一种软磁粉末复合物、磁粉芯材料及基于有机物长成的绝缘包覆方法
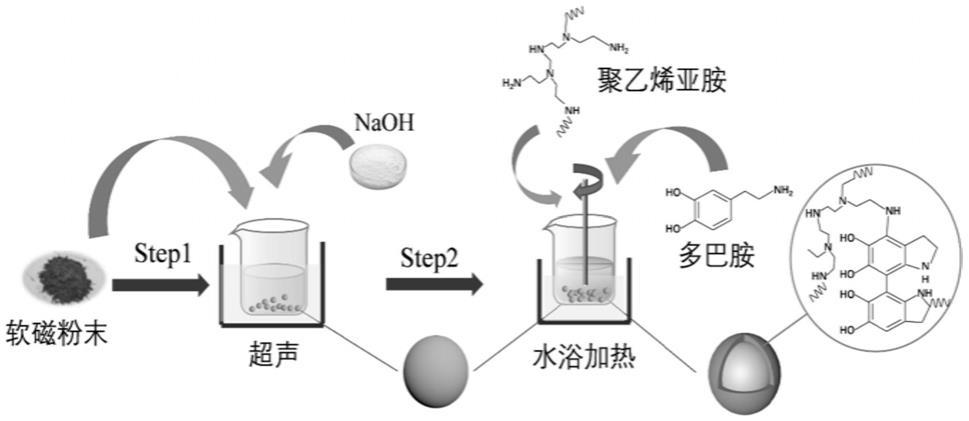
1.本技术具体涉及一种软磁粉末复合物、磁粉芯材料及基于有机物长成的绝缘包覆方法,属于磁粉芯制备技术领域。
背景技术:
2.磁粉芯是由软磁粉末与绝缘介质混合制备而成的一种软磁复合材料。软磁粉末和绝缘介质的压制成型技术很大程度上决定了磁粉芯的致密程度,磁粉和绝缘包覆材料的添加量和体积分数、绝缘包覆膜厚、绝缘包覆层的均匀度、包覆层与基体的结合效果对磁粉芯的电阻率、磁导率、矫顽力、磁滞损耗、涡流损耗等性能至关重要。
3.涡流损耗是以金属及合金粉末等为基体的复合材料中功率损耗的主要原因。涡流损耗与工作频率的二次方成正比,与电阻率成反比。通常,金属粉末及合金粉末在软磁磁粉芯等应用背景下,都需要进行绝缘包覆处理以降低涡流损耗。因此,粉末颗粒的绝缘包覆工艺就要求包覆后粉末具有高电阻率、良好热稳定性、包覆的高完整性及高均匀度等特性。
4.目前常用的包覆方法有有机、无机、有机无机复合包覆以及磁性相包覆等四类。目前大部分包覆方法停留在无机包覆中的磷酸盐包覆和金属氧化物包覆以及有机包覆中的硅树脂包覆,这些现有绝缘包覆方法存在制备难度大,工艺稳定性差、环境污染度高,人体危害性大、包覆层易破裂、温度稳定性低及包覆均匀度差等弊端。
5.(1)现有的有机物包覆方法大多采用溶胶凝胶法和化学气相沉积法,包覆层较厚,且厚度较难调控,使磁导率恶化。有机包覆中常用的有机包覆物为树脂类,包括环氧树脂、酚醛树脂和有机硅树脂等。有机树脂与金属磁粉的极性不同,通常采用硅烷偶联剂对磁粉进行表面预处理,改善树脂与磁粉之间的浸润情况和结合度。
6.有机包覆中热塑性树脂较早得到应用,但其缺点也很快暴露出来,其使用时易溶于工业溶液且熔点较低限制热处理温度等,故而热固性树脂如环氧树脂又得到应用与推广,但其热稳定性也不够高,并且大多数树脂抗老化能力弱,使用时效短。
7.另一方面,从软磁粉末与树脂间亲和力来说,树脂是憎水性,软磁粉末是亲水性的,因而二者之间的表面特性是相反的,减弱了软磁粉末与有机包覆剂之间的润湿,导致有机包覆剂不能完全均匀的包覆在软磁粉末表面,使得有效颗粒尺寸增加,电阻率降低,涡流损耗增加。
8.(2)由于有机包覆剂的熔点普遍较低,无机包覆剂逐渐涌现。目前常用的无机包覆方法有磷酸盐包覆、金属氧化物包覆、铁氧体包覆等,大致可分成两类,一类是用磷酸、铬酸或具有氧化性的硝酸等与磁粉直接反应(钝化),在其表面生成具有高电阻率的绝缘包覆膜;另一类则是磁粉不参与绝缘层形成的溶胶凝胶法,如al(no3)3水解沉淀在fe-si-al磁粉表面生成al(oh)3,经退火热处理后形成了以al2o3为主的包覆层。
9.磷酸盐包覆:该类方法应用较早,但磷酸盐包覆材料的电阻率较低,在高频下涡流损耗大,镀液稳定性差且操作复杂,同时磷酸等有害物质对人体危害性大,不适合大规模工
业生产和应用。
10.金属氧化物包覆:该类方法是目前较为主流的包覆方法,在于其一般有较高的热稳定性与较好的电绝缘能力,满足热处理要求,但其脆性大,在压制过程中绝缘包覆层易破裂,导致包覆效果变差。
11.铁氧体包覆:该类方法能够将包覆材料对基体磁学性能的影响降到最低,兆赫范围内能够保证磁学性能的稳定因而得到广泛应用,但铁氧体包覆均匀度低,压坯致密度低。
12.(3)有机无机复合包覆使无机和有机包覆的优势相结合,近年来被广泛应用与研究,在树脂中加入纳米sio2和znso4,抑制其在固化过程中产生的水分子,从而提高树脂的稳定性,并通过凝胶溶胶法在树脂中原位生成纳米二氧化硅,使树脂和无机纳米粒子之间形成稳定的化学键。
13.(4)磁性相包覆将具有铁磁性磁学性能的材料用来做包覆/粘接剂,利用其磁学特性来增强或保持软磁复合材料中主体粉末颗粒间的软磁耦合效果,以及提高或尽量维持整体饱和磁感应强度等性能调控。
技术实现要素:
14.本技术通过共沉积长成的方式在金属以及合金软磁粉末表面沉积聚多巴胺/聚乙烯亚胺层,对软磁粉末颗粒进行绝缘包覆。其绝缘膜很薄且厚度可控性强,分布均匀性高,可以实现电阻率提高,有效减少磁粉颗粒间涡电流,降低磁粉芯的涡流损耗;同时其高均匀性薄绝缘膜可以减少绝缘粘接工艺中其他非磁性物质的添加比例,可以提高软磁颗粒间的软磁耦合效果,有效提升磁导率和饱和磁感应强度及降低磁滞损耗。
15.根据本技术的一个方面,提供了一种软磁粉末复合物,包括软磁粉末与绝缘包覆层,绝缘包覆层包裹软磁粉末;
16.绝缘包覆层的材料由盐酸多巴胺(以下简称多巴胺)与聚乙烯亚胺交联反应制备获得;软磁粉末选自含有fe元素的金属和/或合金材料。
17.可选地,绝缘包覆层采用多巴胺与聚乙烯亚胺共沉积的方法包裹软磁粉末。
18.可选地,多巴胺的分子量为189.64,聚乙烯亚胺(pei)的重均分子量为600~70000。
19.可选地,绝缘包覆层的厚度小于1000nm。
20.可选地,绝缘包覆层的厚度为100~1000nm。
21.可选的,绝缘包覆层的厚度上限独立地选自900nm、800nm、700nm、600nm、500nm、300nm,下限独立地选自200nm、300nm、500nm、600nm、700nm。
22.可选地,软磁粉末颗粒累积分布为50%的粒径d
50
《75μm。
23.可选地,软磁粉末的粒径为15~45μm。
24.根据本技术的一个方面,提供了一种软磁粉末复合物的制备方法,包括以下步骤:
25.(1)将软磁粉末在酸性或碱性溶液中超声后,用去离子水清洗至中性,增加了软磁粉末表面粗糙度,放入聚乙烯亚胺溶液中超声处理,得预处理后的软磁粉末;
26.(2)配制含有多巴胺和聚乙烯亚胺的tris-hcl缓冲溶液,超声处理,得共沉积溶液a;
27.(3)将预处理后的软磁粉末浸于共沉积溶液a中,25~60℃下恒温搅拌8~30h,得
所述软磁粉末复合物。
28.可选地,软磁粉末在酸性或碱性溶液中超声的时间为30~40min;聚乙烯亚胺溶液的浓度为50mg/ml,放入聚乙烯亚胺溶液中超声处理的时间为30~40min。
29.可选地,tris-hcl缓冲溶液的浓度为0.05~1mol/l,ph值为8.5~10。
30.可选地,步骤(2)中,多巴胺与聚乙烯亚胺的质量比为(0.5~4):1。
31.可选地,步骤(3)中,共沉积溶液a与软磁粉末的配比为10~20ml/g。
32.可选的,共沉积溶液a与软磁粉末的配比上限独立地选自18、17、16、14ml/g,下限独立地选自11、12、13、15ml/g。
33.多巴胺与聚乙烯亚胺的质量比独立地选自0.7:1、1:1、1.2:1、1.5:1、1.8:1、2:1、2.5:1、3:1、3.5:1、4:1,或上述两点间的任意值。
34.可选地,聚乙烯亚胺(pei)的重均分子量为600~70000;软磁粉末的尺寸为15~45μm。
35.根据本技术的一个方面,提供了一种磁粉芯材料,磁粉芯材料包括上述软磁粉末复合物或上述制备方法制备得到的软磁粉末复合物。
36.可选地,磁粉芯材料还包括粘接剂;绝缘粘接剂与软磁粉末复合物的质量比为(0.5~4):100。
37.绝缘粘接剂与软磁粉末复合物的质量比独立地选自3.5:100、3:100、2.5:100、2:100、1.5:100、1:100,或上述两点间的任意值。
38.可选的,绝缘粘接剂选自有机硅树脂、环氧树脂、酚醛树脂、硬脂酸锌中的至少一种。
39.可选地,磁粉芯材料的磁导率在20khz内可稳定在40左右,饱和磁感应强度为0.9t,铁损w
0.1/100k
最低可达511.6mw/cm3。
40.根据本技术的一个方面,提供了一种磁粉芯材料的制备方法,将上述任一软磁粉末复合物或上述任一制备方法制备得到的软磁粉末复合物与绝缘粘接剂混合,压制成型后进行热处理,得所述磁粉芯材料。
41.可选地,压制成型的方式为冷压成型,冷压成型工艺参数为压强1800mpa,保压时间60s;
42.热处理的温度为300~500℃,热处理的时间为1h。
43.作为本技术的一种具体实施方式,一种软磁粉末复合物的制备方法,包括以下步骤:
44.(1)获取软磁粉末;
45.(2)将naoh溶于去离子水中配制待用的0.1~1mol/l naoh溶液;
46.(3)将软磁粉末加入上述naoh溶液中超声30min后取出软磁粉末,并用去离子水清洗直至软磁粉末的水溶液ph值呈中性。
47.(4)将三羟甲基氨基甲烷(tris)溶于去离子水中超声分散配制待用的0.05~0.1mol/l tris溶液;
48.(5)取浓盐酸溶液(hcl)加入去离子水中使之稀释;
49.(6)将上述稀释后的盐酸溶液加入tris溶液调节其ph值至8.5~10;
50.(7)将多巴胺(da)和聚乙烯亚胺(pei)加入上述ph值为8.5~10的tris溶液中,通
过超声分散均匀得到tris-hcl/da+pei溶液;
51.(8)将上述经过naoh溶液处理并清洗后的软磁粉末加入tris-hcl/da+pei溶液中机械搅拌,同时水浴加热并烘干,得到所述软磁粉末复合物。
52.本技术中,多巴胺加入到tris-hcl溶液中后溶解在其中,逐渐长成聚多巴胺聚集体,同时氧化后与聚乙烯亚胺中氨基反应;聚乙烯亚胺本身为白色粘稠状液体,加入tris-hcl溶液后经过超声溶解并完全分散在该溶液中。
53.采用多巴胺与聚乙烯亚胺共沉积包覆方法,多巴胺中酚羟基被氧化成醌基后,聚乙烯亚胺中的氨基能够迅速与多巴胺发生交联反应,其中聚乙烯亚胺既参与多巴胺的交联反应形成共聚包覆层,又给共聚包覆层表面提供氨基,且其一方面能够加速与聚多巴胺的共沉积速率,另一面通过破坏聚多巴胺内部非共价键作用力而抑制其过度聚集,从而得到表面形貌更加均匀平整的共沉积层。
54.本技术中,涉及反应原料时均为多巴胺+聚乙烯亚胺(da+pei),涉及沉积反应及反应后包覆层时均为聚多巴胺/聚乙烯亚胺(pda/pei)。
55.在本技术中,
56.(1)磁损耗(铁损w):金属磁粉芯在交变磁场中工作时,一方面被磁化,一方面将产生能量的损耗,能量损耗总值叫做磁损耗。磁损耗由磁致损耗、涡流损耗、剩余损耗组成。
57.(2)涡流损耗:当外磁场随着频率变化时,由于电磁感应,材料中将产生感应电流,从而引起涡流损耗,交变磁场频率越高,涡流越大。绝缘包覆层能够将涡流阻隔在磁粉颗粒内部从而降低磁粉颗粒间的涡流,进而降低磁粉芯涡流损耗。
58.(3)磁致损耗:交变磁场中对磁性材料进行磁化,由于磁致现象而产生的功率损耗,与磁粉芯矫顽力有关,非磁性物质添加过多则会引起矫顽力上升进而使磁滞损耗增加,故而本技术中纳米及微米级绝缘包覆膜能够保持较低磁滞损耗。
59.(4)剩余损耗:在低频弱场中,剩余损耗主要是磁后效损耗,在高频情况下,剩余损耗主要是尺寸共振损耗、畴壁共振损耗和自然共振损耗。
60.(5)电阻率:反应物质对电流阻碍作用的属性,与材料种类、压力、温度、磁场等因素相关。
61.(6)矫顽力:磁性材料在饱和磁化后,当外磁场退回到零时其磁感应强度b并不退到零。需要在原磁化场相反方向加上一定大小的磁场才能使磁感应强度退回到零,该磁场称为矫顽力。软磁粉末以外物质的种类和添加比例等都将影响磁粉颗粒间的软磁性耦合效果,非磁性物质的添加和增量及压制密度的过小也会提升磁粉芯的矫顽力,引起磁滞损耗的增加。
62.(7)饱和磁感应强度:磁性材料在外加磁场中被磁化时所能够达到的最大磁化强度叫做饱和磁感应强度,为实现电子元器件的小型化提供条件。饱和磁感应强度直接影响了磁芯体的功率输出能力,是作为功率型器件的核心性能,为实现电子元器件的小型化提供条件。对于磁粉芯来说,相同的主体磁粉情况下,参与磁化的主体软磁粉末比例越高,其饱和磁化强度越高。本技术中超薄的绝缘膜厚保持了非晶磁粉芯良好的高饱和磁感应强度。
63.本技术能产生的有益效果包括:
64.本技术采用的绝缘包覆方法能够在一定温度范围内实现高稳定性,绝缘包覆层结
构不发生变化,成膜均匀性高致密性好,膜厚薄且可控(通过调节时间和浓度及溶液ph值等参数实现对磁粉表面膜厚的控制),有效隔绝颗粒间涡流。在提高电阻率的同时,保证基体的高磁导率和高饱和磁化强度等良好的本征软磁特性,同时包覆层粘附力大,能够降低绝缘粘接剂无效添加,易于压制成型,减小非磁性相稀释效应,进一步提高基体磁学性能,且危害性小,制备过程操作简单易重复。
65.将绝缘包覆后磁粉在1800mpa下压制成型,300~500℃去应力退火,扫描电子显微镜下软磁粉末表面可见粗糙包覆层,不同绝缘包覆效果的磁粉颗粒在透射电子显微镜下可以观测到其绝缘层厚度范围为100~1000nm。将得到的磁粉芯磁学性能与同样制备条件下未进行多巴胺/聚乙烯亚胺共沉积绝缘包覆的磁粉相比,铁损w
0.1/50k
降低17~43%(w
0.1/50k
是指在磁感应强度为0.1t、频率为50khz的工况条件下测得的铁损),初始磁导率μ稳定在兆赫兹内。
附图说明
66.图1为本技术多巴胺/聚乙烯亚胺共沉积包覆流程图;
67.图2为原粉及实施例1~4软磁粉末复合物表面形貌sem图;
68.图3为实施例1~4软磁粉末复合物截面形貌fib-sem图。
具体实施方式
69.下面结合实施例详述本技术,但本技术并不局限于这些实施例。
70.如无特别说明,本技术的实施例中的原料均通过商业途径购买。其中,有机硅树脂购于东莞三金顔料有限公司的sj-106聚酯改性有机硅树脂。
71.实施例1
72.将30g的粒径d
50
为25μm的fe-si-b-c-cr合金软磁原粉放入500ml0.5mol/l的naoh溶液中超声处理30min后取出用去离子水清洗至ph值呈中性;
73.将上述取出的软磁粉末放入60ml浓度为50mg/ml的聚乙烯亚胺(分子量1800)溶液中超声处理30min;
74.将多巴胺和聚乙烯亚胺按照质量比为2:1的比例同时放入500ml0.05mol/l的tris-hcl溶液中,其中多巴胺与聚乙烯亚胺的浓度分别8mg/ml与4mg/ml,得到tris-hcl/da+pei溶液,并将上述在聚乙烯亚胺溶液超声处理后软磁粉末加入tris-hcl/da+pei溶液;
75.将上述混合溶液在40℃恒温下加热的同时机械搅拌24h,干燥后得到软磁粉末复合物;
76.其软磁粉末复合物表面形貌如图2中的实施例1图,截面形貌如图3中的实施例1图,聚多巴胺/聚乙烯亚胺绝缘包覆层平均厚度为415
±
100nm,包覆层上层为铂保护层(fib实验中保护有机包覆层不被离子束轰击融化),下层为软磁粉末基体;
77.将粘接剂(有机硅树脂)与上述软磁粉末复合物混合,二者质量比为2:100,并溶于丙酮溶液混合均匀后不断搅拌直至丙酮完全挥发,然后取出混合粉末放入真空干燥箱里60℃干燥3h;
78.将上述干燥后混合粉末放入液压成型机中,使用1800mpa压强并保压60s,压制成外径20.3mm、内径12.7mm的磁环压坯;
79.将上述磁环压坯放入真空热处理炉中450℃保温0.5h得到磁粉芯样品。
80.实施例2
81.将30g的粒径d
50
为25μm的fe-si-b-c-cr软磁原粉放入500ml浓度为0.5mol/l的naoh溶液中超声处理30min后取出用去离子水清洗至ph值呈中性;
82.将上述取出的软磁粉末放入60ml浓度为50mg/ml的聚乙烯亚胺(分子量600)溶液中超声处理30min;
83.将质量比为1:1的多巴胺和的聚乙烯亚胺同时放入0.1mol/l的tris-hcl溶液中,其中多巴胺与聚乙烯亚胺的浓度均为12mg/ml,得到tris-hcl/da+pei溶液,并将上述在聚乙烯亚胺溶液超声处理后的软磁粉末加入tris-hcl/da+pei溶液;
84.将上述混合溶液25℃恒温加热同时机械搅拌30h并干燥后得到软磁粉末复合物;
85.其软磁粉末复合物表面形貌如图2中的实施例2图,截面形貌如图3中的实施例2图,多巴胺/聚乙烯亚胺绝缘包覆层平均厚度为800
±
100nm,下层为软磁粉末基体;
86.将粘接剂(有机硅树脂)与上述软磁粉末复合物混合,其质量百分配比为2:100,并溶于丙酮溶液混合均匀后不断搅拌直至丙酮完全挥发,然后取出混合粉末放入真空干燥箱里60℃干燥3h;
87.将上述干燥后混合粉末放入液压成型机中,使用1800mpa压强并保压60s,压制成外径20.3mm、内径12.7mm的磁环压坯;
88.将上述磁环压坯放入真空热处理炉中480℃保温1h得到磁粉芯样品。
89.实施例3
90.将30g的粒径d
50
为25μm的fe-si-b-c-cr软磁原粉放入500ml浓度为0.5mol/l的naoh溶液中超声处理30min后取出用去离子水清洗至ph值呈中性;
91.将上述取出的软磁粉末放入60ml浓度为50mg/ml的聚乙烯亚胺(分子量600)溶液中超声处理30min;
92.将质量比为2:1的多巴胺和聚乙烯亚胺(分子量600)同时放入0.1mol/l的tris-hcl溶液中,其中多巴胺与聚乙烯亚胺的浓度分别8mg/ml与4mg/ml,得到tris-hcl/da+pei溶液;
93.将上述在聚乙烯亚胺溶液超声处理后软磁原粉加入上述tris-hcl/da+pei溶液60摄氏度恒温加热并机械搅拌8h并干燥后得到软磁粉末复合物;
94.将粘接剂(有机硅树脂)与上述软磁粉末复合物混合,其质量百分配比为2:100,并溶于丙酮溶液混合均匀后不断搅拌直至丙酮完全挥发,然后取出混合粉末放入真空干燥箱里60℃干燥3h;
95.其软磁粉末复合物表面形貌如图2中的实施例3图,截面形貌如图3中的实施例3图,聚多巴胺/聚乙烯亚胺绝缘包覆层平均厚度为680
±
100nm。
96.将上述干燥后混合粉末放入液压成型机中,使用1800mpa压强并保压60s,压制成外径20.3mm、内径12.7mm的磁环压坯;
97.将上述磁环压坯放入真空热处理炉中480℃保温1h得到磁粉芯样品。
98.实施例4
99.将30g的粒径d
50
为25μm的fe-si-b-c-cr软磁原粉放入500ml浓度为0.5mol/l的naoh溶液中超声处理30min后取出用去离子水清洗至ph值呈中性;
100.将上述取出的软磁粉末放入60ml浓度为50mg/ml的聚乙烯亚胺(分子量600)溶液中超声处理30min;
101.将质量比为1:1的多巴胺和的聚乙烯亚胺(分子量600)同时放入0.1mol/l的tris-hcl溶液中,其中多巴胺与聚乙烯亚胺的浓度均为24mg/ml,得到tris-hcl/da+pei溶液,并将上述在聚乙烯亚胺溶液超声处理后的软磁粉末加入tris-hcl/da+pei溶液;
102.将上述混合溶液50℃恒温加热同时机械搅拌20h并干燥后得到软磁粉末复合物;
103.其软磁粉末复合物表面形貌如图2中的实施例4图,截面形貌如图3中的实施例4图,聚多巴胺/聚乙烯亚胺绝缘包覆层平均厚度为600
±
100nm,下层为软磁粉末基体;
104.将粘接剂(有机硅树脂)与上述软磁粉末复合物混合,其质量百分配比为2:100,并溶于丙酮溶液混合均匀后不断搅拌直至丙酮完全挥发,然后取出混合粉末放入真空干燥箱里60℃干燥3h;
105.将上述干燥后混合粉末放入液压成型机中,使用1800mpa压强并保压60s,压制成外径20.3mm、内径12.7mm的磁环压坯;
106.将上述磁环压坯放入真空热处理炉中480℃保温1h得到磁粉芯样品。
107.实施例2与实施例4为不同聚多巴胺/聚乙烯亚胺浓度的对比实验,由于多巴胺/聚乙烯亚胺的浓度差异造成反应溶液中聚多巴胺/聚乙烯亚胺纳米聚集体的颗粒大小,生长数量以及堆砌厚度都有不同,因而造成聚多巴胺/聚乙烯亚胺绝缘包覆层的厚度差异,故而对其相应磁粉芯的软磁性能有一定影响。
108.实施例1与实施例3为不同聚乙烯亚胺分子量的对比实验,当pei分子量增加时,一方面提供相互作用的“锚点”间距离增加,导致其在形成的交联网络中的相对密度有所下降;另一方面,溶液中更容易形成聚多巴胺等枝化分子而非交联网络,故在图2中可见实施例1表面形貌较为粗糙,实施例3包覆层更厚。
109.对比例1
110.将硅酸钠与去离子水混合均匀,再加入醇醚磷酸酯并混合均匀,得到硅酸钠溶液;
111.取颗粒平均粒径为35μm的铁硅粉,将包覆炉炉温升到80℃后,向包覆炉内加入木质素磺酸盐,搅拌30min;
112.将硅酸钠溶液加入到金属软磁粉末中搅拌30min;
113.将包覆炉炉温升至150℃下烘烤60min,得到包覆粉末。
114.向包覆粉末中添加其1%重量的三氧化二铝和1%重量的硬脂酸锌润滑剂并混合均匀;
115.将混合均匀的软磁粉末在成型压力2300mpa下模压成外径27.0mm、内径14.7mm磁环压坯,在800℃的h2气氛保护下保温90min,得到硅酸钠包覆磁粉芯。
116.对比例2
117.将30g的粒径d
50
为25μm的fe-si-b-c-cr软磁原粉与有机硅树脂按照100:2的质量比共同溶于丙酮溶液,均匀混合并不断搅拌至丙酮溶液挥发完全,取出该混合粉末放入真空干燥箱里60℃干燥3h;
118.将上述干燥后混合粉末放入液压成型机中,使用1800mpa压强并保压60s,压制成外径20.3mm、内径12.7mm的磁环压坯。
119.将上述磁环压坯放入真空热处理炉中480℃保温1h得到磁粉芯样品。
120.对比例3
121.取磷酸与去离子水混合均匀,制备出待用的磷酸溶液;
122.取颗粒平均粒径为38μm的铁硅铝粉(铁87.8wt.%、硅6.8wt.%、铝5.4wt.%)软磁粉末放入包覆炉中开启搅拌;
123.将磷酸溶液加入到包覆炉中,继续搅拌30min;加入粒径小于100nm的纳米碳酸钙,继续搅拌30min;
124.包覆炉升温到120℃,继续搅拌至干燥,得到预处理后的磁粉;
125.加入预处理后的磁粉重量的0.3%的硬脂酸锌,搅拌均匀;
126.按2300mpa的压力压制成外径27.0mm、内径14.7mm的磁环压坯;
127.在700℃的n2气氛保护下保温30min,进行退火热处理,得到磷酸和纳米碳酸钙二级包覆磁粉芯。
128.测试例
129.采用湖南联众2335a型宽频能量分析仪测量磁粉芯材料在磁感应强度为0.1t、频率为50khz的工况条件下的铁损,其中线圈匝数n1=20,n2=5;
130.采用安捷伦4294a精密阻抗分析仪在频率范围为1khz~110mhz,外加磁场h≈0.1a/m的工况条件下测量磁粉芯的磁导率,其中线圈匝数n1=20,n2=5。
131.参数铁损w
0.1/50k
(mw/cm3)初始磁导率μ实施例131634实施例222932实施例325646实施例426835对比例189826对比例237139对比例352026
132.由上表可以看出,在励磁为0.1t、频率为50khz的条件下测得的铁损及初始磁导率数据对比中,本发明专利中实施例1、2、3、4比对比例1、2、3显示更低的铁损,同时实施例1、2、3、4比对比例1、3显示更高的初始磁导率。对比例2的初始磁导率为39,依然比本发明专利中的实施例3要低。综上所述,结合本发明专利中的绝缘包覆工艺、粘接包覆工艺和冷压成型工艺在制备磁粉芯应用方面具有明显的优势,可以获得高初始磁导率、低铁损的,初始磁导率并具备优异的高频稳定特性。
133.以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1