一种原位固态化金属有机框架基固态电解质及其制备方法和应用
1.本发明属于钠金属电池领域,涉及一种原位固态化金属有机框架基固态电解质的制备及在钠金属电池中的应用。
背景技术:
2.随着现代社会对便携式电子设备和电动汽车的需求日益增长,人们对高性能储能设备的需求也越来越大。锂金属电池在二次电池中具有最高的能量密度,然而锂元素和正极中的过渡金属如钴元素丰度有限。钠金属电池具有与锂金属电池相似的化学性质,同时钠资源丰富且成本低廉,因此成为目前研究的热点。然而,目前报道的钠金属电池通常采用有机电解液,而这类电解液易泄漏、胀气,甚至起火或爆炸。相比于液态电解质,固态电解质可以有效避免漏液、电池内部短路等问题,是一种高安全性的电解质体系。因此,构筑固态钠金属电池是解决安全问题的重要方式。
3.作为固态储能器件的重要组成部分,固态电解质被广泛用于锂离子电池、钠离子电池、锂-硫电池等诸多电池体系。然而,低离子电导和界面相容性差仍然成为限制固态电解质应用的两大主要因素。无机固态电解质例如li
10
gep2s
12
(advanced materials, 2022, 9, 2200822)即使在室温下也表现出极高的离子电导率(通常为10-2 s cm-1
量级)。然而无机固态电解质与电极的界面稳定性较差。与无机固态电解质相比,聚合物电解质在构建紧密的界面、低成本和良好的柔韧性等方面具有更大的优势。原位固态技术是一种简单高效的聚合物电解质的合成策略,具体为将由单体、引发剂和液体电解质形成前驱体溶液注入电池中,并填充电极中的所有孔隙,待加热聚合后在电极和电解质之间形成紧密的界面。聚(乙二醇)二丙烯酸酯基(pegda)单体价格低廉,原位固化技术工艺简单。因此,pegda已经成为实现聚合物原位固态化的良好选择。然而,目前的研究报道中,原位固态化的pegda基聚合物电解质中阳离子和阴离子与聚合物基体之间具有更大配位作用,因此导致电解质具有较低的离子迁移数(energy environmental materials, doi:10.1002/eem2.12447;cn 111554974 a)。所以,改善pegda基聚合物电解质的迁移数势在必行。
4.金属有机框架材料(mofs)是由金属离子和有机配体通过配位键形成的有机-无机杂化材料。mofs材料具有丰富的金属位点和有机官能团,并应用于气体分离、化学催化、药物传输等多个领域。同时,鉴于特殊的结构,mofs材料在固态电解质领域逐渐发挥重要作用,并在锂离子电池、锂-硫电池等电池体系均有应用。鉴于mofs材料孔结构的多样性和可调节性,mof基电解质可为阳离子提供离子传输的唯一通道,从而促进阳离子的均匀传输与分布(energy storage materials, 2022, 47, 262)。因此,mofs材料与聚合物复合有利于改善聚合物电解质的离子电导率。专利cn 112670565 a制备了含氨基的高比表面积mof基复合凝胶固态电解质,从而来提高聚合物电解质的离子电导率,但是该专利并未解决mof电解质与电极界面相容性差的问题。
技术实现要素:
5.为解决上述技术问题,本发明提出一种原位固态化金属有机框架基固态电解质的制备及在钠金属电池中的应用。
6.本发明的技术方案是这样实现的:一种原位固态化金属有机框架基固态电解质的制备方法,步骤如下:(1)制备mof基体膜:将zrcl4溶于含乙酸的n,n-二甲基甲酰胺(dmf)中,充分搅拌后为溶液a。将2-磺酸基对苯二酸单钠溶于浓盐酸中,搅拌后为溶液b。然后混合a和b溶液,搅拌15分钟后置于120 ℃反应釜中。24小时后取出,并离心干燥后得磺酸功能化mof材料,再与ptfe充分混合,擀制成膜并制成直径为16 mm的圆片;(2)制备前驱体溶液:将pegda加热溶解,并依次加入丁二腈、钠盐和氟代碳酸乙烯酯,加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌;(3)制备原位固态化mof基电解质:将步骤(2)的前驱体溶液注入步骤(1)的圆片中,在烘箱内静置数小时即得原位固态化金属有机框架基固态电解质。
7.进一步的,所述步骤(1)中氯化锆与2-磺酸基对苯二酸单钠的摩尔比为1:(0.5~2),氯化锆与乙酸的摩尔比为1:(10~50),2-磺酸基对苯二酸单钠与浓盐酸摩尔比为1:(0.1~2)。
8.进一步的,所述磺酸功能化mof材料与ptfe的摩尔比为(6~9):1,圆片的直径为16mm,厚度为50~150 μm。
9.上述的磺酸功能化mofs的s=o键和pegda的含氧官能团均对钠离子产生相互作用,而这两种作用存在竞争,进而促使钠离子快速传输。丁二腈具有较大的介电常数,可以促进钠盐的解离,因此丁二腈与钠盐的混合物具有较高的离子电导率。同时,聚乙二醇二丙烯酸酯的原位聚合促使电极与电解质的良好界面相容性。
10.进一步的,所述步骤(2)中钠盐为双三氟甲磺酰亚胺钠、高氯酸钠、六氟磷酸钠、双氟磺酰亚胺钠或四氟硼酸钠中的一种或多种。
11.进一步的,所述聚乙二醇二丙烯酸酯、丁二腈和偶氮二异丁腈的重量比为100:(10~70):(0.1~1);丁二腈和钠盐的摩尔比为1:(10~30),前驱体溶液中氟代碳酸乙烯酯的添加质量百分比为2~10 wt%。
12.上述的方法制备的原位固态化金属有机框架基固态电解质,所述原位固态化金属有机框架基固态电解质在常温下呈现准固态。
13.含有上述的原位固态化金属有机框架基固态电解质的钠金属电池,其在充放电循环100周后的容量保持率达到97.3 %。
14.上述的钠金属电池的制备方法,步骤如下:将正极片、步骤(1)的圆片和负极片依次分层放入电池中,并注入前驱体溶液,封装后置于烘箱内,静置后即得钠金属电池。
15.进一步,所述正极片的活性物质为普鲁士蓝、过渡氧化物、聚阴离子类、硫和硫化聚丙烯腈中的一种或几种;负极片为钠片;钠金属电池为扣式电池或软包电池。
16.优选的,所述的烘箱温度为50~80℃,静置时间为1~12 h。
17.本发明具有以下有益效果:1、本发明选用的磺酸功能化mofs(uio-66-so3h)的孔尺寸为12
ꢀå
,且稍大于电解质阴离子的尺寸。因此,基于空间效应,本发明所选用的mofs可以利用孔结构限制电解质阴
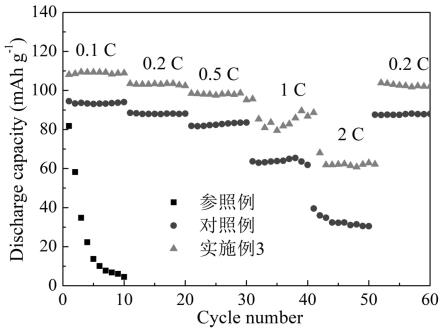
离子的移动,只允许钠离子的快速迁移。同时,如附图1所示,uio-66-so3h材料上具有丰富的磺酸官能团,同时磺酸官能团上的s=o键对钠离子具有强的相互作用。鉴于pegda基聚合物中含氧官能团对钠离子也具有强相互作用,uio-66-so3h材料与pegda基聚合物均对钠离子产生相互作用,这两种相互作用存在竞争,进而促使钠离子快速传输,并提高pegda基聚合物电解质的迁移数。另外,丁二腈可以解离钠盐,并与钠盐复合具有较高的离子电导率。本发明所制备的原位固态化金属有机框架基固态电解质不仅可以促进钠离子均匀分布,而且可以促使钠离子快速传输。因此本发明的固态电解质具备较高的离子电导率。
18.2、本发明选用的原位固态技术是一种简单高效的聚合物电解质的合成策略,具体为将由pegda单体、引发剂和塑晶电解质形成前驱体溶液注入电池中,并填充电极中的所有孔隙,待pegda单体加热聚合后,在电极和电解质之间形成紧密的界面。这种原位固态技术不仅有效避免了mofs颗粒之间界面间隙大的问题,而且促使电极与电解质具有良好的界面稳定性。另外,pegda单体价格低廉,原位固化技术工艺简单,与现有电池制造工艺兼容。结合现有的技术问题,本技术以mofs材料为基体,通过擀制形成mof基体膜,利用mofs材料孔结构,为钠离子提供离子传输的唯一通道。利用原位固态技术将mof基体膜与pegda复合,通过与钠离子的键合作用以期实现高离子电导和良好界面相容性的mof基固态电解质。
19.3、本技术制备的原位固态化金属有机框架基固态电解质,在常温下呈现准固态,因此,使用本发明固态电解质的钠金属电池可以有效避免电池内部短路、电解液泄露等问题,进而显著提高了钠金属电池的安全性。另外,鉴于本发明的原位固态化金属有机框架基固态电解质具有较高的离子电导率,使用此电解质的钠金属电池具有良好的电化学性能。本发明制备方法简单易行,可用于实现工业级制造性能优异的高比能固态电池。
附图说明
20.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
21.图1为本发明电解质中钠离子传输机制示意图。
22.图2实施例3中前驱体溶液在放置12 h后的数码照片图。
23.图3为实施例3中mof基体膜的数码照片图。
24.图4为对照例、参照例和实施例3的电导率测试图。
25.图5为对照例、参照例和实施例3的钠对称电池在0.1 ma cm-2
电流密度下的循环图。
26.图6为对照例、参照例和实施例3的钠金属电池在0.1 c(1c=118 ma g-1
)倍率下的循环图。
27.图7为对照例、参照例和实施例3的钠金属电池的倍率性能图。
具体实施方式
28.下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,
本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
29.实施例1本实施例的原位固态化金属有机框架基固态电解质的制备方法,步骤如下:(1)制备mof基体膜:将0.29 g(1mmol)zrcl4溶于的15 ml dmf中,搅拌后加入570μl(10mmol)乙酸,充分搅拌后为溶液a。将0.165g(0.5 mmol)2-磺酸基对苯二酸单钠溶于20μl(0.1 mmol)浓盐酸中,搅拌后为溶液b。然后混合a和b溶液,搅拌15分钟后置于120 ℃反应釜中。24小时后取出,并离心干燥后得uio-66-so3h材料,再与ptfe以6:1的摩尔比充分混合,擀制成膜并制成直径为16 mm且厚度为50 μm的圆片;(2)制备前驱体溶液:将pegda加热溶解,并依次加入10 wt%丁二腈、高氯酸钠和2wt%氟代碳酸乙烯酯,其中丁二腈和高氯酸钠摩尔比为10:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.1);(3)制备原位固态化mof基电解质和全固态钠金属电池:将步骤(2)的前驱体溶液注入步骤(1)的圆片中,在50 ℃烘箱内静置1小时即得原位固态化mof基电解质。
30.实施例2(1)制备mof基体膜:将0.29 g(1mmol)zrcl4溶于的15 ml dmf中,搅拌后加入1.4 ml(20mmol)乙酸,充分搅拌后为溶液a。将0.33g(1mmol)2-磺酸基对苯二酸单钠溶于0.1 ml(0.5mmol)浓盐酸中,搅拌后为溶液b。然后混合a和b溶液,搅拌15分钟后置于120 ℃反应釜中。24小时后取出,并离心干燥后得uio-66-so3h材料,再与ptfe以8:1的摩尔比充分混合,擀制成膜并制成直径为16 mm且厚度为50 μm的圆片;(2)制备前驱体溶液:将pegda加热溶解,并依次加入50 wt%丁二腈、高氯酸钠和5wt%氟代碳酸乙烯酯,其中丁二腈和高氯酸钠摩尔比为20:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.2)。
31.(3)制备原位固态化mof基电解质和全固态钠金属电池:将步骤(2)的前驱体溶液注入步骤(1)的圆片中,在60 ℃烘箱内静置1小时即得原位固态化mof基电解质。
32.实施例3本实施例的原位固态化金属有机框架基固态电解质的制备方法,步骤如下:(1)制备mof基体膜:将0.29 g(1mmol)zrcl4溶于的15 ml dmf中,搅拌后加入2.1 ml(37 mmol)乙酸,充分搅拌后为溶液a。将0.33 g(1mmol)2-磺酸基对苯二酸单钠溶于0.2 ml(1 mmol)浓盐酸中,搅拌后为溶液b。然后混合a和b溶液,搅拌15分钟后置于120 ℃反应釜中。24小时后取出,并离心干燥后得uio-66-so3h材料,再与ptfe以9:1的摩尔比充分混合,擀制成膜并制成直径为16 mm且厚度为100 μm的圆片;(2)制备前驱体溶液:将pegda加热溶解,并依次加入60 wt%丁二腈、高氯酸钠和5wt%氟代碳酸乙烯酯,其中丁二腈和高氯酸钠摩尔比为20:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.1)。
33.(3)制备原位固态化mof基电解质和全固态钠金属电池:将步骤(2)的前驱体溶液注入步骤(1)的圆片中,在60 ℃烘箱内静置4小时即得原位固态化mof基电解质。
34.其中,图2为实施例3的前驱体溶液。从图中可以得知,前驱体溶液在静置24小时后变为固态,因此所制备的金属有机框架基电解质为固态电解质。图3为mof基体膜的数码照
片图,由图3可以得到本技术制备的膜具有良好的柔性。
35.实施例4本实施例的原位固态化金属有机框架基固态电解质的制备方法,步骤与实施例3相同,不同之处在于将步骤(2)改为:制备前驱体溶液:将pegda加热溶解,并依次加入70 wt%丁二腈、高氯酸钠和5wt%氟代碳酸乙烯酯,其中丁二腈和高氯酸钠摩尔比为20:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.1)。
36.实施例5本实施例的原位固态化金属有机框架基固态电解质的制备方法,步骤与实施例3相同,不同之处在于将步骤(2)改为:制备前驱体溶液:将pegda加热溶解,并依次加入60 wt%丁二腈、六氟磷酸钠和5wt%氟代碳酸乙烯酯,其中丁二腈和六氟磷酸钠摩尔比为20:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.1)。
37.实施例6本实施例的原位固态化金属有机框架基固态电解质的制备方法,步骤如下:(1)制备mof基体膜:将0.29 g(1mmol)zrcl4溶于的15 ml dmf中,搅拌后加入2.6 ml(45mmol)乙酸,充分搅拌后为溶液a。将0.66g(2mmol)2-磺酸基对苯二酸单钠溶于0.3 ml(1.5mmol)浓盐酸中,搅拌后为溶液b。然后混合a和b溶液,搅拌15分钟后置于120 ℃反应釜中。24小时后取出,并离心干燥后得uio-66-so3h材料,再与ptfe以9:1的摩尔比充分混合,擀制成膜并制成直径为16 mm且厚度为150 μm的圆片;(2)制备前驱体溶液:将pegda加热溶解,并依次加入10 wt%丁二腈、高氯酸钠和7wt%氟代碳酸乙烯酯,其中丁二腈和高氯酸钠摩尔比为25:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.8);(3)制备原位固态化mof基电解质和全固态钠金属电池:将步骤(2)的前驱体溶液注入步骤(1)的圆片中,在70 ℃烘箱内静置4小时即得原位固态化mof基电解质。
38.实施例7本实施例的原位固态化金属有机框架基固态电解质的制备方法,步骤如下:(1)制备mof基体膜:将0.29 g(1mmol)zrcl4溶于的15 ml dmf中,搅拌后加入2.9 ml(50mmol)乙酸,充分搅拌后为溶液a。将0.66g(2mmol)2-磺酸基对苯二酸单钠溶于0.4 ml(2mmol)浓盐酸中,搅拌后为溶液b。然后混合a和b溶液,搅拌15分钟后置于120 ℃反应釜中。24小时后取出,并离心干燥后得uio-66-so3h材料,再与ptfe以9:1的摩尔比充分混合,擀制成膜并制成直径为16 mm且厚度为150 μm的圆片;(2)制备前驱体溶液:将pegda加热溶解,并依次加入60 wt%丁二腈、高氯酸钠和10wt%氟代碳酸乙烯酯,其中丁二腈和高氯酸钠摩尔比为30:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:1);(3)制备原位固态化mof基电解质和全固态钠金属电池:将步骤(2)的前驱体溶液注入步骤(1)的圆片中,在80 ℃烘箱内静置4小时即得原位固态化mof基电解质。
39.对照例在手套箱内,以na
3v2
(po4)3为正极材料,金属钠为负极,将玻璃纤维隔膜置于正极
和负极之间并滴加与实施例3相同的前驱体溶液,组装成扣式电池。
40.参照例步骤与对照例相同,不同之处在于将前驱体溶液制备改为:将pegda加热溶解,并依次加入60 wt%丁二腈和高氯酸钠,其中丁二腈和高氯酸钠摩尔比为20:1,待加热搅拌1小时后再加入一定量的偶氮二异丁腈并充分搅拌(pegda与偶氮二异丁腈的重量比为100:0.1)。
41.实施效果例将na
3v2
(po4)3正极片、实施例3中制备的步骤(1)的圆片和钠金属负极片依次分层放入2032型扣式电池中,并注入实施例3中步骤(2)的前驱体溶液,电池封装后置于60 ℃烘箱内,静置4小时后即得固态钠金属电池,与参照例和对照例制备的扣式电池进行电化学测试。
42.测试方法:(1)电导率测试将对照例、参照例和实施例3中的电解质置于两个不锈钢片之间,并组装成纽扣电池。在电压振幅为5mv,频率范围为100 mhz-100khz条件下进行交流阻抗测试,测试结果如表1和图2所示。采用下面公式来计算所有电解质的离子电导率:σ=d/(rs),其中d为基体膜的厚度,r为电解质的本征阻抗,s为电解质的有效面积。
43.表1为对照例、参照例和实施例3的电导率如图4所示,相比于对照例和参照例,实施例3的电解质具有最小的本征阻抗值(r)。同时根据公式来计算离子电导率,结果如表1所示。实施例3的固态电解质具有最高的离子电导率,达4.27 ms/cm,这是一方面由于磺酸功能化mofs的s=o键和pegda的含氧官能团均对钠离子产生相互作用,而这两种作用存在竞争,进而促使钠离子快速传输。另一方面是因为丁二腈与钠盐的混合物具有较高的离子电导率。
44.(2)钠对称电池循环测试将对照例、参照例和实施例3中的电解质置于两个不锈钢片之间,并组装成纽扣电池,在60 ℃烘箱里静置4h。将上述所有电池置于0.1 ma cm-2
电流密度下进行钠沉积/溶解过程。由图5可以看出,参照例的钠对称电池在整个循环过程中电压波动比较大,其相应的电压差也从开始的1.91 v增加到4 v以上。这表明,参照例的电解质与钠的界面稳定性较差。在0.1 ma cm-2
的电流密度下,对照例的钠对称电池相对稳定些,其过电压为0.737 v。然而,实施例3的钠对称电池在整个循环过程中均具有稳定的电压波动,其过电压一直保持在0.246v左右,同时并没有出现短路。这表明实施例3的电解质与钠的稳定性得到了提高。
45.(3)电池循环性能测试将上述所有电池置于0.1 c(1c=118 mah/g)和2.3-3.9 v电压范围下进行充放电循环测试。由图6可以看出,实施例3中的钠金属电池在充放电循环100周后的容量保持率达到97.3 %,而参照例仅能循环10周。这说明使用原位固态化mof基电解质的电池具有优异的循环性能。
46.(4)电池倍率性能测试将上述纽扣电池在不同的倍率下进行充放电循环测试。由图7可知,实施例3的钠金属电池具有优异的倍率性能。在1 c倍率下,实施例3的放电比容量达到62.7 mah/g,而参照例不能在大倍率下正常充放电。
47.以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1