装置。燃料电池由阳极、阴极和两极之间的电解质组成。在阳极一侧通入o2或空气,通过电解质的离子传导,在阴极和阳极发生电子转移,并在两极产生电势差,形成一个电池。联接两级,在外电路中形成电流,便可带动负载工作。作为燃料电池的一种,直接甲醇燃料电池(directmethanolfuelcell,简称dmfc)具有结构紧凑、质量轻、燃料来源丰富、储存携带方便、环境友好等优点,是目前各国政府优先发展的高新技术之一。
对dmfc而言,电催化剂的利用不仅可以加速电极反应、抑止副反应的发生,而且可以提高电池的能量输出效率和整体性能。然而在燃料电池中,甲醇在pt表面产生的各种中间体中有co存在,而pt与co中间体结合能较高,这就会造成pt表面活性位点大面积被占用而失活,即pt中毒,使催化活性大大降低,因此限制了其商业化的发展。目前,dmfc所使用的催化剂无论在阳极还是阴极均以pt系贵金属催化剂为主。
申请号为cn201610208955.5的中国专利公开了一种“金-铂纳米复合材料”,纳米金呈枝状,球状铂颗粒修饰在纳米金表面,au与pt复合后改变了pt的内部原子排列以及费米能级,进而改变pt的表面性能,提高复合催化剂的表面催化活性、抗毒化能力及稳定性。申请号为201610521094.6的中国专利公开了一种“ptpd电催化剂”,该催化剂具备一种新异的空心网状结构,增大催化剂的比表面积,从而提高催化活性,产生优异的电催化氧化甲醇性能。通过加入第二元金属来改变催化剂的结构形貌,或是改变催化剂的晶体结构来提高催化剂的活性以及抗毒化能力,是目前燃料电池催化剂发展的重要手段,然而引入的第二元金属应当在提高催化剂性能的同时,降低催化剂的成本,才能进一步加速燃料电池商业化的进步。申请号为201610168232.7的中国专利公开了“一种pt-mn-石墨烯燃料电池复合催化剂的制备方法”,通过改变制备过程中pt源与mn源的加入量得到性能最佳的配比,并表现出对甲醇优异的催化性能。申请号为201611136275.3的中国专利公开了“一种氧化物@贵金属核壳纳米线催化剂的制备方法”,氧化物包覆在贵金属纳米材料外层,所制备的催化剂具有成本低、抗毒化性强、活性高、稳定性好等优点,申请号为201510028612.6的中国专利公开了“一种直接甲醇燃料电池的阳极催化剂”,具有高比表面积和高导电性。利用了钨的碳化物做辅助催化剂,强化解离吸附在pt上的co的作用,从而对甲醇的催化氧化表现出优异的活性。不论是向催化剂中加入过渡金属、过渡金属氧化物或是过渡金属碳化物助催化剂,都能有效提高对醇类燃料的催化活性,pt和助催化剂之间属于“体相接触”,不能有效利用助催化剂的作用来改变pt价电子结构;也不能使pt和助催化剂的活性位点充分暴露出来,所以导致助催化能力、协同催化作用收到传质距离的限制,催化剂整体活性并不高。
过渡金属碳化物是碳原子进入过渡金属晶格而产生的一类具有金属性质的间充化合物,通常具有高硬度、高熔点、抗腐蚀性以及良好的稳定性等优点。研究发现,过渡金属碳化物主晶面上的原子排布与pt相似,费米能级附近电子态能量密度比较高,由于受到碳原子的影响,过渡金属原子中电子不再全部共有为巡游电子,而只有一部分5d电子像pt一样变成局部电子,这种表面电子结构使得部分过渡金属碳化物具有类pt的催化活性。同时,过渡金属碳化物可以在相对pt较低的电位下分解水,产生oh中间体,与pt表面牢固结合的co反应生成co2脱离pt表面,释放活性位点,提高催化活性。因此制备出pt与碳化物原子级接触的结构能极大地缩短pt与碳化物之间的传质距离,能强有力地提高二者之间的协同作用,进一步提高催化剂的催化活性、以及抗co中毒能力。一元金属碳化物由于其本身固有的电子结构特点,催化活性和助催化能力受到限制,因此,需要通过掺杂改性的方法,即引入其他金属元素来改善一元金属碳化物的价电子结构,提高其催化活性和助催化能力。
技术实现要素:
本发明提供了一种pt/金属碳化物/碳纳米材料催化剂及其制备方法,在降低pt用量的同时,通过控制合成pt与过渡金属碳化物达到原子级接触,成功制备出具有较高催化活性,适用于直接甲醇燃料电池的复合催化剂。
为了达到上述目的,本发明采用以下技术方案实现:
一种pt/金属碳化物/碳纳米材料催化剂,所述pt/金属碳化物/碳纳米材料催化剂中pt原子和金属碳化物分子均匀分布,且两者均匀分散在碳纳米材料的表面;催化剂中pt的质量分数为5%~30%,金属碳化物的质量分数为5%~15%,其余为碳纳米材料。
一种pt/金属碳化物/碳纳米材料催化剂的制备方法,包括如下步骤:
(1)将碳纳米材料加入到浓硫酸与浓硝酸的混酸溶液中,在40~80℃下超声1~3h,然后过滤洗涤,再加入去离子水进行超声分散得到碳源溶液;
(2)向步骤(1)所得到的碳源溶液中加入表面改性剂,超声分散均匀后进行抽滤洗涤,然后将沉淀物加入到去离子水中,再次超声分散得到前驱体材料溶液;
(3)向步骤(2)所得到的前驱体材料溶液中加入可溶于水的铂盐溶液,以及钨盐溶液或钼盐溶液中的一种,搅拌均匀,得到前驱体溶液;
(4)将步骤(3)所得到的前驱体溶液进行冷冻干燥,除去水分,得到粉体材料;
(5)将步骤(4)所得的粉体材料进行微波加热,在弱还原气氛下快速升温加热至900~1200℃,恒温热处理1~3h后自然冷却,得到pt/金属碳化物/碳纳米材料催化剂。
所述碳纳米材料包括碳黑、多壁碳纳米管。
所述铂盐溶液为0.1~0.5mol/l的h2ptcl6水溶液,钨盐溶液包括钨酸钠溶液、钨酸铵溶液;钼盐溶液包括钼酸钠溶液、钼酸铵溶液。
所述弱还原性气氛为n2与h2的混合气体或ar与h2的混合气体,且混合气体中h2的体积百分含量<10%。
所述粉体材料进行微波加热时,从室温以20~60℃/min的速率升温至900~1200℃。
所述混酸溶液中浓硫酸与浓硝酸溶液的体积比为3︰0.9~1.1。
所述表面改性剂为聚二烯基丙二甲基氯化铵质量分数为5%~15%的水溶液,即pdda溶液,pdda溶液与碳源溶液的质量比为1:1~1.2。
与现有技术相比,本发明的有益效果是:
本发明所述pt/金属碳化物/碳纳米材料催化剂实现了原子级接触,可明显提高催化转化效率和催化剂使用寿命;与现有商业etek催化剂材料相比,本发明具以下突出的优点:
1)本发明所述pt/金属碳化物/碳纳米材料催化剂中,pt原子与金属碳化物分子均匀分布、充分接触,达到原子级接触结构,提高了pt与金属碳化物之间的协同作用,提高了催化剂催化活性和抗co中毒能力;
2)本发明所述pt/金属碳化物/碳纳米材料催化剂在制备过程中,采用了分子自组装、冷冻干燥技术和微波加热技术,使pt与过渡金属原位还原和碳化,有效防止了团聚、分相和偏析;
3)制备过程简单、容易控制,没有沉积金属后的过滤洗涤步骤,减少了金属元素的流失,提高了贵金属的利用率。
附图说明
图1为实施例1所制备催化剂的xrd图谱。
图2为实施例2所制备催化剂的xrd图谱。
图3为实施例3所制备催化剂的xrd图谱。
图4为实施例4所制备催化剂的xrd图谱。
图5为实施例1所制备催化剂的sem照片。
图6为实施例2所制备催化剂的sem照片。
图7为实施例3所制备催化剂的sem照片。
图8为实施例4所制备催化剂的sem照片。
图9为实施例1所制备催化剂与商业etek催化剂,在n2饱和的0.5mol/lh2so4溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图10为实施例2所制备催化剂与商业etek催化剂,在n2饱和的0.5mol/lh2so4溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图11为实施例3所制备催化剂与商业etek催化剂,在n2饱和的0.5mol/lh2so4溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图12为实施例4所制备催化剂与商业etek催化剂,在n2饱和的0.5mol/lh2so4溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图13实施例1所制备催化剂与商业etek催化剂在n2除氧后0.5mol/lh2so4+1mol/lch3oh溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图14实施例2所制备催化剂与商业etek催化剂在n2除氧后0.5mol/lh2so4+1mol/lch3oh溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图15实施例3所制备催化剂与商业etek催化剂在n2除氧后0.5mol/lh2so4+1mol/lch3oh溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
图16实施例4所制备催化剂与商业etek催化剂在n2除氧后0.5mol/lh2so4+1mol/lch3oh溶液中的循环伏安曲线,扫速为50mv/s,扫描范围-0.2~+1.04v,室温。
具体实施方式
下面结合附图对本发明的具体实施方式作进一步说明:
本发明所述一种pt/金属碳化物/碳纳米材料催化剂,所述pt/金属碳化物/碳纳米材料催化剂中pt原子和金属碳化物分子均匀分布,且两者均匀分散在碳纳米材料的表面;催化剂中pt的质量分数为5%~30%,金属碳化物的质量分数为5%~15%,其余为碳纳米材料。
一种pt/金属碳化物/碳纳米材料催化剂的制备方法,包括如下步骤:
(1)将碳纳米材料加入到浓硫酸与浓硝酸的混酸溶液中,在40~80℃下超声1~3h,然后过滤洗涤,再加入去离子水进行超声分散得到碳源溶液;
(2)向步骤(1)所得到的碳源溶液中加入表面改性剂,超声分散均匀后进行抽滤洗涤,然后将沉淀物加入到去离子水中,再次超声分散得到前驱体材料溶液;
(3)向步骤(2)所得到的前驱体材料溶液中加入可溶于水的铂盐溶液,以及钨盐溶液或钼盐溶液中的一种,搅拌均匀,得到前驱体溶液;
(4)将步骤(3)所得到的前驱体溶液进行冷冻干燥,除去水分,得到粉体材料;
(5)将步骤(4)所得的粉体材料进行微波加热,在弱还原气氛下快速升温加热至900~1200℃,恒温热处理1~3h后自然冷却,得到pt/金属碳化物/碳纳米材料催化剂。
所述碳纳米材料包括碳黑、多壁碳纳米管。
所述铂盐溶液为0.1~0.5mol/l的h2ptcl6水溶液,钨盐溶液包括钨酸钠溶液、钨酸铵溶液;钼盐溶液包括钼酸钠溶液、钼酸铵溶液。
所述弱还原性气氛为n2与h2的混合气体或ar与h2的混合气体,且混合气体中h2的体积百分含量<10%。
所述粉体材料进行微波加热时,从室温以20~60℃/min的速率升温至900~1200℃。
所述混酸溶液中浓硫酸与浓硝酸溶液的体积比为3︰0.9~1.1。
所述表面改性剂为聚二烯基丙二甲基氯化铵质量分数为5%~15%的水溶液,即pdda溶液,pdda溶液与碳源溶液的质量比为1:1~1.2。
进一步地,本发明所述一种pt/金属碳化物/碳纳米材料催化剂的制备过程及反应原理具体为:
(1)用混酸溶液对碳纳米载体材料进行表面预处理,将称量好的混酸溶液与碳纳米载体材料混合在锥形瓶内,置于超声清洗机内,将温度设定为40~80℃,超声时间为1~3h。该过程中碳纳米载体表面会产生羟基、羧基等负电集团。
(2)向步骤(1)所得酸处理后的溶液在砂芯抽滤瓶上进行抽滤洗涤,洗去过量的酸,直至滤液的ph值为中性后停止洗涤,将抽滤得到的碳纳米材料溶于适量的去离子水中,在超声清洗机内进行超声分散,直至溶液分散均匀,没有明显的黑色颗粒后停止超声。
(3)向步骤(2)所得酸处理后的前驱体分散液中加入表面改性剂,放入搅拌子,在磁力搅拌器上进行搅拌,室温,转速设定为600~700r/min,时间为0.5~1h。表面改性剂在溶液中电离出cl-1,与碳载体表面的负电集团结合。
(4)将步骤(3)所得分散液进行抽滤洗涤,洗去过量的表面改性剂。洗涤2~3次后,将得到的改性后的碳纳米载体材料溶于适量去离子水中,在超声清洗机内进行超声分散,直至溶液分散均匀,没有明显的黑色颗粒后停止超声。
(5)向步骤(4)所得分散均匀的前驱体材料溶液加入铂盐或过渡金属w、mo的金属盐,放入搅拌子,在磁力搅拌器上进行搅拌,室温,转速设定为600~700r/min,时间为1.5~2.5h。在静电引力作用下自组装,使金属盐离子与带正电的表面改性剂结合,定位于碳纳米载体表面,形成具有有序结构的纳米前驱体材料。
(6)将步骤(5)得到的催化剂前体溶液再次超声15min后置于冷冻干燥机内进行冷冻干燥,将纳米前驱体结构固定并去除水及其它溶剂得到前躯体固体粉末。首先对样品进行预冻,8~10h后到达-55℃。冻结后对干燥箱抽真空,真空状态下缓慢升高样品台温度,3~5h样品台温度升至-50℃,5~7h样品台温度升至-40℃,7~9h样品台温度升至-32℃,9~10h样品台温度升至-20℃,10~11h样品台温度升至30℃后保温,直至样品温度达到与载物台相同温度即干燥完成。
(7)将步骤(6)所得前驱体固体材料放入微波管式炉进行微波加热,利用微波快速加热作用使pt和过渡金属元素在碳纳米材料表面快速还原碳化,防止pt和金属碳化物分相和团聚。于ar-h2气氛下程序升温至900~1200℃,升温速率30~40℃/min,恒温热处理1~3h,之后自然冷却至室温,得到原子级接触的pt/金属碳化物/碳纳米催化剂。
以下实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。下述实施例中所用方法如无特别说明均为常规方法。
本发明所采用的设备和原料等均可从市场购得或属本领域常用。以下是各实施例采用的主要试剂:
【实施例1】
本实施例中,一种pt/金属碳化物/碳纳米材料催化剂的制备过程如下:
配置浓硫酸与浓硝酸体积比3:1的混酸溶液,将碳黑加入到混酸溶液中,置于超声仪内设定温度60℃震荡2h。结束后用砂芯漏斗减压抽滤,并用去离子水洗涤3次,将沉淀物加入去离子水中,再次超声分散,按照碳黑与表面活性剂质量比1:1加入表面活性剂pdda溶液,搅拌0.5h后用砂芯漏斗对分散液进行减压抽滤,并用去离子水洗3次,将沉淀物加入一定量的去离子水,再次超声分散,然后向溶液中加入na2wo4·2h2o,加入量按照碳与钨摩尔比80:1确定,再滴入h2ptcl6溶液,按照溶液中pt的质量为所制备的pt/金属碳化物/碳纳米材料催化剂质量的20%加入,搅拌2h,再次超声15min;然后进行冷冻干燥,首先对样品进行预冻,8~10h后到达-55℃。冻结后对干燥箱抽真空,真空状态下缓慢升高样品台温度,3~5h样品台温度升至-50℃,6h样品台温度升至-40℃,8h样品台温度升至-32℃,9h样品台温度升至-20℃,10h样品台温度升至30℃后保温,直至样品温度达到与载物台相同温度即干燥完成。干燥完成后将样品置于微波加热工作站内,ar-h2气氛下1200℃烧结2h,待炉腔温度降至室温取出,即可得到pt与wc达到原子级接触的pt/wc/c催化剂。
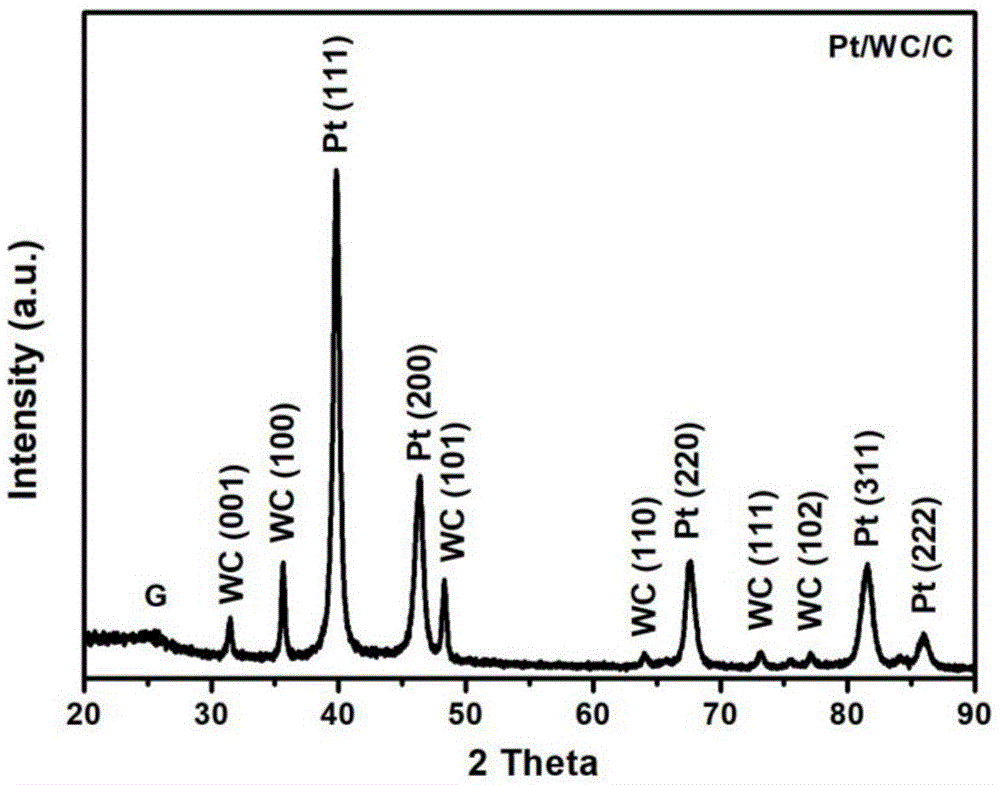
如图1所示,在本实施例所制备pt/wc/c催化剂的xrd图谱中,能观察到明显的pt晶体衍射峰和wc晶体衍射峰,通过谢乐公式计算得到样品中pt晶粒尺寸为10.88nm,wc晶粒尺寸为10.96nm。
如图5所示,在本实施例所制备pt/wc/c催化剂的sem照片中,能明显看到pt与wc颗粒,分布均匀,且颗粒大小与谢乐公式计算结果一致。
如图9硫酸溶液里的循环伏安曲线所示,本实施例所制备pt/wc/c催化剂的氢区活性面积(ecsa)明显大于商业etek催化剂,即ecsa实施例1=93.86m2g-1,ecsaetek=80.21m2g-1;如图13甲醇溶液里的循环伏安曲线所示,本实施例所制备pt/wc/c催化剂的峰电流高出etek催化剂峰电流大约150mamg-1pt,通过计算if/ib,得到if/ib实施例一=1.54,if/ibetek=0.79,本实施例所制备pt/wc/c催化剂的值明显高于etek催化剂的值,说明本实施例所制备pt/wc/c催化剂的抗co中毒能力高于商业etek催化剂。
【实施例2】
本实施例中,一种pt/金属碳化物/碳纳米材料催化剂的制备过程与实施例1类似,加入的碳源为mwcnts,其余步骤相同,经制备得到pt/wc/mwcnts催化剂。
如图2所示,在本实施例所制备pt/wc/mwcnts催化剂的xrd图谱中,能观察到明显的pt晶体衍射峰峰和wc晶体衍射峰,通过谢乐公式计算得到样品中pt晶粒尺寸为11.20nm,wc晶粒尺寸为11.05nm。
如图6所示,在本实施例所制备pt/wc/mwcnts催化剂的sem照片中,能明显看到pt与wc颗粒均匀分布在mwcnts的表面,且颗粒大小与谢乐公式计算结果一致。
如图10硫酸溶液里的循环伏安曲线所示,实施例2所制备pt/wc/mwcnts催化剂的ecsa实施例2=57.06m2g-1;如图14甲醇溶液里的循环伏安曲线所示,本实施例所制备pt/wc/mwcnts催化剂的峰电流高出etek催化剂峰电流大约170mamg-1pt,通过计算if/ib,得到if/ib实施例2=1.16,本实施例所制备pt/wc/mwcnts催化剂的值明显高于etek催化剂的值,说明本实施例所制备pt/wc/mwcnts催化剂的抗co中毒能力高于商业etek催化剂。
【实施例3】
本实施例中,一种pt/金属碳化物/碳纳米材料催化剂的制备过程与实施例1类似,按照碳钨钼摩尔比160:1:1加入药品,其余步骤相同,经制备得到pt/wmoc/c催化剂。
如图3所示,在本实施例所制备pt/wmoc/c催化剂的xrd图谱中,能观察到明显的pt晶体衍射峰,通过谢乐公式计算得到样品中pt晶粒尺寸为8.94nm。
如图6所示,在本实施例所制备pt/wmoc/c催化剂的sem照片中,能明显看到pt与金属碳化物颗粒分布均匀,且颗粒大小与谢乐公式计算结果一致,而在xrd图谱中并未出现金属碳化物的特征峰,证明金属碳化物以一种不定型的形式存在,即pt与碳化物实现原子级接触。
如图11硫酸溶液里的循环伏安曲线所示,本实施例所制备pt/wmoc/c催化剂的ecsa实施例3=98.69m2g-1;如图15甲醇溶液里的循环伏安曲线所示,本实施例所制备pt/wmoc/c催化剂的峰电流高出etek催化剂峰电流大约160mamg-1pt,通过计算if/ib,得到if/ib实施例三=1.24,本实施例所制备pt/wmoc/c催化剂的值明显高于etek催化剂的值,说明本实施例所制备pt/wmoc/c催化剂的抗co中毒能力高于商业etek催化剂。
【实施例4】
本实施例中,一种pt/金属碳化物/碳纳米材料催化剂的制备过程与实施例1类似,加入的碳源为mwcnts,其余步骤相同,经制备得到pt/wmoc/mwcnts催化剂。
如图4所示,在本实施例所制备pt/wmoc/mwcnts催化剂的xrd图谱中,能观察到明显的pt晶体衍射峰,通过谢乐公式计算得到样品中pt晶粒尺寸为10.62nm。
如图6所示,在本实施例所制备pt/wmoc/mwcnts催化剂的sem照片中,能明显看到pt与金属碳化物颗粒分布均匀,且颗粒大小与谢乐公式计算结果一致,而在xrd图谱中并未出现金属碳化物的特征峰,证明金属碳化物以一种不定型的形式存在,即pt与碳化物实现原子级接触。
如图12硫酸溶液里的循环伏安曲线所示,本实施例所制备pt/wmoc/mwcnts催化剂的ecsa实施例4=178.45m2g-1;如图16甲醇溶液里的循环伏安曲线所示,本实施例所制备pt/wmoc/mwcnts催化剂的峰电流高出etek催化剂峰电流大约190mamg-1pt,通过计算if/ib,得到if/ib实施例四=1.04,本实施例所制备pt/wmoc/mwcnts催化剂的的值明显高于etek催化剂的值,说明本实施例所制备pt/wmoc/mwcnts催化剂的的抗co中毒能力高于商业etek催化剂。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!