用于活化“干扰素基因刺激因子”-依赖性信号传导的组合物和方法与流程
发明背景
提供以下对发明背景的讨论仅仅是为了有助于读者理解本发明,并不承认描述或构成本发明的现有技术。
人体免疫系统总体上可分为两个分支,称为“先天免疫”和“适应性免疫”。免疫系统的先天分支通过包括补体系统和趋化因子/细胞因子系统在内的多种可溶性因子;和包括肥大细胞、巨噬细胞、树突细胞(DC)和自然杀伤细胞在内的多种特化细胞类型来主要负责初始炎症应答。相比之下,适应性免疫分支涉及延迟的和持续时间更长的抗体应答,以及CD8+和CD4+T细胞应答,所述应答在针对抗原的免疫记忆中起关键作用。免疫系统的第三分支可确定为涉及γδT细胞和具有有限T细胞受体谱系的T细胞(如NKT细胞和MAIT细胞)。
对于针对抗原的有效免疫应答而言,抗原呈递细胞(APC)必须加工抗原并且在适当MHC情境下将抗原展示给T细胞,然后将引起细胞毒性细胞和辅助T细胞的T细胞刺激。在抗原呈递之后,APC和T细胞上必须发生共刺激分子的成功相互作用,否则活化就会中止。GM-CSF和IL-12在许多肿瘤模型中充当有效促炎分子。例如,GM-CSF诱导骨髓前体细胞来增殖并分化成树突细胞(DC),但需要额外的信号来将其活化成熟为活化T细胞所必需的有效抗原呈递细胞。有效免疫疗法的障碍包括可限制适当大小和功能的细胞毒性CD8T细胞的诱导的对于靶抗原的耐受性、所产生的T细胞到达恶性细胞位点的输送不佳以及所诱导的T细胞应答的持久性不佳。
吞噬肿瘤细胞碎片的DC对用于主要组织相容性复合物(MHC)呈递的材料进行加工、上调共刺激分子的表达,并且迀移至局部淋巴节来刺激肿瘤特异性淋巴细胞。此途径导致与肿瘤相关抗原起反应的CD4+和CD8+T细胞的增殖和活化。实际上,此类细胞可常常在患者的血液、淋巴组织和恶性病灶中被检测到。
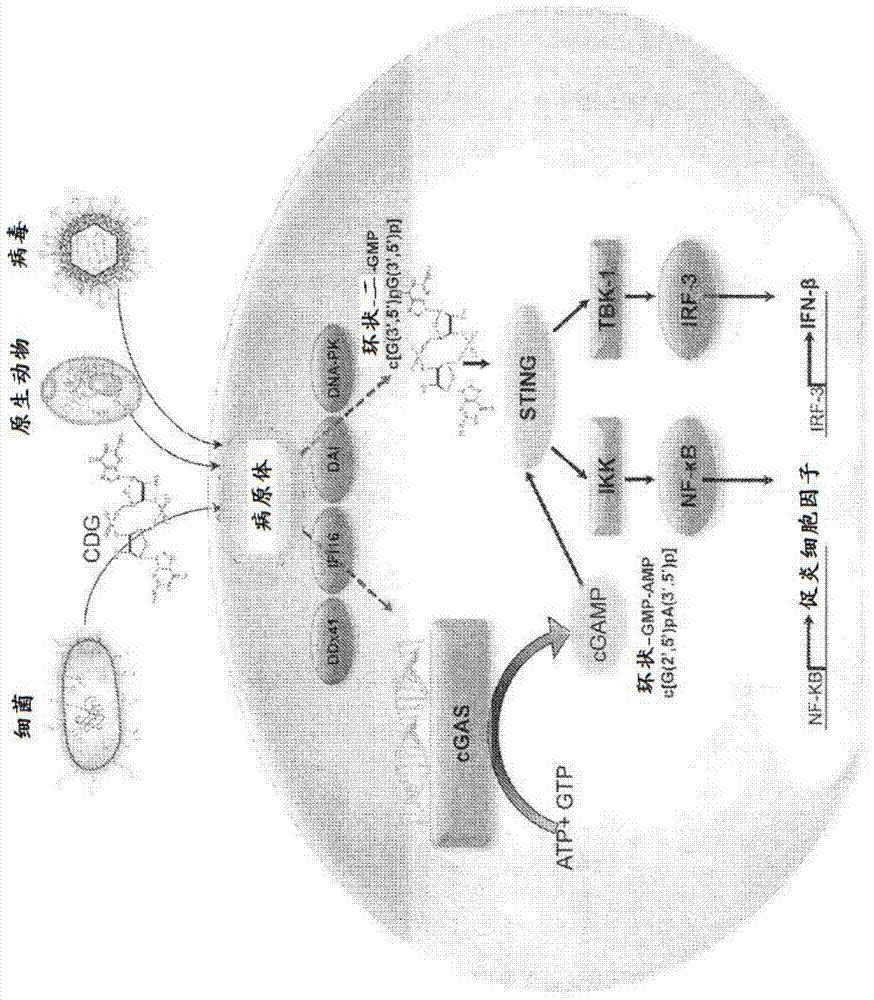
免疫逃避的潜在机制的新见解,以及通过与免疫检查点抑制剂或其它疗法进行组合来直接或间接地增强治疗性疫苗接种的效力的组合治疗方案已用作开发诱导有效抗肿瘤免疫的疫苗的基础。CDN环状-二-AMP(由单核细胞增多性李斯特菌(Listeria monocytogenes)产生)及其类似物环状-二-GMP(由侵肺军团菌(Legionella pneumophila))被宿主细胞识别为PAMP(病原体相关分子模式),其与称为STING的PRR(病原体识别受体)结合。STING是宿主哺乳动物细胞的细胞质中的衔接蛋白质,其活化TANK结合激酶(TBK1)—IRF3信号传导轴,导致诱导IFN-β和强烈活化先天免疫的其它IRF-3依赖性基因产物。现在认识到STING是宿主胞质监测途径的组分,其感测细胞内病原体的感染并且作为响应来诱导IFN-β的产生,从而导致显现适应性保护病原体特异性免疫应答,所述免疫响应由抗原特异性CD4和CD8T细胞以及病原体特异性抗体组成。环状嘌呤二核苷酸的实例相当详细地描述于例如美国专利No.7,709458和No.7,592,326;WO2007/054279;以及Yan等的Bioorg.Med.Chem Lett.18:5631(2008)中,其各自据此以引用的方式并入。
仍然需要用于治疗可能对传统治疗方法具有顽固性的疾病如癌症的免疫策略的改进组合物和方法。
发明概述
本发明的目的在于提供调节对疾病的免疫应答的组合物。
在第一方面,本发明提供组合物,其包含:
一种或多种环状嘌呤二核苷酸(“CDN”),其诱导干扰素基因刺激因子(“STING”)-依赖性I型干扰素产生。如下文所述,多种CDN可用于本发明中。优选的环状嘌呤二核苷酸包括但不限于c-di-AMP、c-di-GMP、c-di-IMP、c-AMP-GMP、c-AMP-IMP、c-GMP-IMP及其类似物中的一种或多种。此清单并非旨在进行限制。
根据本发明的环状嘌呤二核苷酸的通式结构如下:
其中R1和R2各自为嘌呤,并且结构旨在反映磷酸键可连接至核糖上的2’或3’位,并且未参与环键的2’或3’位的其余部分为–OH。本发明涵盖2’,5’,2’,5’CDN和2’,5’,3’,5’CDN。举例而言,具有2’-5’键的c-di-GMP是指上述的其中R1和R2各自为鸟嘌呤并且各磷酸键为2’-至-5’的分子。
出于本发明的目的,此通式结构经进一步修饰以引入赋予结合STING能力的取代基并且诱导STING-依赖性信号传导级联(最优选地诱导人STING-依赖性信号传导级联),从而诱导STING-依赖性I型干扰素产生以及其它共调控基因。举例而言,本发明提供组合物,其包含以下化合物:
其中各X独立地为O或S,并且R3和R4各自独立地为H或任选取代的1至18个碳和0至3个杂原子的直链烷基、任选取代的1至9个碳的烯基、任选取代的1至9个碳的炔基,或任选取代的芳基,其中取代在存在时可独立地选自C1-6烷基直链或支链、苄基、卤素、三卤代甲基、C1-6烷氧基、–NO2、–NH2、–OH、=O、其中R’为H或低级烷基的–COOR’、–CH2OH和–CONH2,其中R3和R4不同时为H。
在优选的实施方案,R3和R4中的一者或两者独立地包含通过细胞酯酶移除的前药离去基团。在某些实施方案中,R3和R4中的一者或两者为C6至C18脂肪酸酯。在某些实施方案中,R3和R4中的一者或两者选自肉豆蔻酰基、戊酰基、己酰基、庚酰基等。
在某些实施方案中,各X为S。在优选的实施方案中,当各X为S时,所述组合物包含一种或多种基本上纯的Sp,Sp、Rp,Rp、Sp,Rp或Rp,Sp立体异构体。
在某些实施方案中,R1和R2各自独立地选自腺嘌呤、鸟嘌呤、肌苷和黄嘌呤或其类似物。优选地,R1和R2各自独立地为腺嘌呤或鸟嘌呤。
如下文所述,根据本发明的环状嘌呤二核苷酸组合物与具有3’-5’键的c-di-GMP相比时,可诱导至少2倍、更优选5倍或10倍或更多倍的STING-依赖性I型干扰素产生。如本文中所提到的,最优选地,STING为人STING。在优选的实施方案中,根据本发明的基本上纯的环状嘌呤二核苷酸组合物活化人STING,但仅具有双-(3’,5’)键的环状嘌呤二核苷酸不会活化人STING。
在它们作为佐剂的作用中,在某些实施方案中,本发明的组合物可用作采用疫苗的治疗剂或预防性策略中的佐剂。因此,本发明的基本上纯的CDN或其前药或药学上可接受的盐可与一种或多种疫苗一起使用,所述疫苗经选择以刺激对一种或多种预定抗原的免疫应答。本发明的基本上纯的CDN或其前药或药学上可接受的盐可与此类疫苗一起提供或组合提供。
此类疫苗可包含含有目标抗原的灭活或减毒细菌或病毒、纯化抗原、经重组工程化以表达和/或分泌抗原的活病毒或细菌递送载体、载有抗原或转染有包含编码抗原的核酸的组合物的细胞的抗原呈递细胞(APC)载体、脂质体抗原递送媒介物或编码抗原的裸核酸载体。此清单并非旨在进行限制。举例而言,此类疫苗还可包含表达和分泌GM-CSF、CCL20、CCL3、IL-12p70、FLT-3配体中的一种或多种的灭活肿瘤细胞。
本发明的基本上纯的CDN或其前药或药学上可接受的盐可以包含药学上可接受的载体、佐剂和媒介物的制剂形式通过多种肠胃外和非肠胃外途径来施用给有需要的个体。优选的途径是肠胃外,并且包括但不限于皮下、静脉内、肌内、动脉内、皮内、鞘内和硬膜外施用中的一种或多种。还优选的是瘤内途径。特别优选的是通过皮下施用来施用。优选的药物组合物被配制为水性的或水包油乳液。
本发明的组合物可包含以下组分或与以下组分一起施用:一种或多种额外的药学活性组分如佐剂、脂质、双层间交联多层囊泡、可生物降解的聚(D,L-乳酸-共-乙醇酸)[PLGA]基或聚酸酐基纳米粒子或微粒以及纳米多孔粒子支撑的脂质双层、CTLA-4和PD-1途径拮抗剂、PD-1途径阻断剂、诱导先天免疫的灭活细菌(例如,灭活或减毒的单核细胞增多性李斯特菌)、通过Toll样受体(TLR)、(NOD)-样受体(NLR)、维甲酸诱导基因型(RIG)-I-样受体(RLR)、C-型凝集素受体(CLR)来介导先天免疫活化的组合物、病原体相关分子模式(“PAMP”)、化学治疗剂等。
在一个有关方面,本发明涉及诱导、刺激或辅助个体的免疫应答的方法。这些方法包括将本发明的基本上纯的CDN或其前药或药学上可接受的盐施用给个体。优选的施用途径为肠胃外。如上文所提到的,特别优选的是此类环状嘌呤二核苷酸的硫代磷酸酯衍生物。
在某些实施方案中,所述方法为癌症治疗方法。举例而言,本发明的基本上纯的CDN或其前药或药学上可接受的盐可单独提供,或与本领域中已知的一种或多种癌疫苗组合物一起提供或附加提供。接受此治疗的患者可患有选自以下的癌症:结直肠癌细胞、呼吸-消化道鳞状癌细胞、肺癌、脑癌、肝癌、胃癌、肉瘤、白血病、淋巴瘤、多发性骨髓瘤、卵巢癌、子宫癌、乳腺癌、黑素瘤、前列腺癌、胰腺癌细胞和肾癌。在其它实施方案中,所述方法为诱导、刺激或辅助对病原体的免疫应答的方法。
关于治疗患有癌症的哺乳动物,本文中所述的方法可包括在向哺乳动物施用有效量的本发明的基本上纯的CDN或其前药或药学上可接受的盐,任选地在向哺乳动物施用基本疗法以移除或杀灭表达癌抗原的癌细胞之前或之后。本发明的组合物可作为新辅助疗法施用;然而在优选的实施方案中,本发明的组合物是在基本疗法之后施用。在各个实施方案中,基本疗法包括移除哺乳动物的癌细胞的外科手术、杀灭哺乳动物体内癌细胞的放射疗法,或外科手术和放射疗法两者。
在其它实施方案中,本文中所述的方法可包括向哺乳动物施用有效量的本发明的基本上纯的CDN以治疗其中Th1至Th2免疫的转移可赋予临床益处的病症。细胞介导免疫(CMI)与产生细胞因子IL-2、干扰素(IFN)-γ和肿瘤坏死因子(TNF)-α的TH1CD4+T淋巴细胞相关。相比之下,体液免疫与产生IL-4、IL-6和IL-10的TH2CD4+T淋巴细胞相关。对于TH1应答的免疫偏离通常产生细胞毒性T淋巴细胞(CTL)、自然杀伤(NK)细胞、巨噬细胞和单核细胞的活化。一般而言,Th1应答对细胞内病原体(宿主细胞内的病毒和细菌)和肿瘤更有效,而Th2应答对细胞外细菌、寄生物(包括蠕虫和毒素)更有效。此外,预期先天免疫的活化可使T-辅助细胞1型和2型(Th1/Th2)免疫系统平衡正常化并且可抑制Th2型应答的过度反应,该过度反应会引起免疫球蛋白质(Ig)E-依赖性变态反应和变应性哮喘。
附图简述
图1描绘环状嘌呤二核苷酸(“CDN”)介导的信号传导。CDN(例如,c-di-AMP或c-di-GMP)诱导IFN-β的产生,通过与胞质受体STING(干扰素基因刺激因子)结合,和通过TBK-1/IRF-3途径来诱导信号传导,通过与IFN受体结合以及后续信号传导来导致DC的自分泌和旁分泌活化。
图2A描绘c-[G(2’,5’)pG(3’,5’)p]和二硫代衍生物的合成方案。
图2B描绘c-[A(2’,5’)pA(3’,5’)p]和二硫代衍生物的合成方案。
图2C描绘化合物10、20、21、22和23的结构。
图3A描绘化合物9a的1H-NMR结果。
图3B描绘化合物9a的COSY(3.5-6.0ppm 1H-轴)结果。
图3C描绘化合物9a的HMBC(3.0-5.5ppm 1H-轴)结果。
图3D描绘化合物21的1H-NMR结果。
图3E描绘化合物21的COSY(3.5-6.0ppm 1H-轴)结果。
图3F描绘化合物21的HMBC(0-9.5ppm 1H-轴)结果。
图3G描绘化合物21的HMBC(3.5-5.5ppm 1H-轴)结果。
图3H描绘化合物19b的分析型HPLC(2-20%ACN/10mM TEAA缓冲液–20min)结果。
图4描绘c-[G(2’,5’)pG(3’,5’)p]和二硫代核糖O-取代衍生物。
图5描绘c-[A(2’,5’)pA(3’,5’)p]和二硫代核糖O-取代衍生物。
图6描绘c-[G(2’,5’)pA(3’,5’)p]和二硫代核糖O-取代衍生物。
图7描绘在用多种环状二核苷酸分子刺激后,THP-1细胞中的1型干扰素产生
图8描绘在用多种环状二核苷酸分子刺激后,来自独立供体的人PBMC的1型干扰素和干扰素γ的归一化RNA表达水平
图9(A-C)描绘在用多种环状二核苷酸分子刺激后,来自独立供体的人PBMC的1型干扰素α和β蛋白质以及干扰素γ蛋白质的水平。
图10描绘在用多种环状二核苷酸分子治疗后,作为佐剂效力标志的人细胞中的IFN-β诱导。
图11(a)-(c)描绘在用多种环状二核苷酸分子治疗后,作为免疫活化量度的分别在自然杀伤(NK)细胞、CD4+和CD8+T细胞上的表面CD69表达的上调。
图12描绘多种CDN衍生物对磷酸二酯酶处理的抗性。
图13描绘多种已知的STING变体。
图14描绘通过测量IFNβ-LUC报道基因的诱导倍数而得到的编码人STING变体等位基因的HEK293细胞系的刺激。
图15A描绘受刺激的人树突细胞对MHC I类(HLA-ABC)、CD80、CD83和CD86的表面表达。
图15B为在LPS或CDN刺激后,人DC中CD80、CD86、CD83和MHC I类(HLA-ABC)表达的代表性直方图。
图16描绘在用环状二核苷酸佐剂化OVA蛋白质进行疫苗接种后7天,C57BL/6小鼠的PBMC中的OVA-特异性CD8T细胞免疫。
图17描绘在用环状二核苷酸佐剂化OVA蛋白质进行疫苗接种后7天,C57BL/6或goldentickt(STING-/-)小鼠的PBMC中的OVA-特异性CD8T细胞免疫。
图18描绘在用多种环状二核苷酸分子治疗后,B16黑素瘤模型中的肿瘤体积。
图19描绘在用多种环状二核苷酸分子治疗后,CT26肿瘤模型中的存活曲线。
图20A描绘野生型C57BL/6小鼠在施用ML RR-CDN后与接受HBSS和CpG二核苷酸的对照小鼠相比的肿瘤抑制。
图20B描绘在STING缺陷小鼠中获得的结果。
图21A描绘在施用ML RR-CDN后,对已确立的CT26结肠癌的排斥。
图21B描绘来自用ML RR-CDA治疗的小鼠的IFN-γ诱导。
图22A描绘在施用ML RR-CDN后,对已确立的4T1乳腺癌的排斥。
图22B描绘防止CT26肿瘤细胞再激发的保护。
图23描绘CT26(A)和4T1(B)荷瘤动物在施用ML RR-CDA后与HBSS媒介物对照相比的经治疗的原发性肿瘤的抑制。
图24A描绘在施用ML RR-CDA后,B16黑素瘤中的经治疗的原发性肿瘤的抑制。
图24B和C描绘在施用ML RR-CDA后,与图形形式(B)中的HBSS媒介物对照和照片形式(C)中的肺组织本身相比的远端肺肿瘤结节的生长抑制。
发明详述
本发明涉及新颖和高度活性的环状-二-核苷酸(CDN)免疫刺激因子的用途,所述免疫刺激因子通过最近发现的被称为STING(干扰素基因刺激因子)的胞质受体来活化DC。具体而言,本发明的CDN以组合物的形式提供,所述组合物包含诱导STING-依赖性I型干扰素产生的一种或多种环状嘌呤二核苷酸,其中存在于所述组合物种的环状嘌呤二核苷酸为基本上纯的2’,5’,2’,5’和2’,5’,3’,5’CDN。
设计和开发佐剂的新见解基于以下基本理解:被称为病原体相关分子模式(PAMP)的保守微生物结构被宿主细胞模式识别受体(PRR)所感测,从而触发下游信号传导级联,这导致诱导细胞因子和趋化因子和引发特异性的适应性免疫应答。微生物的PAMP补体如何接合先天免疫系统会影响适应性应答的发展,所述适应性应答适于对抗入侵病原体以防止引起疾病。佐剂设计的目的在于选择限定的PAMP或对指定PRR具有特异性的合成分子以引发所需应答。诸如单磷酰脂质A(MPL)和CpG的佐剂是被Toll受体(TLR)所识别的PAMP,其是通过MyD88和Trif衔接分子来传导信号并且介导对NF-kB依赖性促炎细胞因子的诱导的一类跨膜PRR。MPL(TLR-4激动剂)和CpG(TLR-9激动剂)是临床上领先的佐剂,并且是FDA已批准或待批准的疫苗组分。虽然存在于细胞表面上的TLR(如TLR-4)和核内体(如CpG)感测细胞外和空泡病原体,但包括病毒和细胞内细菌在内的多种病原体的生殖性生长循环在胞质中进行。细胞外、空泡和胞质PRR的区室化已导致以下假说:先天免疫系统通过监测胞质来区分病原性和非病原性微生物。对本领域技术人员应显而易见的是,对PRR具有特异性的激动剂包含胞质监测途径,该胞质监测途径引发对抗细胞内病原体的保护性免疫的形成并且与疫苗设计有关。这些相同的靶向配体对于开发已知需要肿瘤特异性CD4+和CD8+T细胞的靶向恶性肿瘤的有效疫苗而言将是必需的。
胞质监测途径(CSP)的活化对于形成针对细胞内病原体的保护性免疫而言必不可少。CSP检测出细菌、病毒和原生动物病原体,从而导致TANK结合激酶(TBK-1)/IRF-3信号传导轴的活化以及IFN-β和其它共调控基因的诱导。病毒和细菌核酸均活化此途径,并且IFN-β的诱导不依赖于MyD88和Trif。虽然I型干扰素通常被认为主要作为宿主抗病毒应答,但IFN-β的诱导是感染细胞内细菌单核细胞增多性李斯特菌(Lm)的巨噬细胞中胞质生长的标志。小鼠李斯特菌病模型的熟知的二分法是,尽管野生型Lm会引发保护小鼠免于细菌攻击的强效CD4和CD8T细胞免疫,但用李斯特菌素O(LLO)-缺失的Lm进行疫苗接种不会引发功能性T细胞或诱导保护性免疫。这种差别证明需要通过Lm来表达宿主细胞基因和胞质通路以引发功能T细胞介导的保护性免疫。受感染宿主细胞中的IFN-β水平由Lm多药外排泵(MDR)调控,所述Lm多药外排泵分泌结构上不相关的小分子,包括抗生素。IFN-β在局限于吞噬溶酶体的Lm LLO突变体所感染的宿主细胞中未被诱导。在于所有TLR介导的信号传导方面缺陷的受感染MyD88-/-Trif-/-巨噬细胞中诱导正常水平的IFN-β。这些数据证实,尽管Lm响应于野生型Lm感染而接合TLR,但需要宿主细胞CSP来形成与IFN-β的诱导相关联的保护性免疫。
环状-二-核苷酸(CDN)通过与胞质PRR,即STING的直接结合来活化胞质监测途径。对Lm和其它细胞内细菌感染的I型干扰素应答源于c-di-AMP或其相关环状二核苷酸(CDN)c-di-GMP的分泌,及其与DDX41和DEAD(天冬氨酸-谷氨酸-丙氨酸-天冬氨酸)框解旋酶以及STING(干扰素基因刺激因子)(最近定义的胞质监测途径的受体)的直接结合。CDN是由大多数细菌表达的第二信使并且调控各种过程,包括生物膜的游动和形成。除了活化TBK-1/IRF-3信号传导途径之外,还响应于结合CDN STING而活化IkB激酶,导致NF-kB转录因子易位至细胞核,从而活化多种促炎基因的表达。
直到最近,STING如何感测胞质DNA仍不难以理解。与直接结合dsDNA的AIM2不同,STING缺乏任何明显的DNA结合域。其它候选DNA传感器如DDX41、DNA-PK和DAI激酶是否为通过STING进行dsDNA信号传导的必要介导物尚不明确。这个难题随着环状GMP-AMP合酶(cGAS)的发现而得以解决,环状GMP-AMP合酶是一种宿主细胞核苷酸转移酶,其响应于结合dsDNA而合成第二信使环状二-GMP-AMP,该环状二-GMP-AMP直接结合STING并通过TBK-1/IRF-3轴引发信号传导级联,从而导致IFN的诱导。另外,cGAS先天免疫DNA传感器产生活化STING信号传导的非典型环状二-核苷酸。与由细菌产生的环状二核苷酸第二信使(其中核苷酸间磷酸桥键通过双-(3’,5’)键连接)不同,由cGAS合成的环状-GMP-AMP中的核苷酸间磷酸桥键通过非典型2’,5’和3’,5’键连接(代表为c[G(2’,5’)pA(3’,5’)p])。因此,STING(干扰素基因刺激因子)已出现作为感测胞质病原体核酸的中心途径,感测方式是通过直接结合细胞内细菌分泌的环状二核苷酸(CDN)6,或通过结合由宿主细胞环状GMP-AMP合酶(cGAS)响应于结合胞质病原体核酸而合成的c-GMP-AMP第二信使。
天然CDN分子对存在于宿主细胞例如抗原呈递细胞中的磷酸二酯酶的降解是敏感的,所述细胞吸收包含所述天然CDN分子的疫苗制剂。所限定的佐剂的效力可被此类降解所减弱,因为佐剂将无法结合和活化其限定的PRR靶标。较低量的佐剂效力可例如通过较低量的先天免疫的标志分子(如IFN-β)的诱导表达(与较弱的疫苗效力相关)来测量,如通过测量的抗原特异性免疫应答的量级所限定。
在本发明中,提供了基本上纯的2’,5’,2’,5’和2’,5’,3’,5’CDN,特别是2’,5’,2’,5’和2’,5’,3’,5’c-di-AMP以及c-di-GMP的二硫代-二磷酸衍生物。所述c-di-AMP和c-di-GMP分子的二硫代-二磷酸衍生物的合成方法导致产生非对映体的混合物,包括c-di-AMP和c-di-GMP分子的Rp,Rp、Sp,Sp、SpRp和Rp,Sp二硫代-二磷酸衍生物。这些个体物质可被分离,并且在其药学特性方面表现出显著的差异。
定义
“施用”在本文中关于人类、哺乳动物、哺乳动物受试者、动物、兽医受试者、安慰剂受试者、研究受试者、实验受试者、细胞、组织、器官或生物流体使用时是指但不限于使外源配体、试剂、安慰剂、小分子、药剂、治疗剂、诊断剂或组合物接触受试者、细胞、组织、器官或生物流体等。“施用”可指例如治疗、药代动力学、诊断、研究、安慰剂和实验方法。细胞的处理涵盖使试剂接触细胞,以及使试剂接触流体,其中流体与细胞接触。“施用”还涵盖例如通过试剂、诊断、结合组合物或通过另一细胞对细胞的体外和离体处理。“一起施用”不意味着暗指两种或更多种药剂以单一组合物来施用。尽管本发明涵盖以单一组合物来施用,但此类药剂可单独施用来递送至单一受试者,这些施用可在相同或不同的时间进行,并且可通过相同施用途径或不同的施用途径进行。
“激动剂”在涉及配体和受体时包括刺激受体的分子、分子的组合、复合物或试剂的组合。例如,粒细胞-巨噬细胞集落刺激因子(GM-CSF)的激动剂可涵盖GM-CSF、GM-CSF的突变蛋白质或衍生物、GM-CSF的肽模拟物、模拟GM-CSF生物功能的小分子或刺激GM-CSF受体的抗体。
“拮抗剂”在涉及配体和受体时包括对受体起到抑制、对抗、下调和/或脱敏作用的分子、分子的组合或复合物。“拮抗剂”涵盖抑制受体的组成型活性的任何试剂。组成型活性是在不存在配体/受体相互作用的情况下显现的活性。“拮抗剂”还涵盖抑制或防止受体的刺激(或调控)活性的任何试剂。举例而言,GM-CSF受体的拮抗剂包括但不限于结合到配体(GM-CSF)并防止其结合到受体的抗体,或结合到受体并防止配体结合到该受体的抗体,或其中抗体将受体锁定在无活性构象。
关于本发明的CDN的“基本上纯化的”意指按存在于组合物中的CDN活性物的重量计,指定的物质的量占至少50%,更往往占至少60%,典型地占至少70%,更典型地占至少75%,最典型地占至少80%,通常占至少85%,更通常占至少90%,最通常占至少95%,以及常规地占至少98%或更多。水、缓冲剂、盐、洗涤剂、还原剂、蛋白酶抑制剂、稳定剂(包括添加的蛋白质如白蛋白)和赋形剂的重量一般不用于确定纯度。
“特异性”或“选择性”结合在涉及配体/受体、核酸/互补核酸、抗体/抗原或其它结合对(例如,细胞因子与细胞因子受体)(各自总体上在本文中称为“靶标生物分子”或“靶标”)时,指示与蛋白质和其它生物制剂的异质群体中的靶标的存在相关的结合反应。特异性结合可意指例如从所涵盖方法的抗体的抗原结合位点衍生的结合化合物、核酸配体、抗体或结合组合物以比与非靶标分子的亲和力往往大至少25%、更往往大至少50%、最往往大至少100%(2倍)、通常大至少十倍、更通常大至少20倍并且最通常大至少100倍的亲和力结合至它的靶标。
“配体”是指结合至靶标生物分子的小分子、核酸、肽、多肽、糖类、多糖、聚糖、糖蛋白、糖脂或其组合。虽然此类配体可为受体的激动剂或拮抗剂,但配体也涵盖并非激动剂或拮抗剂,并且不具有激动或拮抗性质的结合剂。配体对于其同源靶标的特异性结合往往以“亲和力”表示。在优选的实施方案中,本发明的配体以介于约104M-1和约108M-1之间的亲和力结合。亲和力计算为Kd=koff/kon(koff是解离速率常数,Kon是缔合速率常数并且Kd是平衡常数)。
亲和力可通过测量多种浓度(c)下的标记配体的结合分率(r)来在平衡状态下测定。数据使用Scatchard方程来作图:r/c=K(n-r):其中r=平衡状态下的结合配体的摩尔数/受体的摩尔数;c=平衡状态下的游离配体浓度;K=平衡缔合常数;并且n=每个受体分子的配体结合位点的数目。通过图解分析,r/c相对于X轴上的r来在Y轴上绘制,由此产生Scatchard绘制。通过Scatchard分析的亲和力测量在本领域中是熟知的。参见例如van Erp等,J.Immunoassay 12:425-43,1991;Nelson和Griswold,Comput.Methods Programs Biomed.27:65-8,1988。在替代方案中,亲和力可通过等温滴定量热法(ITC)来测量。在典型的ITC实验中,将配体溶液滴定到至同源靶标的溶液中。随时间的推移监测其相互作用时所释放的热量(ΔH)。由于连续量的配体滴定到ITC细胞中,吸收或释放的热量与结合量成正比。随着体系达到饱和,热量信号减弱直至只观察到稀释热。然后,根据来自每次注射的热量相对于细胞中配体与结合搭配物的比率的绘图来获得结合曲线。结合曲线用适当结合模型进行分析以确定KB、n和ΔH。注意KB=1/Kd。
如本文所用的术语“受试者”是指人类或非人生物体。因此,本文所述的方法和组合物适用于人类和兽医疾病两者。在某些实施方案中,受试者为“患者”,即正在接受疾病或病症的医疗护理的活人。这包括正在研究其病理学体征的无确定疾病的人。优选的是目前被诊断患有由本发明的组合物和方法靶向的特定癌症的受试者。用本文所述的组合物治疗的优选癌症包括但不限于前列腺癌、肾癌、黑素瘤、胰癌、宫颈癌、卵巢癌、结肠癌、头颈癌、肺癌和乳腺癌。
“治疗有效量”定义为足以显示出患者有益效果即导致所治疗的病症的症状减轻、防止或改善的试剂或药物组合物的量。当药剂或药物组合物包括诊断剂时,“诊断有效量”定义为足以产生信号、图像或其它诊断参数的量。药物制剂的有效量将根据多种因素变化,诸如个体的易感性程度、个体的年龄、性病和体重、以及个体的特异反应。“有效量”涵盖但不限于可改善、逆转、缓解、预防或诊断医疗病况或病症的症状或体征或其致病过程的量。除非另外明确地或通过上下文指明,否则“有效量”不限于足以改善病症的最低量。
“治疗”(关于病症或疾病)是获得有益或所需结果(包括并优选临床结果)的方法。对于本发明,关于疾病的有益或所需结果包括但不限于以下一者或多者:预防疾病、改善与疾病相关的病况、治愈疾病、减轻疾病的严重性、延迟疾病的进展、缓解与疾病相关的一种或多种症状、提高患病者的生活质量和/或延长生存时间。同样,对于本发明,关于病症的有益或所需结果包括但不限于以下一者或多者:预防病症、改善病症、治愈病症、减轻病症的严重性、延迟病症的进展、缓解与病症相关的一种或多种症状、提高患病者的生活质量和/或延长生存时间。例如,在其中本文所述的组合物用于治疗癌症的实施方案中,有利的或所需的结果包括但不限于以下一个或多个:减少赘生或癌细胞的增殖(或对其进行破坏)、减少在癌症中发现的赘生细胞的转移、缩小肿瘤的尺寸、减少由癌症产生的症状、增加患有癌症的人的生活质量、减少治疗疾病所需要的其它药物的剂量、延缓癌症的进程和/或延长患有癌症的患者的生存时间。根据上下文,“治疗”受试者可暗示受试者需要治疗,例如,在受试者具有预期通过施用试剂来改善的病症的情况中。
“疫苗”涵盖预防性疫苗。疫苗还涵盖治疗性疫苗,例如施用给患有病况或病症的哺乳动物的疫苗,该病况或病症与该疫苗提供的抗原或表位相关。
环状嘌呤二核苷酸
原核和真核细胞利用多种小分子进行细胞信号传导和细胞内及细胞间通信。已知诸如cGMP、cAMP的环状核苷酸在原核和真核细胞中具有调控和引发活性。与真核细胞不同,原核细胞还使用环状嘌呤二核苷酸作为调控分子。在原核生物中,两个GTP分子的缩合通过酶双鸟苷酸环化酶(DGC)来催化以得到c-diGMP,其代表细菌中的重要调控因子。
最近的研宄表明环状diGMP或其类似物也可刺激或增强患者的免疫或炎症应答或可通过充当哺乳动物中的佐剂来增强对于疫苗的免疫应答。病原体衍生DNA的胞质检测需要通过TANK结合激酶1(TBK1)及其下游转录因子IFN-调控因子3(IRF3)来进行信号传导。称为STING的跨膜蛋白质(IFN基因的刺激因子;也称为MITA、ERIS、MPYS和TMEM173)用作这些环状嘌呤二核苷酸的信号传导受体,导致刺激TBK1-IRF3信号传导轴和STING依赖性I型干扰素应答。参见例如图1。Burdette等,Nature 478:515-18,2011证实STING直接结合至环状双鸟苷酸单磷酸,但不结合至其它不相关的核苷酸或核酸。
用作获得本发明CDN的前体的环状嘌呤二核苷酸相当详细地描述于例如Gao等,Cell(2013)153:doi:10.1016/j.cell.2013.04.046;美国专利No.7,709458和No.7,592,326;WO2007/054279;和Yan等,Bioorg.Med.Chem Lett.18:5631(2008)中,其各自据此以引用的方式并入。可使用有机化学技术对CDN进行修饰以便产生本发明的CDN。
优选的嘌呤包括但不限于腺嘌呤、鸟嘌呤、肌苷、次黄嘌呤、黄嘌呤、异鸟嘌呤等。本发明的CDN优选地为硫代磷酸类似物,最优选地为其基本上纯的Sp,Sp、Rp,Rp、SpRp或Rp,Sp立体异构体。
如在结构中所示,每个核糖包含可被取代的2’或3’羟基。如下文所述,本发明的CDN可在这些2’或3’羟基中的一者或两者处包含取代(其并非环键的一部分),所述取代提供前药离去基团或影响活性、溶解度、生物利用率等的其它修饰。如本文中所用,“前药”是指对所涵盖的化合物的修饰,其中经修饰的化合物表现出较低的药理学活性(与经修饰的化合物相比),并且其中经修饰的化合物通过酶促或非酶促反应在体内(如在靶细胞或靶器官中)转化回未经修饰的形式。在某些实施方案中,一个核糖上的羟基包含前药离去基团。前药可改变药物的物理化学、生物药学和药代动力学性质。传统的前药被分类为通过体内转化形成活性药物的药物。开发前药的原因通常是由于与母体药物相关的不良水溶性、化学不稳定性、低口服生物利用率、缺乏血脑屏障渗透性和高首过代谢。合适的前药部分描述于例如“Prodrugs and Targeted Delivery,”J.Rautico,Ed.,John Wiley&Sons,2011中。
优选的环状嘌呤二核苷酸为硫代磷酸类似物,其在本文中被称为“硫代磷酸”。硫代磷酸为其中未桥接氧中的一个被硫取代的正常核苷酸的变体。核苷酸间键的硫化会急剧降低内切核酸酶和外切核酸酶(包括5'至3'和3'至5'DNA POL 1外切核酸酶、核酸酶S1和P1、核糖核酸酶、血清核酸酶和蛇毒磷酸二酯酶)的作用。此外,跨越脂质双层的可能性增加。
硫代磷酸键是固有手性的。技术人员将认识到此结构中的磷酸可以R或S形式存在。因此,Rp,Rp、Sp,Sp、Sp,Rp和Rp,Sp形式是可能的。
如上文所提到的,本发明的环状嘌呤二核苷酸包含CDN,特别是CDN硫代磷酸的2’-O-和3’-O-取代基形式。额外的稳定性和生物利用率可通过取代核糖部分的2’-OH来提供。本文适合的取代基团包括但不限于:卤素、羟基、烷基、烯基、炔基、酰基(-C(O)Raa)、羧基(-C(O)0-Raa)、脂族基团、脂环族基团、烷氧基、取代的氧基(-0-Raa)、芳基、芳烷基、杂环基、杂芳基、杂芳基烷基、氨基(-N(Rbb)(Rcc))、亚氨基(=NRbb)、酰胺基(-C(O)N(Rbb)(RcC)或-N(Rbb)C(O)Raa)、叠氮基(-N3)、硝基(-N02)、氰基(-CN)、氨基甲酰胺基(-OC(O)N(Rbb)(Rcc)或-N(Rbb)C(O)ORaa)、脲基(-N(Rbb)C(O)-N(Rbb)(Rcc))、硫脲基(-N(Rbb)C(S)N(Rbb)(Rcc))、胍基(-N(Rbb)C(=NRbb)N(Rbb)(Rcc))、脒基(-C(=NRbb)N(Rbb)(RcC)或-N(Rbb)C(=NRbb)(Raa))、硫醇(-SRbb)、亚磺酰基(-S(O)Rbb)、磺酰基(-S(O)2Rb)以及磺酰胺基(-S(O)2N(Rbb)(RcC)或-N(Rbb)S(O)2Rbb)。其中每个Raa、Rbb和Rcc独立地为H、任选连接的化学官能团或进一步的取代基团,优选的列表包括但不限于H、烷基、烯基、炔基、脂族基团、烷氧基、酰基、芳基、芳烷基、杂芳基、脂环族基团、杂环基团和杂芳基烷基。本文所述化合物内所选的取代基以递归程度(recursive degree)存在。
如本文所用的术语“烷基”是指包含最多24个碳原子的饱和直链或支链烃基。烷基的实例包括但不限于甲基、乙基、丙基、丁基、异丙基、正己基、辛基、癸基、十二烷基等。烷基典型地包含1至约24个碳原子,更典型地包含1至约12个碳原子,其中包含1至约6个碳原子是更优选的。如本文所用的术语“低级烷基”包含1至约6个碳原子。如本文所用的烷基可任选地包含一个或更多个进一步的取代基团。
如本文所用的术语“烯基”是指包含最多24个碳原子并具有至少一个碳-碳双键的直链或支链烃基。烯基的实例包括但不限于乙烯基、丙烯基、丁烯基、1-甲基-2-丁烯-1-基、二烯诸如1,3-丁二烯等。烯基典型地包含2至约24个碳原子,更典型地包含2至约12个碳原子,其中包含2至约6个碳原子是更优选的。如本文所用的烯基可任选地包含一个或更多个进一步的取代基团。
如本文所用的术语“炔基”是指包含最多24个碳原子并具有至少一个碳-碳三键的直链或支链烃基。炔基的实例包括但不限于乙炔基、1-丙炔基、1-丁炔基等。炔基典型地包含2至约24个碳原子,更典型地包含2至约12个碳原子,其中包含2至约6个碳原子是更优选的。如本文所用的炔基可任选地包含一个或更多个进一步的取代基团。
如本文所用的术语“酰基”是指通过从有机酸中移除羟基而形成的基团,并具有通式-C(O)-X,其中X典型地为脂族、脂环族或芳族的。实例包括脂族羰基化合物、芳族羰基化合物、脂族磺酰基化合物、芳族亚磺酰基化合物、脂族亚磺酰基化合物、芳族磷酸酯、脂族磷酸酯等。如本文所用的酰基可任选地包含进一步的取代基团。
术语“脂环族”是指其中环为脂族的环系。该环系可包含一个或多个环,其中至少一个环是脂族的。优选的脂环族化合物包含环中具有约5至约9个碳原子的环。如本文所用的脂环族可任选地包含进一步的取代基团。
如本文所用的术语“脂族”是指包含最多24个碳原子的直链或支链烃基,其中任何两个碳原子之间的饱和度为单键、双键或三键。脂族基团优选地包含1至约24个碳原子,更典型地包含1至约12个碳原子,其中包含1至约6个碳原子是更优选的。脂族基团的直链或支链可由一个或多个包括氮、氧、硫和磷的杂原子中断。此类被杂原子中断的脂族基团包括但不限于多烷氧基化合物诸如聚亚烷基二醇类、聚胺类和聚亚胺类。如本文所用的脂族基团可任选地包含进一步的取代基团。
如本文所用的术语“烷氧基”是指在烷基和氧原子之间形成的基团,其中氧原子用于将烷氧基连接到母体分子。烷氧基的实例包括但不限于甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基、新戊氧基和正己氧基等等。如本文所用的烷氧基可任选地包含进一步的取代基团。
如本文所用的术语“氨基烷基”是指氨基取代的C\-Cn烷基。该基团的烷基部分与母体分子形成共价键。氨基可位于任何位置,并且氨基烷基可在烷基和/或氨基部位被进一步的取代基团取代。
如本文所用的术语“芳烷基”和“芳基烷基”是指共价连接到C\-Cn烷基的芳族基团。所得的芳烷基(或芳基烷基)的烷基部分与母体分子形成共价键。实例包括但不限于苄基、苯乙基等。如本文所用的芳烷基可任选地包含连接到烷基、芳基或两者上形成基团的进一步的取代基团。
如本文所用的术语“芳基”和“芳族”是指具有一个或多个芳环的单环或多环的碳环系基团。芳基的实例包括但不限于苯基、萘基、四氢萘基、茚满基、茚基(idenyl)等。优选的芳基环系在一个或多个环中具有约5至约20个碳原子。如本文所用的芳基可任选地包含进一步的取代基团。
如本文所用的术语“卤代”或“卤素”是指选自氟、氯、溴和碘的原子。
如本文所用的术语“杂芳基”和“杂芳族”是指包含单环或多环芳环、环系或稠合环系的基团,其中这些环的至少一个为芳族的并且包含一个和或多个杂原子。杂芳基还意在包括稠合环系,包括其中稠环的一个或多个不含杂原子的体系。杂芳基通常包含一个选自硫、氮或氧的环原子。杂芳基的实例包括但不限于:吡啶基、吡嗪基、嘧啶基、吡咯基、吡唑基、咪唑基、噻唑基、噁唑基、异噁唑基、噻二唑基、噁二唑基、苯硫基、呋喃基、喹啉基、异喹啉基、苯并咪唑基、苯并噁唑基、喹噁啉基等。杂芳基可直接地或通过连接部分诸如脂族基团或杂原子连接到母体分子上。如本文所用的杂芳基可任选地包含进一步的取代基团。
如本文所用的术语“杂芳基烷基”是指进一步包含共价连接的C1-C12烷基的如先前定义的杂芳基。所得杂芳基烷基的烷基部分能够与母体分子形成共价键。实例包括但不限于吡啶基甲基、吡啶基乙基、萘啶基丙基(napthyridinylpropyl)等。如本文所用的杂芳基烷基可在杂芳基或烷基部分的一者或两者上任选地包含进一步的取代基团。
如上文所提到的,优选的环状嘌呤二核苷酸还包括CDN,特别是CDN硫代磷酸的前药形式。前药可改变药物的物理化学、生物药学和药代动力学性质。传统的前药被分类为通过体内转化形成活性药物的药物。开发前药的原因通常是由于与母体药物相关的不良水溶性、化学不稳定性、低口服生物利用率、缺乏血脑屏障渗透性和高首过代谢。合适的前药部分描述于例如“Prodrugs and Targeted Delivery,”J.Rautico,Ed.,John Wiley&Sons,2011中。
如本文关于环状嘌呤二核苷酸所用的术语“基本上纯的”是指Rp,Rp或Rp,Sp形式相对于上图中指示的手性中心处的其它可能的立体化学构型而言为至少75%纯的。举例而言,“基本上纯的Rp,Rpc-di-GMP硫代磷酸”相对于c-di-GMP硫代磷酸的Rp,Sp和Sp,Sp形式而言将为至少75%纯的。在优选的实施方案中,基本上纯的环状嘌呤二核苷酸为至少85%纯的、至少90%纯的、至少95%纯的、至少97%纯的以及至少99%纯的。虽然本发明的基本上纯的环状嘌呤二核苷酸制剂为“立体化学上纯的”,但这并非意在指示制剂内的在这些手性中心处具有特定立体化学构型的所有CDN在其它方面是相同的。例如,基本上纯的环状嘌呤二核苷酸制剂可包含Rp,Rp c-di-GMP硫代磷酸和Rp,Rp c-di-AMP硫代磷酸的组合,并且仍为基本上纯的环状嘌呤二核苷酸制剂。此类制剂还可包含后文所述的对患者治疗有利的其它组分,条件是制剂内的所有CDN在这些手性中心处具有特定立体化学构型。
本文描述的CDN组合物可单独或联合药学上可接受的赋形剂以足以诱导、修改或刺激适当免疫应答的量施用给宿主。免疫应答可包括但不限于特异性免疫应答、非特异性免疫应答、特异性和非特异性应答、先天应答、初次免疫应答、适应性免疫、二次免疫应答、记忆免疫应答、免疫细胞活化、免疫细胞增殖、免疫细胞分化和细胞因子表达。在某些实施方案中,CDN组合物与一种或多种额外的组合物联合施用,所述组合物包含旨在刺激对一种或多种预定抗原的免疫应答的疫苗;佐剂;CTLA-4和PD-1途径拮抗剂、脂质、脂质体、化学治疗剂、免疫调节细胞系等。
CDN组合物可在额外的治疗性或预防性组合物或方式之前、之后和/或与其同时施用。这些包括而不限于B7共剌激分子、白介素-2、干扰素-γ、GM-CSF、CTLA-4拮抗剂、OX-40/OX-40配体、CD40/CD40配体、沙莫司亭(sargramostim)、左旋咪唑(levamisol)、痘苗病毒、卡介苗(Bacille Calmette-Guerin,BCG)、脂质体、明矾、氟氏完全或不完全佐剂、解毒的内毒素、矿物油、表面活性物质如脂卵磷脂(Iipolecithin)、普卢兰尼克(pluronic)多元醇、聚阴离子、肽及油或烃乳液。相对于抗体应答而言优先剌激溶细胞性T细胞应答的诱导T细胞免疫应答的载体是优选的,但也可使用刺激这两种类型的应答的那些载体。在药剂为多肽的情况下,可施用多肽本身或编码多肽的多核苷酸。载体可为细胞,例如抗原呈递细胞(APC)或树突细胞。抗原呈递细胞包括诸如巨噬细胞、树突细胞和B细胞的细胞类型。其他专门的抗原-呈递细胞包括单核细胞、边缘区Kupffer细胞、小胶质细胞、朗氏细胞(Langerhans'cell)、指突状树突细胞、滤泡树突细胞和T细胞。还可以使用兼性抗原-呈递细胞。兼性抗原呈递细胞的实例包括星形细胞、滤泡细胞、内皮和成纤维细胞。载体可以为细菌细胞,其经转化以表达多肽或递送随后在免疫接种个体的细胞中表达的多核苷酸。可添加诸如氢氧化铝或磷酸铝的佐剂以提高疫苗触发、增强或延长免疫应答的能力。诸如以下的额外材料也是潜在的佐剂:细胞因子、趋化因子以及类似CpG的细菌核酸序列、toll-样受体(TLR)9激动剂以及针对TLR2、TLR4、TLR5、TLR7、TLR8、TLR9的额外激动剂,包括脂蛋白、LPS、单磷酰脂质A、脂膜酸、咪喹莫特(imiquimod)、瑞喹莫德(resiquimod)和单独或联合所述组合物使用的诸如聚I:C的另外的维甲酸诱导基因I(RIG-I)激动剂。佐剂的其它代表性实例包括合成佐剂QS-21,其包含从皂皮树(Quillaja saponaria)的树皮和短小棒状杆菌(Corynebacterium parvum)中纯化的均质皂草苷(McCune等,Cancer,1979;43:1619)。应当理解,佐剂经受优化。换句话讲,技术人员可进行常规实验来确定要使用的最佳佐剂。
与额外的治疗剂共同施用的方法是本领域中熟知的(Hardman等(编)(2001)Goodman and Gilman’s The Pharmacological Basis of Therapeutics,第10版,McGraw-Hill,New York,NY;Poole和Peterson(编)(2001)Pharmacotherapeutics for Advanced Practice:A Practical Approach,Lippincott,Williams&Wilkins,Phila.,PA;Chabner和Longo(编)(2001)Cancer Chemotherapy and Biotherapy,Lippincott,Williams&Wilkins,Phila.,PA)。
由于本发明化合物的佐剂性质,其使用可联合其它治疗方式,包括其它疫苗、佐剂、抗原、抗体和免疫调节剂。下文提供实例。
佐剂
除以上所述的环状嘌呤二核苷酸之外,本发明的组合物还可包含一种或多种额外物质,所述额外物质由于其性质可起作用来刺激或以其它方式利用免疫系统,从而对存在于灭活肿瘤细胞上的癌抗原进行应答。此类佐剂包括但不限于脂质、脂质体、诱导先天免疫的灭活细菌(例如,灭活或减毒的单核细胞增多性李斯特菌)、通过Toll样受体(TLR)、(NOD)-样受体(NLR)、维甲酸诱导基因型(RIG)-I样受体(RLR)和/或C-型凝集素受体(CLR)来介导先天免疫活化的组合物。PAMP的实例包括但不限于脂蛋白、脂多肽、肽聚糖、酵母聚糖、脂多糖、奈瑟菌孔蛋白、鞭毛蛋白、profillin、半乳糖基神经酰胺、胞壁酰二肽。肽聚糖、脂蛋白质和脂磷壁酸是革兰氏阳性菌的细胞壁组分。脂多糖由大多数细菌表达,MPL是一个实例。鞭毛蛋白是指由病原性和共生菌分泌的细菌鞭毛的结构组分。α-半乳糖基神经酰胺(α-GalCer)是自然杀伤细胞T(NKT)细胞的活化剂。胞壁酰二肽是所有细菌共有的生物活性肽聚糖基序。此清单并非限制性的。下文描述优选的佐剂组合物。
CTLA-4和PD-1途径拮抗剂
CTLA-4被认为是适应性免疫应答的重要负性调控因子。活化的T细胞上调CTLA-4,其以比CD28高的亲和力结合抗原呈递细胞上的CD80和CD86,从而抑制T细胞刺激、IL-2基因表达和T细胞增殖。CTLA4阻断的抗肿瘤效应已在结肠癌、转移性前列腺癌和转移性黑素瘤的鼠科模型中观察到。
伊匹单抗(lpilimumab)(YervoyTM)和曲美木单抗(tremelimumab)是结合至人CTLA4并且防止其与CD80和CD86的相互作用的人源化单克隆抗体。使用伊匹单抗和曲美木单抗的I期和II期研究已证实在癌症患者中的临床活性。可通过类似策略来靶向的其它负性免疫调控因子包括程序性细胞死亡1、B和T淋巴细衰减因子、转化生长因子ββ、白介素-10和血管内皮生长因子。
PD-1是在活化T细胞上表达的适应性免疫应答的另一种负性调控因子。PD-1结合至B7-H1和B7-DC,并且PD-1的接合抑制T细胞活化。已证实PD-1途径阻断的抗肿瘤效应。BMS-936558、MK3475、CT-011、AMP-224和MDX-1106在文献中被报道是可用于本发明的PD-1途径阻断剂的实例。
TLR激动剂
如本所用的术语“Toll样受体”(或“TLR”)是指感测微生物产物和/或引发适应性免疫应答的Toll样受体家族蛋白质或其片段的成员。在一个实施方案中,TLR活化树突细胞(DC)。Toll样受体(TLR)是最初被鉴定为辨识微生物病原体的先天免疫系统的传感器的模式识别受体家族。TLR包含保守性跨膜分子家族,其包含富含亮氨酸重复序列的胞外域、跨膜域和细胞内TIR(Toll/IL-1R)域。TLR识别微生物中的独特结构,通常被称为“PAMP”(病原体相关分子模式)。配体与TLR结合引起细胞内信号传导途径的级联,所述细胞内信号传导途径诱导涉及炎症和免疫的因子的产生。
在人体内,已经鉴定十种TLR。在细胞表面上表达的TLR包括TLR-1、-2、-4、-5和-6,而TLR-3、-7/8和-9通过ER区室表达。人树突细胞子集可基于独特TLR表达模式来鉴定。举例而言,骨髓或“常规”子集的DC(mDC)在被刺激时表达TLR1-8,并且产生活化标志物(例如CD80、CD86、MHC I类和II类、CCR7)、促炎细胞因子和趋化因子的级联。此刺激和所产生表达的结果是抗原特异性CD4+和CD8+T细胞触发。这些DC获得吸收抗原并且将其以适当形式呈递给T细胞的增强能力。相比之下,类浆细胞子集的DC(pDC)在活化后只表达TLR7和TLR9,并且导致NK细胞以及T细胞的活化。由于死亡肿瘤细胞可不利地影响DC功能,因此提出用TLR激动剂活化DC可有利于以治疗癌症的免疫疗法途径来触发抗肿瘤免疫。还提出使用辐射和化疗来成功治疗乳腺癌需要TLR4活化。
本领域中已知并且可用于本发明的TLR激动剂包括但不限于以下:
Pam3Cys,TLR-1/2激动剂;
CFA,TLR-2激动剂;
MALP2,TLR-2激动剂;
Pam2Cys,TLR-2激动剂;
FSL-1,TLR-2激动剂;
Hib-OMPC,TLR-2激动剂;
聚肌苷酸:聚胞苷酸(聚I:C),TLR-3激动剂;
聚腺苷酸-聚尿苷酸(聚AU),TLR-3激动剂;
用聚-L-赖氨酸和羧甲基纤维素稳定的聚肌苷酸-聚胞苷酸TLR-3激动剂;
单磷酰脂质A(MPL),TLR-4激动剂;
LPS,TLR-4激动剂;
细菌鞭毛蛋白,TLR-5激动剂;
唾液酸化-Tn(STn),与多种人癌细胞上的MUC1粘蛋白相关的碳水化合物和TLR-4激动剂;
咪喹莫特,TLR-7激动剂;
瑞喹莫德,TLR-7/8激动剂;
洛索立宾(loxoribine),TLR-7/8激动剂;和
非甲基化CpG二核苷酸(CpG-ODN),TLR-9激动剂。
由于其辅助性质,TLR激动剂优选地与其它疫苗、佐剂和/或免疫调节剂联合使用,并且可以各种组合来组合。因此,在某些实施方案中,如本文所述的结合至STING并诱导STING-依赖性TBK1活化的环状嘌呤二核苷酸和表达并分泌刺激树突细胞诱导、募集和/或成熟的一种或多种细胞因子的灭活肿瘤细胞可与用于治疗目的的一种或多种TLR激动剂一起施用。
抗体治疗剂
抗体依赖性细胞介导的细胞毒性(ADCC)是细胞介导的免疫防御机制,由此免疫系统的效应细胞主动溶解膜表面抗原已被特异性抗体结合的靶细胞。其是抗体作为体液免疫反应的一部分可起作用以限制和牵制感染的机制之一。经典ADCC由自然杀伤(NK)细胞介导;巨噬细胞、嗜中性粒细胞和嗜酸性粒细胞也可介导ADCC。ADCC为治疗性单克隆抗体(包括曲妥单抗和利妥昔单抗)对抗肿瘤的重要作用机制。本发明的化合物可增强ADCC。
以下是可与本发明的化合物一起使用的抗体的示例性清单。
莫罗单抗(Muromonab)-CD3:用于防止器官如肾移植的急性排斥。人源化形式显示出抑制1型糖尿病中β细胞的自身免疫破坏的前景。
英夫利昔单抗(Infliximab)和阿达木单抗(adalimumab)结合至肿瘤坏死因子-α(TNF-α)。用于一些炎性疾病如类风湿性关节炎、牛皮癣、克隆氏疾病(Crohns disease)中。
奥马珠单抗(Omalizumab)结合至IgE,从而防止IgE与肥大细胞结合。针对变应性哮喘使用。
达克珠单抗(Daclizumab)结合至IL-2受体在活化T细胞表面上暴露的部分。用于防止移植肾脏的急性排斥。
利妥昔单抗(商品名=)。结合至发现于大多数B细胞上的CD20分子并用于治疗B细胞淋巴瘤。
替伊莫单抗(Ibritumomab)(商品名=)。其为对抗B细胞(和淋巴瘤)上与同位素缀合的CD20分子的单克隆抗体。给予补以Rituxan的淋巴瘤患者。
托西莫单抗(Tositumomab)其为对抗CD20和放射性同位素碘-131(131I)的单克隆抗体的缀合物。
西妥昔单抗(Cetuximab)阻断HER1,一种发现于一些肿瘤细胞(一些乳腺癌、淋巴瘤)上的表皮生长因子(EGF)的受体。
曲妥单抗阻断HER2,一种在大约20%的乳腺癌中过表达的生长因子受体。
结合CD30的单克隆抗体缀合物,CD30是一种由一些淋巴瘤细胞表达但在需要补充骨髓的正常干细胞中未被发现的细胞表面分子。
阿仑单抗(Alemtuzumab)结合至CD52,一种发现于淋巴细胞上并贫化T细胞和B细胞的分子。已表现出对慢性淋巴细胞性白血病的完全缓解并且显示出防止肾移植排斥的前景。
Lym-1结合至可在淋巴瘤细胞上以高水平表达的HLA-DR编码的组织相容性抗原。
伊匹单抗用于增强身体本身对肿瘤的免疫应答。
Vitaxin。结合至发现于肿瘤血管上但不在供给正常组织的血管上的血管整联蛋白(α-v/β-3)。在II期临床试验中,Vitaxin已显示出使实体瘤缩减而无有害副作用的一些前景。
贝伐单抗(Bevacizumab)结合至血管内皮生长因子(VEGF),从而防止其结合至其受体。用于治疗结肠直肠癌。
阿昔单抗(Abciximab)通过结合血小板表面上的与纤维蛋白原有关的受体来抑制血小板聚集。有助于防止经历过血管成形术的患者的冠状动脉再堵塞。
递送剂
脂质体是由一个(“单层”)或多个(“多层”)磷脂层形成的囊泡。由于磷脂结构单元的两亲特性,脂质体通常包含呈现出亲水外表面并封闭亲水核心的亲水层。脂质体在结合亲水/疏水组分中的多能性、它们的无毒性质、生物降解性、生物相容性、佐剂性、诱导细胞免疫、持续释放特性以及被巨噬细胞的迅速摄取使得它们成为引人注目的用于递送抗原的候选物。
以引用方式并入整体并入本文中的WO2010/104833描述了合适的脂质体制剂。以上提及的具有或不具有“免疫原性多肽或碳水化合物”的此类脂质体制剂在本文中称为(Molecular Express,Inc.),可包含一种或多种额外的组分,例如肽聚糖、脂肽、脂多糖、单磷酰脂A、脂膜酸、瑞喹莫德、咪喹莫特、鞭毛蛋白、含非甲基化CpG基序的寡核苷酸、β-半乳糖基神经酰胺、胞壁酰二肽、全反式维甲酸、双链病毒RNA、热休克蛋白质、二十八烷基二甲基溴化铵、阳离子表面活性剂、Toll样受体激动剂、二肉豆蔻酰基三甲基铵丙烷和nod样受体激动剂。有利地,这些脂质体制剂可用于递送根据本发明的一种或多种环状嘌呤二核苷酸。
此外,虽然以上论述的脂质体制剂使用“类固醇衍生物”作为将免疫原性多肽或碳水化合物连接至脂质体的锚定剂,但类固醇可仅以非缀合类固醇如胆固醇形式来提供。
由脂质混合物制备脂质体的合适方法是本领域熟知的。参见例如Basu&Basu,Liposome Methods and Protocols(Methods in MolecularBiology),Humana Press,2002;Gregoriadis,Liposome Technology,第3版,Informa HealthCare,2006。优选的方法包括其中所述的挤出、匀化和超声方法。制备用于本发明的脂质体的示例性方法描述于WO2010/104833中,该方法包括干燥脂质体混合物,然后在水性媒介物中水合并超声处理以形成脂质体。
在某些实施方案中,提供在特定平均大小范围内的脂质体。脂质体大小可例如通过将包含脂质体的水性媒介物挤出通过具有预选孔径的膜并收集流过膜的材料而加以选择。在优选的实施方案中,将脂质体的直径选择为基本上在50与500nm之间,更优选地直径基本上在50与200nm之间,最优选地直径基本上在50与150nm之间。如本文在此背景下所用的术语“基本上”意指至少75%、更优选80%、最优选90%的脂质体在指定的范围内。
可用于本发明的其它脂质和脂质样佐剂包括水包油(o/w)乳液(参见例如Muderhwa等,J.Pharmaceut.Sci.88:1332–9,1999)),TLR(Molecular Express,Inc.)、毛地黄皂苷(参见例如美国专利5,698,432)和葡萄吡喃糖基脂质(参见例如美国专利申请20100310602)。
纳米粒子还代表适于大多数施用途径的药物递送体系。近年来,已探究了多种用于制备纳米粒子的天然和合成聚合物,其中聚(乳酸)(PLA)、聚(乙醇酸)(PGA)以及它们的共聚物(PLGA)因其生物相容性和生物降解性而已被深入研究。纳米粒子和其它纳米载体用作若干类药物如抗癌剂、抗高血压剂、免疫调节剂和激素;以及大分子如核酸、蛋白质、肽和抗体的载体。参见例如Crit.Rev.Ther.Drug Carrier Syst.21:387-422,2004;Nanomedicine:Nanotechnology,Biology and Medicine 1:22-30,2005。
化学治疗剂
在附加实施方案中,所述方法还包括向受试者施用有效量的一种或多种化疗治疗剂作为附加治疗。在某些实施方案中,所述一种或多种化疗治疗剂选自醋酸阿比特龙、六甲蜜胺、脱水长春碱、奥里斯他汀(auristatin)、贝沙罗汀(bexarotene)、比卡鲁胺、BMS 184476、2,3,4,5,6-五氟-N-(3-氟-4-甲氧基苯基)苯磺酰胺、博来霉素、N,N-二甲基-L-缬氨酰-L-缬氨酰-N-甲基-L-缬氨酰-L-脯氨酰-1-L脯氨酸-叔-丁酰胺、恶病质素、西马多丁(cemadotin)、苯丁酸氮芥、环磷酰胺、3',4'-双脱氢-4'-脱氧-8'-降长春花碱、多西紫杉醇、多西他赛(doxetaxel)、环磷酰胺、卡铂、卡莫司汀(carmustine)、顺铂、念珠藻素(cryptophycin)、环磷酰胺、阿糖胞苷、达卡巴嗪(dacarbazine)(DTIC)、更生霉素、柔红霉素、丁西他滨多拉司他汀(decitabine dolastatin)、多柔比星(阿霉素)、鬼臼乙叉苷、5-氟尿嘧啶、非那雄胺(finasteride)、氟他胺、羟基脲和羟基脲紫杉烷、异环磷酰胺、利阿唑(liarozole)、氯尼达明(lonidamine)、洛莫司汀(lomustine)(CCNU)、MDV3100、二氯甲二乙胺(氮芥)、美法仑、米伏布林羟乙基磺酸盐(mivobulin isethionate)、根霉素、sertenef、链脲霉素、丝裂霉素、氨甲喋呤、紫杉烷类、尼鲁米特(nilutamide)、奥那司酮(onapristone)、紫杉醇、泼尼莫司汀(prednimustine)、丙卡巴肼(procarbazine)、RPR109881、磷酸雌莫司汀(stramustine phosphate)、他莫西芬(tamoxifen)、他索纳明(tasonermin)、紫杉酚、维A酸、长春花碱、长春新碱、硫酸长春地辛和长春氟宁。
免疫调节细胞系
所谓“灭活肿瘤细胞”是指经过处理以防止细胞分裂的肿瘤细胞(对于患者为“自体的”或“同种异体的”)。出于本发明的目的,此类细胞保持其免疫原性和其代谢活性。作为癌症疗法的一部分,此类肿瘤细胞经过基因修饰以表达在患者体内表达的转基因。因此,本发明的组合物或疫苗包括赘生性(例如,肿瘤)细胞,其对于经历治疗的患者为自体或同种异体的,并且最优选地是与困扰患者的细胞相同的一般类型的肿瘤细胞。例如,通常向患有黑素瘤的患者施用从黑素瘤获得的基因修饰细胞。用于本发明中的使肿瘤细胞灭活的方法(例如使用放射)是本领域所熟知的。
本发明的灭活肿瘤细胞与一种或多种共刺激分子或药剂一起施用给患者。优选共刺激剂包括刺激树突细胞诱导、募集和/或成熟的一种或多种细胞因子。评估此类共刺激剂的方法在文献中是熟知的。DC的诱导和成熟通常通过在刺激之后某些膜分子如CD80和CD86的增加的表达,和/或如IL-12和I型干扰素的促炎细胞因子的分泌来评估。
在优选的实施方案中,对灭活肿瘤细胞本身进行修饰以表达并分泌刺激树突细胞诱导、募集和/或成熟的一种或多种细胞因子。本发明以示例性的方式描述关于GM-CSF的用途。因此,举例而言,肿瘤细胞可表达编码GM-CSF的转基因,如美国专利No.5,637,483、No.5,904,920、No.6,277,368和No.6,350,445以及美国专利公布No.20100150946中所述,所述专利各自以引用方式明确并入本文。用于治疗胰癌的GM-CSF-表达性基因修饰癌细胞形式或“细胞因子表达性细胞疫苗”描述于美国专利No.6,033,674和No.5,985,290中,所述专利均以引用方式明确并入本文。
代替或与GM-CSF一起,可由此类灭活肿瘤细胞和/或旁邻细胞表达的其它合适细胞因子包括但不限于CD40配体、IL-12、CCL3、CCL20和CCL21中的一个或多个。此清单并非旨在进行限制。
虽然优选的是施用给受试者的灭活肿瘤细胞表达一种或多种目标细胞因子,但肿瘤细胞系可伴随有灭活旁邻细胞系,其表达并分泌刺激树突细胞诱导、募集和/或成熟的一种或多种细胞因子。旁邻细胞系可提供刺激树突细胞诱导、募集和/或成熟的所有细胞因子,或可补充由灭活肿瘤细胞表达并分泌的刺激树突细胞诱导、募集和/或成熟的细胞因子。举例而言,免疫调节细胞因子表达性旁邻细胞公开于美国专利No.6,464,973和No.8,012,469,Dessureault等,Ann.Surg.Oncol.14:869-84,2007以及Eager and Nemunaitis,Mol.Ther.12:18-27,2005,所述专利各自以引用方式明确并入本文。
所谓“粒细胞-巨噬细胞集落刺激因子(GM-CSF)多肽”是指具有免疫调节活性并且与GenBank登录号AAA52122.1具有至少约85%氨基酸序列同一性的细胞因子或其片段。
疫苗
在某些实施方案中,CDN组合物与一种或多种疫苗联合施用,所述疫苗旨在刺激对一种或多种预定抗原的免疫应答。可用于本发明中的靶抗原的实例在下表中列出。靶抗原还可为包含表格所列抗原的免疫活性部分的片段或融合多肽。此清单并非旨在进行限制。
表1.抗原。
对于其合适抗原在本领域中已知的其它生物包括但不限于沙眼衣原体(Chlamydia trachomatis)、酿脓链球菌(Streptococcus pyogenes)(A群链球菌)、无乳链球菌(Streptococcus agalactia)(B群链球菌)、肺炎链球菌(Streptococcus pneumonia)、金黄色葡萄球菌(Staphylococcus aureus)、大肠杆菌(Escherichia coli)、流感嗜血杆菌(Haemophilus influenzae)、脑膜炎奈瑟氏菌(Neisseria meningitidis)、淋病奈瑟氏菌(Neisseria gonorrheae)、霍乱弧菌(Vibrio cholerae)、沙门氏菌(Salmonella)种(包括伤寒沙门氏菌(typhi)、鼠伤寒沙门氏菌(typhimurium))、肠道沙门氏菌(enterica)(包括幽门螺杆菌志贺氏杆菌(Helicobactor pylori Shigella flexneri)和其它D群志贺氏杆菌种)、鼻疽伯克氏菌(Burkholderia mallei)、类鼻疽伯克氏菌(Burkholderia pseudomallei)、克雷伯氏肺炎杆菌(Klebsiella pneumonia)、梭状芽孢杆菌(Clostridium)种(包括艰难梭状芽胞杆菌(C.difficile))、副溶血弧菌(Vibrio parahaemolyticus)和创伤弧菌(V.vulnificus)。此清单并非旨在进行限制。
药物组合物
如本文使用的术语“药物”是指旨在用于治愈、治疗或预防疾病并且经历美国食品和药物管理局(或其非美国的相应部门)成为处方或非处方药物品的批准过程的化学物质。配制并施用此类组合物的技术的详细内容可见于Remington,The Science and Practice of Pharmacy,第21版(Mack Publishing Co.,Easton,PA)以及Nielloud和Marti-Mestres,Pharmaceutical Emulsions and Suspensions:第2版(Marcel Dekker,Inc,New York)。
出于本公开目的,药物组合物可通过包括口服、肠胃外、吸入喷雾、局部或经直肠的各种手段以包含药学上可接受的载体、佐剂和媒介物的制剂形式来施用。如本文使用的术语肠胃外包括但不限于使用各种输注技术来皮下、静脉内、肌内、动脉内、皮内、鞘内和硬膜外注射。如本文使用的动脉内和静脉内注射包括通过导管来施用。也涵盖通过冠状动脉内支架和冠状动脉内储集器来施用。本发明化合物的肿瘤内施用可直接局部活化浸润的DC、直接促进肿瘤细胞凋亡或使肿瘤细胞对细胞毒性剂敏化。如本文使用的术语口服包括但不限于口服摄取,或通过舌下或口腔途径来递送。口腔施用包括流体饮料、能量棒以及丸剂制剂。
药物组合物可呈适合于预期施用方法的任何形式。例如,当用于口服使用时,可制备例如片剂、含片、锭剂、水性悬浮液或油性悬浮液、可分散粉末或颗粒剂、乳液、硬或软胶囊、糖浆或酏剂。可根据本领域已知的用于制备药用组合物的任意方法制备预期用于口服使用的组合物,并且此类组合物可包含一种或多种试剂(包括甜味剂、调味剂、着色剂和防腐剂)以提供适口的制剂。包含与适合于制备片剂的药学上可接受的无毒赋形剂掺合的药物化合物的片剂是可接受的。这些赋形剂可为例如惰性稀释剂,例如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠;成粒剂和崩解剂(例如,玉米淀粉或藻酸);粘合剂(例如,淀粉、明胶或阿拉伯胶);以及润滑剂;例如硬脂酸镁、硬脂酸或滑石粉。片剂可为未经包衣或通过已知技术进行包衣的,所述技术包括肠溶包衣、结肠包衣或微囊封以延缓胃肠道内的崩解和吸收和/或提供较长时间内的持续作用。例如,可采用延时材料如单独或与蜡一起的单硬脂酸甘油酯或二硬脂酸甘油酯。
用于口服使用的制剂也可作为硬明胶胶囊存在,其中所述药物化合物与惰性固体稀释剂(例如磷酸钙或高岭土)混合,或作为软明胶胶囊存在,其中所述活性成分与水或油介质(例如花生油、液体石蜡或橄榄油)混合。
药物组合物可配制成与适合于制备水性悬浮液的赋形剂掺合的水性悬浮液。此类赋形剂包括悬浮剂,例如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、海藻酸钠、聚乙烯吡咯烷酮、黄蓍胶和阿拉伯胶,以及分散剂或湿润剂,例如天然存在的磷脂(例如卵磷脂)、氧化烯与脂肪酸的缩合产物(例如聚氧乙烯硬脂酸酯)、氧化乙烯与长链脂族醇的缩合产物(例如十七亚乙基氧基鲸蜡醇)、氧化乙烯与衍生自脂肪酸和己糖醇酐的偏酯的缩合产物(例如聚氧乙烯脱水山梨糖醇单油酸酯)。水性混悬剂还可以含有一种或多种防腐剂如对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯、一种或多种着色剂、一种或多种调味剂以及一或多种甜味剂如蔗糖或糖精。
可通过将活性成分悬浮在植物油(例如花生油、橄榄油、芝麻油或椰子油)或矿物油(例如液体石蜡)中来配制油性悬浮液。口服悬浮液还可含有增稠剂,例如蜂蜡、硬石蜡或鲸蜡醇。可添加甜味剂(例如上文所述的那些)和调味剂,以提供适口的口服制剂。可以通过添加抗氧化剂(例如抗环血酸)来保存这些组合物。
适合用于通过添加水来制备水性悬浮液的本公开的可分散粉末和颗粒剂提供了与分散剂或润湿剂、悬浮剂以及一种或多种防腐剂掺合的活性成分。通过以上已经提到的那些来例示合适的分散剂或润湿剂和悬浮剂。还可以存在额外的赋形剂,例如甜味剂、调味剂和着色剂。
本公开的药物组合物也可以水包油乳液的形式存在。油相可为植物油如橄榄油或花生油,矿物油如液体石蜡,或这些的混合物。合适的乳化剂包括天然存在的树胶,例如阿拉伯树胶和黄蓍树胶;天然存在的磷脂,例如大豆卵磷脂,由脂肪酸衍生的酯或偏酯;和己糖醇酐,例如脱水山梨糖醇单油酸酯;以及这些偏酯与环氧乙烷的缩合物,例如聚氧乙烯脱水山梨糖醇单油酸酯。所述乳液还可包含甜味剂和调味剂。
可以使用诸如甘油、山梨醇或蔗糖的甜味剂来配制糖浆和酏剂。此类制剂还可包含缓和剂、防腐剂、调味剂或着色剂。
本公开的药物组合物可呈无菌可注射制剂的形式,例如作为无菌可注射水性或油性悬浮液。可根据已知技术,使用上述那些合适的分散剂或润湿剂以及悬浮剂来配制所述悬浮液。无菌可注射制剂还可为处于肠胃外可接受的无毒稀释剂或溶剂中的无菌可注射溶液或悬浮液,例如处于1,3-丁二醇中的溶液或制备为冻干粉末。可以采用的可接受的媒介物和溶剂包括水、林格溶液和等渗氯化钠溶液。另外,无菌的非挥发性油类可在常规上用作溶剂或悬浮介质。出于此目的,可使用包括合成甘油一酯或甘油二酯的任何温和的非挥发性油。此外,脂肪酸(例如油酸)可类似地用于注射物的制备。
可与载体材料此组合以产生单一剂型的活性成分的量将根据所治疗的主体和特定的施用模式而改变。例如,旨在对人口服施用的延时释放制剂可包含大约20至500mg的与合适且适宜量的载体材料相混合的活性材料,所述载体材料可在总组合物的约5%至约95%变化。优选的是制备提供便于施用的容易测量的量的药物组合物。通常,全身施用的有效量为约0.1mg/kg至约100mg/kg并且取决于许多因素,包括例如受试者(例如,哺乳动物如人)的年龄和体重、需要治疗的确切病症和其严重性、施用途径并且最终由主治医师或兽医酌处。然而,应理解,任何特定患者的具体剂量水平取决于各种因素,包括所使用的具体化合物的活性,所治疗的个体的年龄、体重、一般健康状况、性别和饮食;施用时间和途径;排泄速率;以前施用的其它药物;和经历疗法的特定病症的严重性,如本领域的技术人员所充分理解的。
如上文所提到的,适于口服施用的本文公开的制剂可提供为诸如各自包含预定量的活性成分的胶囊、扁囊剂或片剂的离散单位;粉末或颗粒剂;在水性液体或非水性液体中的溶液或悬浮液;或水包油液体乳液或油包水液体乳液。药物组合物还可以作为大丸剂、药糖剂或糊剂施用。
片剂可通过压制或模制而制备,任选地含有一种或多种辅助成分。压制的片剂可通过在合适的机器中对自由流动形式(如粉末或颗粒剂)的活性成分进行压制来制备,所述活性成分任选与粘合剂(例如,聚维酮、明胶、羟丙乙基纤维素)、润滑剂、惰性稀释剂、防腐剂、崩解剂(例如,羟乙酸淀粉钠、交联聚维酮、交联羧甲基纤维素钠)表面活性剂或分散剂混合。模制的片剂可以通过在合适的机器中对用惰性液体稀释剂润湿的粉末状化合物的混合物进行模制来制备。片剂可任选地包衣或刻痕并且可配制以便提供其中活性成分的缓慢或控制释放,使用例如不同比例的羟基丙基甲基纤维素来提供所需释放特征。片剂可任选提供有肠溶或结肠包衣以在肠的部分而非胃内提供释放。当式1化合物易于发生水解时,这对于此类化合物是特别有利的。
适于在口中局部施用的制剂包括锭剂,其在调味的基料(通常是蔗糖和阿拉伯胶或黄蓍胶)中包含活性成分;软锭剂(pastille),其在惰性基料如明胶和甘油或蔗糖和阿拉伯胶中包含活性成分;以及漱口剂,其在合适的液体载剂中包含活性成分。
直肠施用的制剂可具有包含例如可可脂或水杨酸盐的合适基料的栓剂形式呈现。
适用于经阴道施用的制剂可作为阴道栓剂、卫生棉条、霜剂、凝胶剂、糊剂、泡沫剂或喷雾制剂提供,它们除了活性成分外还包含本领域已知合适的载体。
用于肠胃外施用的制剂包括水性和非水性等渗无菌注射溶液,其可包含抗氧化剂、缓冲剂、抑菌剂和使得制剂与预期接受者的血液等渗的溶质;以及水性和非水性无菌悬浮液,其可包括悬浮剂和增稠剂。可将制剂提供于单位剂量或多剂量容器(例如密封安瓿及小瓶)中,并且可储存于冷冻干燥(冻干)条件下,仅需要在即将使用之前添加无菌液体载体(例如注射用水)。注射液和悬浮液可由先前所述种类的无菌粉末、颗粒剂和片剂制备。
如本文使用,药学上可接受的盐包括但不限于:醋酸盐、吡啶、铵、哌嗪、二乙胺、烟酰胺、甲酸盐、尿素、钠、钾、钙、镁、锌、锂、肉桂酸盐、甲氨基、甲磺酸盐、苦味酸盐、酒石酸盐、三乙氨基、二甲氨基和三(羟甲基)氨基甲烷。额外的药学上可接受的盐是本领域技术人员已知的。
特定患者的有效量可取决于诸如以下的多种因素:所治疗的病症、患者的总体健康状况、施用途径和剂量以及副作用的严重性。可获得治疗和诊断方法的指导(参见例如Maynard等(1996)A Handbook of SOPs for Good Clinical Practice,Interpharm Press,Boca Raton,FL;Dent(2001)Good Laboratory and Good Clinical Practice,Urch Publ.,London,UK)。
有效量可以一个剂量给出,但是不限于一个剂量。因此,施用可为2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20次或更多次施用药物组合物。当在本发明方法中存在药物组合物的不止一次施用时,施用可隔开1分钟、2分钟、3、4、5、6、7、8、9、10分钟或更多分钟的时间间隔,间隔约1小时、2小时、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24小时等。在小时的语境中,术语“约”意指±30分钟内的任何时间间隔。施用还可隔开1天、2天、3天、4天、5天、6天、7天、8天、9天、10天、11天、12天、13天、14天、15天、16天、17天、18天、19天、20天、21天以及它们的组合的时间间隔。本发明不限于隔开相等时间的给药间隔,而是涵盖间隔不等的给药。
例如一次/周、两次/周、三次/周、四次/周、五次/周、六次/周、七次/周、每两周一次、每三周一次、每四周一次、每五周一次等的给药方案可用于本发明。给药方案涵盖例如1周、2周、3周、4周、5周、6周、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、12个月的总给药时段。
提供上述给药方案的循环。循环可以约例如每7天;每14天;每21天;每28天;每35天;42天;每49天;每56天;每63天;每70天;等等来重复。非给药的间隔可存在于循环之间,其中间隔可为约例如7天;14天;21天;28天;35天;42天;49天;56天;63天;70天;等等。在此语境中,术语“约”意指±1天、±2天、±3天、±4天、±5天、±6天或±7天。
额外的治疗剂共同施用的方法是本领域中熟知的(Hardman等(编)(2001)Goodman and Gilman’s The Pharmacological Basis of Therapeutics,第10版,McGraw-Hill,New York,NY;Poole和Peterson(编)(2001)Pharmacotherapeutics for Advanced Practice:A Practical Approach,Lippincott,Williams&Wilkins,Phila.,PA;Chabner和Longo(编)(2001)Cancer Chemotherapy and Biotherapy,Lippincott,Williams&Wilkins,Phila.,PA)。
如所提到的,本发明的组合物优选地配制成供肠胃外或肠道递送的药物组合物。典型的供施用给动物的药物组合物包含药学上可接受的媒介物,例如水溶液、无毒赋形剂,包括盐、防腐剂、缓冲剂等。参见例如Remington's Pharmaceutical Sciences,第15版,Easton编,Mack Publishing Co.,第1405-1412和1461-1487页(1975);The NationalFormulary XIV,第14版,,American Pharmaceutical Association,Washington,DC(1975)。非水性溶剂的实例为丙二醇、聚乙二醇、植物油以及可注射的有机酯如油酸乙酯。水性载体包括水、醇/水溶液、盐水溶液、肠胃外媒介物如氯化钠、林格氏葡萄糖等。静脉内媒介物包括流体和营养补充剂。防腐剂包括抗微生物剂、抗氧化剂、螯合剂和惰性气体。药物组合物的各种组分的pH和精确浓度根据本领域的常规技能进行调整。
反复施用特定的疫苗(同源加强)已证实可有效加强体液应答。这样的方法在加强细胞免疫方面可能无效,原因是在免疫前载体往往损害稳固的抗原呈递并生成适当的炎性信号。一种解决此问题的方法是按顺序施用使用不同抗原递送体系的疫苗(异源加强)。在异源加强的方案中,至少一个初免或加强递送包括递送本文所述的灭活肿瘤细胞/环状嘌呤二核苷酸组合物。方案的异源分支可包括使用以下策略中的一种或多种来递送抗原:
包含目标抗原的灭活或减毒的细菌或病毒,其为已使用一些变性条件来处理以使得其在发动病原入侵方面无效或低效的粒子;
纯化抗原,其通常为从病原体的细胞培养物纯化的天然产生抗原或包含病原体的组织样品,或其重组型式;
活病毒或细菌递送载体,其重组工程化以在受试者的宿主细胞中表达和/或分泌抗原。这些策略依赖于对病毒或细菌载体进行减毒(例如,通过基因工程),使其成为非病原性和无毒的;
抗原呈递细胞(APC)载体,例如树突细胞(DC)载体,其包括负载有抗原,或转染有包含编码抗原(例如,用于治疗去势难治性转移性前列腺癌的(Dendreon Corporation))的核酸的组合物的细胞;
脂质体抗原递送媒介物;和
裸DNA载体和裸RNA载体,其可通过基因枪、电穿孔、细菌膜影(bacterial ghost)、微球、微粒、脂质体、聚阳离子纳米粒子等来施用。
初免疫苗和加强疫苗可通过以下途径的任一种或组合进行施用。在一个方面,初免疫苗和加强疫苗通过相同的途径施用。在另一个方面,初免疫苗和加强疫苗通过不同的途径施用。术语“不同的途径”涵盖但不限于身体上的不同位点,例如为口腔、非口腔、肠道、肠胃外、直肠、节间(淋巴结)、静脉内、动脉、皮下、肌内、肿瘤内、肿瘤周围、肿瘤内、输注、粘膜、鼻腔、脑脊髓空间或脑脊液中等的位点,以及通过不同的模式,例如口腔、静脉内和肌内。
有效量的初免或加强疫苗可以一个剂量给予,但不限于一个剂量。因此,施用可为2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20次或更多次施用疫苗。如果存在不止一次疫苗施用,则施用可隔开1分钟、2分钟、3、4、5、6、7、8、9、10分钟或更多分钟的时间间隔,间隔约1小时、2小时、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24小时等。在小时的语境中,术语“约”意指±30分钟内的任何时间间隔。施用还可隔开1天、2天、3天、4天、5天、6天、7天、8天、9天、10天、11天、12天、13天、14天、15天、16天、17天、18天、19天、20天、21天以及它们的组合的时间间隔。本发明不限于隔开相等时间的给药间隔,而是涵盖间隔不等的给药,例如初免计划由第1天、4天、7天和25天的施用组成,这只是给出非限制性实例。
实施例
以下实施例用于阐述本发明。这些实施例绝对无意限制本发明的范围。
实施例1.一般方法
购买适于溶液相寡核苷酸合成的无水溶剂和试剂并使用无水技术在干燥氩气或氮气下进行处理。亚酰胺偶联反应和环化在干燥氩气或氮气下于无水乙腈或吡啶中进行。无水吡啶中所有反应的起始物料通过从吡啶浓缩(三次)来干燥。使用Fluka 60A高纯度级或Merck级9385二氧化硅以甲醇在二氯甲烷中的梯度来进行制备型硅胶快速色谱法。在配备有于254nm处监测的ProStar 330光二极管阵检测器的Varian ProStar 210HPLC系统上使用Varian Microsorb 10微米C18250×4.6mm或Varian 3微米C18 100×4.6mm柱和10mM TEAA和乙腈梯度来进行分析型HPLC。在配备有于254nm处监测的SPD-20A UV/Vis检测器的Shimadzu制备型LC20-AP HPLC系统上使用Varian Microsorb 60-8C-18 41.6×250mm柱以及10mM TEAA和乙腈的梯度以50ml/min的流速进行制备型HPLC。使用C-18Sep-Pak(Waters)以3%(wt/wt)的负载进行固相萃取。在配有PDA、MS和ELSD检测的单四极Shimadzu 2010EV仪上使用Shimadzu LC20D分析型HPLC来获得LC/MS(ESI/APCI)。高分辨率FT-ICR质谱仪购自耶鲁大学(New Haven,CT)的WM Keck基金生物技术资源实验室(Foundation Biotechnology Resource Laboratory)和加利福尼亚大学伯克利分校(UC Berkeley)的QB3/Chemistry Mass Spect Lab。
1H、31P、1H-1H COSY(2D NMR相关光谱学)、1H-31P HMBC(异核多键相关光谱学)光谱在具有10uL D2O的d6-DMSO中(D2O添加后16小时的延迟)在45℃下于Varian INOVA-500NMR光谱仪上获得,所述光谱仪针对1H以500MHz操作并且针对31P以202MHz操作。将所得FID转移至PC并使用来自Acorn NMR Inc.的NUTS NMR处理软件进行处理。化学位移参考DMSO溶剂,2.50ppm(1H)。根据参考NMR光谱1的IUPAC建议,31P化学位移使用“统一标度(unified scale)”至0ppm的绝对1H频率来参考。一些1H和31P光谱在JEOL ECX-400NMR光谱仪上获得,所述光谱仪针对1H以400MHz操作并且针对31P以162MHz操作。
使用绝对值方式以直接维度的2048个数据点和间接维度的256个时间点获得梯度COSY光谱。使用正弦钟形平方函数对这两种维度进行变迹。将间接维度用零填充以得到2048×2048个点的最终矩阵大小以及在两个维度上3.91赫兹/数据点的分辨率。
磷酸二酯键处的区域化学结构(regiochemistry)分配:1H-1H COSY结合1H-31P HMBC(并且在一些情况中,磷解偶联)实验用于提供磷酸二酯键的区域化学结构为2’,5’-3’,5’的直接证据(参见针对9a的实验讨论和图3A-G)。类似的1H-31P HMBC实验确认在最终的甲硅烷基脱保护或离子交换后所有合成的环状二核苷酸的磷酸二酯键处的非典型区域化学结构(2’,5’-3’,5’)
RR-和RS-非对映体(合成序列的主要CDN产物)的分配遵循文献方法(Zhao等,Nucleosides,Nucleotides and Nucleic Acids289:352-378,2009)。
所有CDN产物(图2A-2C)是≥95%纯的,如C18反相HPLC分析(254nm处的UV检测)所指示
缩写和首字母缩略:鸟嘌呤=G.异丁酰基鸟嘌呤=Gib.4,4-二甲氧基三苯甲基=DMT.OCH2CH2CN=CEO.叔丁基二甲基甲硅烷基=TBS.腺嘌呤=A.苯甲酰基腺嘌呤=ABz.环状-[A(2’,5’)pA(3’,5’)p]=ML-CDA=19a(TEA盐).二硫代-[RP,RP]-环状-[A(2’,5’)pA(3’,5’)p]=ML-RR-CDA=19b(TEA盐);21(钠盐);22(铵盐).二硫代-[RP,SP]-环状-[A(2’,5’)pA(3’,5’)p]=ML-RS-CDA=19c(TEA盐).环状-[G(2’,5’)pG(3’,5’)p]=ML-CDG=9a(TEA盐).二硫代-[RP,RP]-环状-[G(2’,5’)pG(3’,5’)p]=ML-RR-CDG=9b(TEA盐).二硫代-[RP,SP]-环状-[G(2’,5’)pG(3’,5’)p]=ML-RS-CDG=9c(TEA盐).环状[G(2’,5’)pA(3’,5’)p]=ML-cGAMP.二硫代-[RP,RP]-环状-[G(2’,5’)pA(3’,5’)p]=ML-RR-cGAMP=20(TEA盐).一硫代-环状-[A(2’,5’)pA(3’,5’)Rp]=ML-3’,5’-R-CDA=19d(TEA盐).2’-O-肉豆蔻酰基-环状-[G(2’,5’)pG(3’,5’)p]=C14-ML-CDG=10(TEA盐).ML-cGAMP=2’,3’-cGAMP=环状-[G(2’,5’)pA(3’,5’)p]=23(TEA盐)
ML-cGAMP(图2c中的结构23)以酶促方式由细胞cGAS制备并通过制备型HPLC纯化。
实施例2.针对ML-CDG系列的一般实验(图2a):环状[G(2’,5’)pG(3’,5’)p]9a的合成。
1)3的制备。向4.87g(5.0摩尔)N2-异丁酰基-5'-O-(4,4'-二甲氧基三苯甲基)-2'-O-叔丁基二甲基甲硅烷基-3'-O-[(2-氰乙基)-N,N-二异丙基氨基膦基]鸟苷(1)在25ml乙腈中的溶液添加0.18ml(10摩尔)水和1.23g(6摩尔)三氟乙酸吡啶鎓。在5分钟室温搅拌后,添加25ml叔丁胺,在室温下搅拌反应15分钟。减压下除去溶剂,得到呈泡沫物的2,然后将其与乙腈(2×50ml)共蒸发。向2在60ml二氯甲烷中的溶液添加0.9ml(50摩尔)水和60ml 6%(v/v)二氯乙酸在二氯甲烷中的溶液(44摩尔)。10分钟后,在室温下通过添加吡啶(7.0ml,87摩尔)来淬灭反应。将反应混合物浓缩成油状物,然后通过与40ml无水乙腈共蒸发三次来干燥,最后得到12ml体积的3。
2)4的无水溶液的制备。将N2-异丁酰基-5'-O-(4,4'-二甲氧基三苯甲基)-3'-O-叔丁基二甲基甲硅烷基-2'-O-[(2-氰乙基)-N,N-二异丙基氨基膦基]鸟苷(4,6.33g,6.5摩尔)溶解于40ml无水乙腈中,通过与40ml无水乙腈共蒸发三次来干燥,最后得到20ml。添加十个分子筛,然后将溶液在氩气下储存直至使用。
3)3和4偶联以在氧化和脱三苯甲基后得到2’,5’线性二聚体6a。将20ml乙腈中的共沸干燥的4(6.5摩尔)通过注射器添加到3(5.0毫摩尔)中。在室温下搅拌5分钟后,添加2.37ml(15摩尔)5.5M叔丁基过氧化氢在癸烷中的溶液,然后在室温下搅拌反应30分钟。然后将反应冷却至0℃,添加1.25g NaHSO3在2.5ml水中的溶液,除去冰浴,搅拌反应5分钟。浓缩反应成泡沫物,将其溶于80ml二氯甲烷中。添加0.9ml水和80ml 6%(v/v)二氯乙酸在二氯甲烷中的溶液,在室温下搅拌反应10分钟。添加50ml吡啶以淬灭二氯乙酸。减压下除去溶剂,得到呈固体的粗6a。
4)对6a进行环化以得到7a。将6a溶解于50ml无水吡啶中,并将5ml(总反应的1/10th,大约0.5摩尔)通过注射器转移到150ml无水吡啶中。将其浓缩至体积为大约100ml。然后添加2-氯-5,5-二甲基-1,3,2-二氧杂磷杂己烷-2-氧化物(DMOCP,0.35g,1.8摩尔),在室温下搅拌反应30分钟。立即添加0.32ml水,然后添加0.16g碘,在室温下搅拌反应5分钟。然后将反应混合物倾入350ml含0.1g NaHSO3的水中并在室温下搅拌5分钟。在搅拌的同时缓慢添加2g NaHCO3,然后倾入分液漏斗中,用400ml 1:1乙酸乙酯:二乙醚萃取。将水层用400ml 1:1乙酸乙酯:二乙醚再次萃取,合并有机层,经硫酸钠干燥,减压下浓缩,得到0.75g含7a的混合物,为完全受保护的环状-[G(2’,5’)pG(3’,5’)p]。
5)将粗7a用甲胺脱保护以得到粗8a。向750mg 7a中添加18ml甲胺在无水乙醇中的溶液(33重量%),搅拌混合物90分钟,此时HPLC分析指示反应完成。将反应混合物浓缩得到油状物,其在用10ml己烷/乙酸乙酯(50:50)处理时得到灰白色固体。倾析出研磨/洗涤溶剂,减压下除去残余溶剂,得到240mg灰白色固体。
6)粗8a的制备型HPLC。将120mg份的粗8a溶于5ml CH3CN/10mM三乙基乙酸铵水溶液(20/80)中。0.45微米PTFE过滤后,将注射样品施加至C-18Dynamax柱(40×250mm)。用乙腈和10mM三乙基乙酸铵水溶液的梯度进行洗脱(20%至50%CH3CN,以50ml/min流速进行20分钟)。合并来自两次HPLC操作的含有纯8a的HPLC级分,蒸发除去CH3CN,冻干除去大部分的水和挥发性缓冲液,在与乙腈(3×4ml)共沸干燥后得到42mg为双-三乙基铵盐的纯8a。(还可延缓制备型HPLC纯化直至最后一个步骤)。HRMS(FT-ICR)m/z:[M-H]-计算值C32H51N10O14P2Si2917.2606;实测值917.2622。1H NMR(DMSO-d6+痕量D2O)45℃δ8.22(1H,s),7.85(1H,s),5.76-5.79(2H,dd),5.21(1H,m),4.85(1H,m),4.58(1H,t),4.49(1H,d),4.31(1H,m),4.21(1H,m),3.97(1H,d),3.83(3H,m),2.94(12H,m),1.12(18H,t),0.90(9H,s),0.72(9H,s),0.14(6H,d),0.09(3H,s),-0.02(3H,s)。31P NMR(DMSO-d6+痕量D2O)45℃.δ-1.26,-2.02(图3a-3c)。
7)用三乙胺三氢氟酸盐对8a的TBS基团脱保护,用TEAB中和,用C-18Sep-Pak进行固相萃取,得到为双-三乙基铵盐的纯9a。向40mg 8a中添加1.0ml三乙胺三氢氟酸盐。将混合物在室温下搅拌30小时。通过分析型HPLC确认反应完成后,样品通过逐滴添加至12ml激冷的1M三乙基碳酸氢铵中进行中和。将中和的溶液在Waters C-18Sep-Pak上脱盐,产物用CH3CN/10mM三乙基乙酸铵水溶液(1:1)洗脱。将CH3CN在减压下蒸发,将剩余水溶液冷冻和冻干过夜。从甲醇(3×3ml)进行多次蒸发并且从50%乙腈在甲醇中的溶液(1×3ml)进行最终蒸发,得到为29.3mg双-三乙基铵盐的环状-[G(2’,5’)pG(3’,5’)p](9a)。HRMS(FT-ICR)m/z:[M-H]-计算值C20H23N10O14P2689.0876;实测值689.0874。1H NMR(DMSO-d6+痕量D2O)45℃δ7.92(1H,s),7.90(1H,s),5.82(1H,d),5.80(1H,d),4.97(1H,m),4.85(1H,m),4.68(1H,m),4.31(1H,d),4.21(1H,t),4.10(2H,m),3.79(3H,m),2.91(14H,m),1.13(22H,t)。31P NMR(DMSO-d6)45℃.δ1.80,-1.05。
9a的HPLC保留时间为7.25分钟,相比于c-di-GMP的9.3分钟,使用10mM三乙基乙酸铵中2%至20%CH3CN的梯度在C-18柱(3微米,100×4.6mm,0.6ml/min.)上历经20分钟。HRMS(FT-ICR)确认预期的元素式:[M-H]-计算值C20H23N10O14P2689.0876;实测值689.0874。9a的31-P NMR显示在2.03ppm和-0.95ppm处有两个峰(积分1:1),符合2’,5’/3’,5’混合键(c[G(3’,5’)pG(3’,5’)p]和c[G(2’,5’)pG(2’,5’)p]例如由于对称性将只发出一个31-P NMR信号)。磷酸二酯键区域化学结构的直接证据通过1H-1H COSY结合磷解偶联实验以及1H-31P HMBC二维NMR(图3b和图3c)来获得。异头(H-1)质子在5.82ppm处以重叠的双二重峰出现。“A”标号给予低磁场异头(H-1)质子,“B”给予相对其稍高的磁场。以“A”和“B”核糖中的异头质子开始,1H-1H COSY实验(图3b)允许H-2A(4.96ppm)、H-3A(4.31ppm)以及H-2B(4.67ppm)和H-3B(4.84ppm)的分配。低磁场磷(2.03ppm)的放射将H-3B转化成双峰,而高磁场磷(-0.95ppm)的放射导致H-2A的复杂多重峰简化。在两次解偶联实验中,还观察到5’核糖亚甲基多重峰的简化。二维1H-31P HMBC确认解偶联实验的结果。1H-1H COSY结果结合磷解偶联和1H-31P HMBC实验从而提供磷酸二酯键的区域化学结构为2’,5’/3’,5’并且9a为环状[G(2’,5’)pG(3’,5’)p]的直接证据。
实施例3.针对ML-CDA系列的一般实验(图2b):环状[A(2’,5’)pA(3’,5’)p]钠盐21(参见化合物图2c)的合成。
1)13的制备。
向5g(5.15摩尔)N6-苯甲酰基-5'-O-(4,4'-二甲氧基三苯甲基)-2'-O-叔丁基二甲基甲硅烷基-3'-O-[(2-氰乙基)-N,N-二异丙基氨基膦基]腺苷(11)在25ml乙腈中的溶液添加0.18ml(10摩尔)水和1.20g(6.2摩尔)三氟乙酸吡啶鎓。在5分钟室温搅拌后,添加25ml叔丁胺,在室温下搅拌反应15分钟。减压下除去溶剂,得到呈泡沫物的12,然后将其与乙腈(2×50ml)共蒸发,然后溶解于60ml二氯甲烷中。向此溶液中添加水(0.9ml,50摩尔)和60ml 6%(v/v)二氯乙酸(44摩尔)在二氯甲烷中的溶液。10分钟后,在室温下将反应通过添加吡啶(7.0ml,87摩尔)来淬灭,浓缩成油状物,然后通过与40ml无水乙腈共蒸发三次来干燥,最后得到12ml体积的13。
2)14的无水溶液的制备。
N6-苯甲酰基-5'-O-(4,4'-二甲氧基三苯甲基)-3'-O-叔丁基二甲基甲硅烷基-2'-O-[(2-氰乙基)-N,N-二异丙基氨基膦基]腺苷(14,6.4g,6.6摩尔)溶解于40ml无水乙腈中,通过与40ml无水乙腈共蒸发三次来干燥,最后得到20ml。添加十个分子筛,然后将溶液在氩气下储存直至使用。
3)2’,5’-一硫代-线性二聚体16的制备。
将20ml乙腈中的共沸干燥的14(6.4g,6.6摩尔)通过注射器添加到13(5.15摩尔)在12ml无水乙腈的溶液中。在室温下搅拌5分钟后,添加1.14g(5.6摩尔)3-((N,N-二甲基氨基亚甲基)氨基)-3H-1,2,4-二噻唑-5-硫酮(DDTT),然后在室温下搅拌反应30分钟。浓缩反应,将残余油状物溶解于80ml二氯甲烷中。添加水(0.9ml,50摩尔)和80ml 6%(v/v)二氯乙酸(58摩尔)在二氯甲烷中的溶液,在室温下搅拌反应10分钟。添加50ml吡啶以淬灭二氯乙酸。减压下除去溶剂,得到呈固体的粗16b。
4)对16b进行环化和硫化以得到受保护的环状-二硫代非对映异构体17b和17c。
将16b溶解于150ml无水吡啶中,将其浓缩降至大约100ml的体积。然后添加2-氯-5,5-二甲基-1,3,2-二氧杂磷杂己烷-2-氧化物(DMOCP,3.44g,18摩尔),在室温下搅拌反应5分钟。立即添加3.2ml水,然后添加3-H-1,2-苯并二硫醇-3-酮(1.3g,7.7摩尔),在室温下搅拌反应5分钟。然后将反应混合物倾入700ml含20g NaHCO3的水中并在室温下搅拌5分钟,然后倾入分液漏斗中,用800ml 1:1乙酸乙酯:二乙醚萃取。用600ml 1:1乙酸乙酯:二乙醚再次萃取水层。合并有机层,减压下浓缩,得到大约11g含非对映异构体17b和17c的油状物。
5)含17b和17c的粗混合物的硅胶柱色谱法。
将以上粗混合物溶解于二氯甲烷中并施加至250g硅胶柱。使用甲醇在二氯甲烷中的梯度(0-10%)从柱洗脱得到所需非对映异构体。将含所需非对映异构体17b和17c的级分合并且浓缩,得到2.26g大约50%17b和50%17c。
6)完全受保护的环状非对映异构体17b和17c至粗18b和18c的脱保护。
将来自硅胶柱的2.26g粗17b和17c转移到厚壁玻璃压力管中。添加60ml甲醇和60ml浓氨水,将管在搅拌的同时于50℃油浴中加热16h(最近的操作自起始物料于此时开始消耗时为12h)。将反应混合物冷却至接近环境温度,用氮气流鼓泡30分钟,然后转移到大的圆底烧瓶中。减压下小心除去大部分挥发性物质以避起泡沫和崩沸。如果仍存在水,则将残余物冷冻和冻干至干燥。
7)对粗18b和18c进行制备型HPLC纯化以得到纯18b。
将含18b和18c的冻干粗混合物溶于大约50ml CH3CN/10mM三乙基乙酸铵水溶液(60/40)中。0.45微米PTFE过滤后,将4-5ml样品份施加至C-18Dynamax柱(40×250mm)。用乙腈和10mm三乙基乙酸铵水溶液的梯度进行洗脱(30%至50%CH3CN,以50ml/min流速进行20分钟)。合并来自制备型HPLC操作的含有纯18b的级分,蒸发除去CH3CN,冻干得到360mg为双-三乙基铵盐的纯18b(RPRP-非对映异构体)。
8)用三乙胺三氢氟酸盐对18b的两个TBS基团脱保护,用TEAB中和,用C-l8Sep-Pak进行固相萃取,冻干得到为双-三乙基铵盐的纯19b。
8a)向270mg(0.24摩尔)18b中添加5.0ml纯的三乙胺三氢氟酸盐。将反应混合物在室温下搅拌大约40h。通过分析型HPLC确认反应完成后,样品通过逐滴添加至45ml激冷搅拌的1M三乙基碳酸氢铵中进行中和。将中和的溶液在Waters C-18Sep-Pak上脱盐,产物用CH3CN/10mM三乙基乙酸铵水溶液(5:1)洗脱。将CH3CN在减压下蒸发,将剩余水溶液冷冻和冻干。从水进行多轮冻干,得到122mg(57%)为双-三乙基铵盐的二硫代-(Rp,Rp)-[环状-A(2’,5’)pA(3’,5’)p](19b)。
8b)将90mg(0.08摩尔)18b与10ml无水乙腈共蒸发三次。将干燥的残余物溶于0.4ml无水吡啶中。将带有孔针的烧瓶置于50℃油浴中,向搅拌的混合物中同时添加0.62ml三乙胺三氢氟酸盐和1.0ml三乙胺。将混合物在50℃下搅拌两小时。通过分析型HPLC确认反应完成后,样品通过逐滴添加至25ml激冷搅拌的1M三乙基碳酸氢铵中进行中和。将中和的溶液在Waters C-18Sep-Pak上脱盐,产物用CH3CN/10mM三乙基乙酸铵水溶液(1:4)洗脱。将CH3CN在减压下蒸发,将剩余水溶液冷冻和冻干。从水进行多轮冻干,得到54mg(76%)为双-三乙基铵盐的二硫代-(Rp,Rp)-[环状-A(2’,5’)pA(3’,5’)p](19b)。
8c)通过在45℃下于纯TEA-HF中加热,然后用TEAB中和,进行Sep-Pak脱盐和冻干而进行的TEA-HF脱保护的变型
将TEA·3HF(1mL,6.1摩尔)添加到配备有孔针的烧瓶中的18b(41mg,0.04摩尔)中,将混合物在45℃下搅拌。反应过程通过LC监测,在起始物料和单-TBS类似物(~2小时)消耗时,将混合物冷却至室温。在0℃下,将混合物缓慢吸移到1M TEAB(4.9ml)和TEA(1.6ml)的溶液中,通过pH试纸确认略呈碱性的pH。将中和的溶液在Waters C-18Sep-Pak(10g)上脱盐,产物用15%CH3CN/10mM三乙基乙酸铵水溶液洗脱。冻干得到21mg(64%)呈白色固体的19b(双-三乙基铵盐)。通过分析型HPLC(2-20%乙腈/10nM TEAA缓冲液–20分钟)分析显示出>95%纯度(图3h)。1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ8.58(s,1H),8.41(s,1H),8.18(s,1H),8.15(s,1H),6.12(d,J=8.0,1H),5.92(d,J=7.0,1H),5.30(td,J=8.5,4.0,1H),5.24-5.21(m,1H),5.03(dd,J=7.5,4.5,1H),4.39(d,J=4,1H),4.23(dd,J=10.5,4.0,1H),4.18(s,1H),4.14-4.08(m,2H),3.85-3.83(m,1H),3.73(d,J=12.0,1H),3.06(q,J=7.5,12H),1.15(t,J=7.5,1H);31P NMR(200MHz,45℃,(CD3)2SO-15μL D2O)δ58.81,52.54;HRMS(FT-ICR)m/z计算值C20H24O10N10P2S2(M–H)–689.0521,实测值689.0514。
8d)通过Gaffney等(2010)所述的丙酮沉淀对TEA-HF反应进行后处理也是可能的,但我们已使用以上章节8a-8c所述的修改获得了一定程度上更纯净的产物。
10)向钠盐的转化
ML-RR-CDA双-TEA盐(19b)通过下文所述的离子交换易于转化成药学上可接受的钠盐(21)。
ML-RR-CDA·2Na+(21)。将BT50W-X2树脂(100-200目,氢型)(100mg)用去离子水浆料填充到Bio-spin柱中。通过重力使过量去离子水排干。使3个柱床体积的1M NaOH(1ml)通过重力穿过柱,然后是5个柱床体积的去离子水(2ml)。通过重力使过量去离子水排干后,将ML-RR-CDA·2TEA(19b,10mg)在去离子水(1ml)中的溶液加载到柱上。将柱用5个柱床体积的去离子水(2ml)洗脱,收集级分并通过TLC板和UV灯来检查UV活性。将级分合并,冷冻并且冻干过夜,定量得到ML-RR-CDA·2Na+。1H NMR(500MHz,45℃,(CD3)2SO-30μL D2O)δ8.54(s,1H),8.40(s,1H),8.17(s,1H),8.167(s,1H),6.09(d,J=8.0,1H),5.92(d,J=8.0,1H),5.26(td,J=8.5,4.5,1H),5.21-5.19(m,1H),5.01(dd,J=7.5,4.5,1H),4.42(d,J=4,1H),4.23(dd,J=10.5,5.0,1H),4.17(s,1H),4.15-4.00(m,2H),3.90-3.82(m,1H),3.73-3.70(m,1H);31P NMR(200MHz,45℃,(CD3)2SO-30μL D2O)δ58.85,51.53(图3d-3g);HRMS(FT-ICR)m/z计算值C20H23O10N10P2S2(M–H)–689.0521,实测值689.0503。
类似地按照ML-CDG系列实验([19段])通过1H-1H COSY结合1H-31P HMBC二维NMR(图3e–3g)来获得磷酸二酯键区域化学结构的直接证据。
ML-RR-CDG(9b).化合物9b类似地按照ML-CDG合成,遵循ML-CDG系列实验的工序(段),但做以下修改(图2a):e)DDTT;h)3-H-1,2-苯并二硫醇-3-酮;n)以TEA盐形式获得,无需离子交换。
1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ7.98(s,1H),7.94(s,1H),5.85(d,J=9.0,1H),5.80(d,J=7.5,1H),5.25-5.23(m,1H),5.12(dd,J=8.5,4.5,1H),4.73(dd,J=8.0,4.5,1H),4.42(d,J=4.0,1H),4.22(t,J=7.5,1H),4.14-4.10(m,2H),3.94-3.90(m,2H),3.77-3.73(m,1H),3.05(q,J=7.0,12H),1.160(t,J=7.0,1H);31P NMR(200MHz,45℃,(CD3)2SO-15μL D2O)δ59.09,50.37;HRMS(FT-ICR)m/z计算值C20H23O12N10P2S2(M–H)–721.0419,实测值721.0410。
ML-RS-CDG(9c).化合物9c类似地按照ML-CDG合成,遵循ML-CDG系列实验的工序(段),但做以下修改(图2a):e)DDTT;h)3-H-1,2-苯并二硫醇-3-酮;k)收集[Rp,Sp]非对映体8c;n)以TEA盐形式获得,无需离子交换。
1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ8.01(s,1H),7.98(s,1H),5.86(d,J=8.5,1H),5.79(d,J=8.0,1H),5.29(dd,J=8.5,4.0,1H),5.20-5.19(m,1H),4.68(dd,J=8.5,4.0,1H),4.21-4.18(m,2H),4.10-4.05(m,3H),3.71-3.68(m,2H),2.96(q,J=7.0,12H),1.13(t,J=7.0,18H);31P NMR(200MHz,45℃,(CD3)2SO-15μL D2O)δ59.89,57.17;HRMS(FT-ICR)m/z计算值C20H24O12N10P2S2(M–H)–721.041904,实测值721.04143。
C14-ML-CDG(10):化合物10(图2c)类似地按照ML-CDG合成,遵循ML-CDG系列实验的工序(段),但做以下修改(图2a):n)肉豆蔻酸酐,DMF。
向9a的双-三乙胺盐(0.260g,0.291摩尔)中添加3.7ml DMF、0.3ml吡啶和128mg(0.292摩尔)肉豆蔻酸酐。将反应混合物在60℃下加热共5h,冷却至室温,然后用100ul MeOH淬灭。LC迹线指示向主要新产物的转化率为25%,物质的其余部分出现在起始物料的保留时间范围内。通过负模式下的LC/MS确认主要产物的物质为C14-酰化产物,m/z(M-1)为899(计算值C34H49N10O15P2-:889.3)。在蒸发后,将残余物溶于2ml CH3CN、3ml 0.1M TEAA和足量的MeOH中以使得大部分物料进入溶液中。在通过离心短暂下旋(spin down)以除去少量的颗粒物质后,通过C18-prep HPLC使用25%->50%CH3CN的梯度,在10mm TEAA中历经20分钟来纯化溶液。将含所需产物的级分合并且冻干至干燥,得到呈白色固体的36mg C14-ML-CDG 10(三乙基铵盐)。
1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ8.00(s,1H),7.90(s,1H),5.98(d,J=7.5,1H),5.83(d,J=8.5,1H),5.76(dd,J=7.5,4.5,1H),5.15-5.10(m,1H),4.90-4.85(m,1H),4.36(d,J=4.5,1H),4.30-4.27(m,1H),4.07(s,1H),3.94-3.90(m,3H),3.82-3.78(m,1H),3.04(q,J=7.0,12H),2.37-2.23(m,2H),1.51-1.43(m,2H),1.28-1.14(m,38H)。0.85(t,J=7.0,3H);31P NMR(200MHz,45℃,(CD3)2SO-15μLD2O)δ-1.36,-2.12;HRMS(FT-ICR)m/z计算值C34H49O15N10P2(M–H)–899.2860,实测值899.2834。
ML-CDA(19a).化合物19a类似地按照ML-RR-CDA合成,遵循ML-CDA系列实验的工序(段),但做以下修改(图2b):e)t-BuOOH;h)I2/H2O;n)以TEA盐形式获得,无需离子交换。
1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ8.44(s,1H),8.37(s,1H),8.16(s,1H),8.14(s,1H),6.08(d,J=8.0,1H),5.90(d,J=7.5,1H),5.10-5.0(m,3H),4.30(d,J=4.5,1H),4.3-4.19(m,1H),4.14(d,J=1.5,1H),4.05(q,J=11.5,2H),3.78-3.75(m,2H),2.90(q,J=7.5,18H),1.08(t,J=7.0,27H);31P NMR(200MHz,45℃,(CD3)2SO-15μLD2O)δ1.67,-0.47;HRMS(FT-ICR)m/z计算值C20H24O12N10P2(M–H)–657.097763,实测值657.09680。
ML-RS-CDA(19c).化合物19c类似地按照ML-RR-CDA合成,遵循ML-CDA系列实验的工序([0020]段),但做以下修改(图2b):k)收集[Rp,Sp]非对映体18c;n)以TEA盐形式获得,无需离子交换。
1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ8.52(s,1H),8.37(s,1H),8.16(s,1H),8.15(s,1H),6.10(d,J=8.5,1H),5.90(d,J=7.5,1H),5.45(dd,J=8.5,4.5,1H),5.31-5.26(m,1H),5.00(dd,J=8.5,4.5,1H),4.41-4.36(m,1H),4.22(d,J=5.0,1H),4.14-4.07(m,3H),3.70-3.67(m,3H),2.84(q,J=7.0,19H),1.08(t,J=7.5,29H);31P NMR(200MHz,45℃,(CD3)2SO-15μL D2O)δ59.98,57.35;HRMS(FT-ICR)m/z计算值C20H24O10N10P2S2(M–2H+Na)–711.0340,实测值711.0316。
ML-3’-5’-R-CDA(19e).化合物19e类似地按照ML-RR-CDA合成,遵循ML-CDA系列实验的工序(段),但做以下修改(图2b):e)t-BuOOH;h)3-H-1,2-苯并二硫醇-3-酮;n)以TEA盐形式获得,无需离子交换。
1H NMR(500MHz,45℃,(CD3)2SO-15μL D2O)δ8.49(s,1H),8.38(s,1H),8.17(s,1H),8.14(s,1H),6.09(d,J=8.5,1H),5.90(d,J=7.5,1H),5.23(dd,J=8.0,5.0,1H),5.12-5.04(m,2H),4.31(d,J=4.5,1H),4.21-4.14(m,3H),4.10(q,J=11.0,1H),3.80-3.71(m,2H),2.85(q,J=7.0,18H),1.08(t,J=7.5,27H);31P NMR(200MHz,45℃,(CD3)2SO-15μL D2O)δ59.32,-0.37;HRMS(FT-ICR)m/z计算值C20H23O11N10P2S(M–H)–673.0749,实测值673.0729。
呈氨盐形式的ML-RR-CDA(22)。化合物22似地按照ML-RR-CDA合成,遵循ML-CDA系列实验的工序([0020]段),但做以下修改(图2b):n)BT AG50W-X2树脂(100-200目,氢型),1MNH4OH。1H NMR(500MHz,45℃,(CD3)2SO-30μL D2O)δ8.80(s,1H),8.44(s,1H),8.39(s,2H),6.45(d,J=10.0,1H),6.34(s,1H),5.50(td,J=10.5,4.5,1H),5.21-5.15(m,1H),5.02(d,J=4.0,1H),4.92(d,J=4.5,1H),4.61-4.49(m,2H),4.30-4.27(m,2H);1HRMS(FT-ICR)m/z计算值C20H23O10N10P2S2(M–H)–689.0521,实测值689.0504。
ML-RR-cGAMP(20).化合物20(图2c)类似地按照ML-RR-CDA合成,遵循ML-CDA系列实验的工序(段),但做以下修改(图2b):d)pyr,4;n)以TEA盐形式获得,无需离子交换。
1H NMR(500MHz,45℃,(CD3)2SO-30μL D2O)δ8.34(s,1H),8.15(s,1H),8.01(s,1H),5.91(d,J=7.5,1H),5.86(d,J=8.5,1H),5.29-5.23(m,1H),5.17-5.14(m,1H),5.02(dd,J=7.5,4.0,1H),4.41(d,J=4.5,1H),4.25(dd,J=5.0,10.5,1H),4.13-4.03(m,3H),3.95-3.85(m,1H),3.78-3.74(m,1H),2.84(q,J=7.5,18H),1.08(t,J=7.5,28H);31P NMR(200MHz,45℃,(CD3)2SO-30μL D2O)δ58.81,50.91;HRMS(FT-ICR)m/z计算值C20H23O11N10P2S2(M–H)–705.0470,实测值705.0451。
实施例4.核糖2’-和3-取代的衍生物
可用于本发明中的衍生物的实例在图4-6中示出。
实施例5.CDN-诱导的I型干扰素表达
为了测定人细胞中通过天然分子和衍生物分子的每一者所诱导的作为佐剂效力标志的I型干扰素的相对水平,将4×105个THP1-BlueTMISG细胞(用IRF-诱导分泌型胚胎碱性磷酸酶报道基因(Invivogen)转染的人单核细胞细胞系,其在由五个IFN刺激应答元件构成的启动子的控制下表达碱性磷酸酶)用100μM环状[G(3’,5’)pG(3’,5’)p](CDG)、环状[G(2’,5’)pG(3’,5’)p](混合键或ML-CDG)或HBSS在37℃和5%CO2下孵育30分钟。在30分钟后,将细胞洗涤并铺板于含有10%FBS的RPMI培养基中的96孔培养皿上,在37℃和5%CO2下进行孵育。在过夜孵育后收集来自每个样品的细胞培养上清液,将20μL细胞培养上清液添加至180μL QUANTI-Blue试剂(Invivogen)中并孵育45分钟,以评价I型干扰素蛋白质水平。使用Versa Max动态分光光度计(Molecular Diagnostics)每3分钟取吸光度655nm处的读数。
如图7所述,在整个宽的时间点范围内,环状[G(2’,5’)pG(3’,5’)p](ML-CDG)诱导的IFN-β水平显著高于环状[G(3’,5’)pG(3’,5’)p]。这些结果证实,环状[G(2’,5’)pG(3’,5’)p]的纯化制剂与人单核细胞细胞系中的环状[G(3’,5’)pG(3’,5’)p]相比更显著地活化先天免疫应答。
为了测定环状[G(2’,5’)pG(3’,5’)p](ML-CDG)与环状[G(3’,5’)pG(3’,5’)p]相比所诱导的作为活化先天免疫的效力标志的IFN-α、-β和-γ的水平,在37℃和5%CO2下,将分离自独立人供体的1×106个原代人PBMC在96孔U形底板上用5或0.5μM环状[G(3’,5’)pG(3’,5’)p](CDG)或环状[G(2’,5’)pG(3’,5’)p](ML-CDG)、1μg/ml干扰素刺激DNA(ISD)或4μg/ml聚(I:C)孵育30分钟,其中利用Effectene转染试剂(Qiagen)将分子转移到PBMC中。ISD(干扰素刺激DNA)为非TLR依赖性的(Stetston,D.B.等,Immunity 24,93–103,2006年1月)并通过cGAS传导信号,因此是STING-依赖性的,而聚(I:C)可通过TLR3和RIG-I途径来传导信号,因此是非STING-依赖性的。在30分钟后,洗涤细胞并且更换含有10%FBS的RPMI培养基,并在37℃和5%CO2下孵育。在6小时的孵育后,收获细胞的一部分并通过实时定量RT-PCR来评估I型细胞因子干扰素α2(IFNA2)和干扰素β1(IFNB1)以及II型细胞因子基因干扰素γ(IFNG)的基因表达。通过实时定量RT-PCR,使用PrimePCR RNA纯化和cDNA分析系统并且在CFX96基因循环仪(all BioRad)上进行操作来测定基因表达。确定每一者的归一化表达,其解释靶(E靶)和参考(E参考)的PCR扩增具有不同的效率,并且将对数尺度的原始数据单位循环阈值(CT)转化成归一化表达的线性单位。使用的参考基因为GUSB和PGK1,即被确认为变异系数(CV)小于0.5并且M值小于1的基因,因此不会因处理条件不同而发生变化。为了评估这些细胞因子的相关分泌蛋白质水平,在24小时孵育后收获来自剩余细胞的上清液,并且通过细胞计数珠阵列(Cytometric Bead Array)(CBA,BD Biosciences)测定IFN-α和-γ水平,同时通过ELISA(PBL)测定IFN-β水平。
如图8中所示,5μM环状[G(2’,5’)pG(3’,5’)p]与5μM环状[G(3’,5’)pG(3’,5’)p]相比,在所有四个供体中干扰素α2(IFNA2)的基因表达显著更高。类似地,5μM环状[G(2’,5’)pG(3’,5’)p]与5μM环状[G(3’,5’)pG(3’,5’)p]相比,在所有四个供体中干扰素β1(IFNB1)的基因表达显著更高。5μM环状[G(2’,5’)pG(3’,5’)p]与环状[G(3’,5’)pG(3’,5’)p]相比,在所有四个供体中干扰素γ(IFNG)的基因表达水平显著更高。这些数据证实在诱导多种人供体中的先天免疫细胞因子的基因表达方面,环状[G(2’,5’)pG(3’,5’)p]与环状[G(3’,5’)pG(3’,5’)p]相比效力增加。
如图9(a)中所示,5μM环状[G(2’,5’)pG(3’,5’)p]与相同剂量或更低剂量的环状[G(3’,5’)pG(3’,5’)p]相比,在所有四个供体中所诱导的原代人PBMC中的分泌IFN-α的水平显著更高。在图9(b)中,在所有四个供体中,5μM环状[G(2’,5’)pG(3’,5’)p]所诱导的IFN-β的水平(如通过ELISA所评估)也高于环状[G(3’,5’)pG(3’,5’)p]诱导的水平,并且高于ISD和聚I:C对照所诱导的水平。图9(c)证实对于IFN-γ的分泌具有类似的研究结果,如通过CBA所评估。在所有四个供体中,5μM和0.5μM环状[G(2’,5’)pG(3’,5’)p]诱导的IFN-γ水平高于相同剂量或更高剂量的环状[G(3’,5’)pG(3’,5’)p]所诱导的水平,并且高于ISD和聚I:C对照所诱导的水平。这些数据证实在宽泛采样的人供体中,在I型和II IFN产生(对于诱导先天免疫至关重要)方面,环状[G(2’,5’)pG(3’,5’)p]与环状[G(3’,5’)pG(3’,5’)p]相比效力增加。
为了测定人细胞中通过天然分子和衍生物分子的每一者所诱导的作为佐剂效力标志的IFN-β的相对水平,将4×105个THP1-Blue细胞(用IRF-诱导分泌型胚胎碱性磷酸酶报道基因(Invivogen)转染的人单核细胞细胞系)用50μM环状[G(3’,5’)pG(3’,5’)p](CDG)、环状[G(2’,5’)pG(3’,5’)p](混合键或ML-CDG)、Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)(相比于[A(3’,5’)pA(3’,5’)p](CDA)、环状[A(2’,5’)pA(3’,5’)p](混合键或ML-CDA)、Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)或培养基对照)在37℃和5%CO2下孵育30分钟。在30分钟后,将细胞洗涤并铺板于含有10%FBS的RPMI培养基中的96孔培养皿上,在37℃和5%CO2下进行孵育。在过夜孵育后收集来自每个样品的细胞培养上清液,将20μL细胞培养上清液添加至180μL QUANTI-Blue试剂(Invivogen)中并孵育45分钟。使用光谱Max分光光度计(Molecular Diagnostics)取15分钟时取吸光度655nm处的读数。
如图10中所示,Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](MLRR-CDG)衍生物所诱导的IFN-β水平显著高于未经修饰的环状c-di-GMP(CDG)或经修饰的CDG分子所诱导的水平。类似地,Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)分子所诱导的IFN-β水平显著高于未经修饰的CDA或ML CDA分子所诱导的水平。这些结果证实,ML RR-CDN衍生物的纯化制剂与人单核细胞细胞系中的亲本CDN分子相比更显著地活化先天免疫应答。
为了测定衍生物分子活化免疫应答的相对能力,将CDN化合物以50μM、5μM和0.5μM的剂量通过尾根部皮下注射施用给6-8周龄雌性BALB/c小鼠(总体积为100μL,在HBSS中)。24小时后,通过荧光活化细胞分选(FACS)评估与IgG1同种型对照相比小鼠的淋巴细胞免疫细胞活化对自然杀伤(NK)细胞、CD4+和CD8+T细胞上的表面CD69表达的上调。
如图11(a-c)中所示,Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](MLRR-CDG)分子以剂量依赖性方式诱导NK细胞和T细胞的强效免疫活化。Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)分子还诱导NK细胞和T细胞活化,但程度低于ML RR-CDG分子。在所有剂量下,两种ML RR-CDN分子均比ML CDN分子诱导更多的免疫细胞活化。这些数据证实ML RR-CDN分子与ML CDN分子相比具有增强的免疫活化性质,更具体地ML RR-CDG分子诱导强效免疫细胞活化的能力。
实施例6.Rp,Rp二硫代CDN对磷酸二酯酶的增强的抗性
测量人细胞中I型干扰素的诱导以评价未经处理的和经磷酸二酯酶处理的氧代、Rp一硫代和Rp,Rp二硫代衍生物的效力。将五种化合物(环状[A(3’,5’)pA(3’,5’)p](CDA)、环状[A(2’,5’)pA(3’,5’)p](ML-CDA)、Rp一硫代(Rp,一硫代环状[A(2’,5’)pA(3’,5’)p](MLR-CDA)、Rp,Rp二硫代(Rp,Rp二硫代环状[A(3’,5’)pA(3’,5’)p](RR-CDA)和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)用160μg来自东部菱背响尾蛇(Crotalus adamanteus)的蛇毒磷酸二酯酶(SVPD)(Sigma)、2.5mU来自桔青霉(Penicillium citrinum)的核酸酶P1(NP1)(Sigma)处理或经模拟处理。将7μg各化合物稀释于SVPD缓冲液(1X PBS和0.6mm MgCl2)、NP1缓冲液(30mm Na Acetate,pH 5.3,2mM ZnCl2)中,或不做处理,然后在37℃下孵育2小时,接着煮沸10分钟以使核酸酶灭活。将4×105个THP1-BlueTMISG细胞(用IRF-诱导分泌型胚胎碱性磷酸酶报道基因(Invivogen)转染的人单核细胞细胞系,其在由五个IFN刺激应答元件构成的启动子的控制下表达碱性磷酸酶)用50μM模拟处理的、SVPD处理的或NP1处理的分子孵育。在30分钟后,将细胞洗涤并铺板于含有10%FBS的RPMI培养基中的96孔培养皿上,在37℃和5%CO2下进行孵育。在16小时孵育后收集来自每个样品的细胞培养上清液,将20μL细胞培养上清液添加至180μL QUANTI-Blue试剂(Invivogen)中并孵育25分钟,以评价I型干扰素蛋白质水平。使用Versa Max分光光度计(Molecular Diagnostics)测量吸光度655nm处的读数。
如图12中所示,未经处理的Rp,Rp二硫代化合物Rp,Rp二硫代环状[A(3’,5’)pA(3’,5’)p](RR-CDA)和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)与氧代(环状[A(3’,5’)pA(3’,5’)p](CDA)和环状[A(2’,5’)pA(3’,5’)p](ML-CDA)以及Rp一硫代相比是强效的I型干扰素诱导物。(Rp,一硫代环状[A(2’,5’)pA(3’,5’)p](ML R-CDA)CDN衍生物分子。我们在用裂解2’-5’和3’-5’磷酸二酯键的磷酸二酯酶SVPD处理或用选择性消解3’-5’磷酸二酯键的NP1(Pino等,(2008)Journal of Biological Cheimistry,283,36494-36503)处理后,评价CDN衍生物的活性。图12示出Rp,Rp二硫代化合物Rp,Rp二硫代环状[A(3’,5’)pA(3’,5’)p](RR-CDA)和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)在用SVPD和NP1处理后保持其效力,而氧代(环状[A(3’,5’)pA(3’,5’)p](CDA)和环状[A(2’,5’)pA(3’,5’)p](ML-CDA)在用SVPD和NP1消解后丧失其活性。在3’-5磷酸二酯键处包含单硫代取代的Rp一硫代衍生物(Rp,一硫代环状[A(2’,5’)pA(3’,5’)p](ML R-CDA)在NP1消解后保持活性,但易感于裂解2’-5’磷酸二酯键的SVPD处理。氧代、Rp一硫代和Rp,Rp二硫代衍生物对SVPD或NP1消解的不同易感性确认了Rp一硫代和Rp,Rp二硫代衍生物的结构。这些结果还证实了Rp,Rp二硫代衍生物的效用,因为其对存在于血清和/或宿主细胞中的磷酸二酯酶的消解具有抗性,从而导致对先天免疫信号传导的更强效的活化以及提高的体内治疗性抗肿瘤功效,如本文中所示。
实施例7.合成CDN衍生物分子强效活化所有人STING等位基因的信号传导
为了测定五种天然人STING变体(称为WT、REF、HAQ、AQ和Q)对天然和衍生物分子的应答性,生成一组表达人STING等位基因的人胚肾(HEK)293T细胞系。亲本HEK 293T细胞系不表达内源性STING,因此可对外源表达的STING等位基因的应答性进行评价。编码hSTING(REF)-GFP、hSTING(WT)-GFP、hSTING(HAQ)-GFP、hSTING(Q)-GFP和mSTING(WT)-GFP的MSCV2.2质粒获自加利福尼亚大学伯克利分校的Vance实验室(Vance Laboratory)。使用QuickChange定点诱变试剂盒(Stratagene)从hSTING(Q)-GFP获得hSTING(AQ)-GFP。hSTING(REF)等位基因的序列也称为Barber等位基因(Ishikawa,H.和Barber,G.N.(2008).Nature 455,674–678),并且具有NCBI参考序列NP_938023.1。hSTING(REF)与其它WT、HAQ、AQ和Q人STING等位基因的氨基酸差异在图13中示出,其改适自Yi等,Plos One 8:e77846(2013)。使用加利福尼亚大学伯克利分校的癌症研究实验流式细胞术研究部门(Cancer Research Laboratory Flow Cytometry Facility)的Mo Flo细胞分选仪,通过GFP阳性细胞的FACS分选产生表达个体人STING等位基因中每一个的稳定HEK 293T衍生细胞系。将1×104个HEK293T STING细胞接种于96孔板中,用50ng表达位于荧光素酶报道基因上游的人IFN-β启动子的人IFN-β报道基因质粒(pLuc-IFN-β)和10ng用于归一化的TK-renilla进行瞬时转染(采用Lipofectamine 2000)。24小时后,用天然和合成CDN衍生物分子刺激细胞,其中使用毛地黄皂苷透化以确保均匀摄取。用25ul毛地黄皂苷缓冲液(50mm HEPES、100mm KCL、3mm MgCl2、0.1mm DTT、85mm蔗糖、0.2%BSA、1mm ATP、0.1mm GTP、10ug/ml毛地黄皂苷)中的10μM环状[G(3’,5’)pA(3’,5’)p](cGAMP)、环状[G(2’,5’)pA(3’,5’)p](ML-cGAMP)、Rp,Rp二硫代环状[G(2’,5’)pA(3’,5’)p](ML RR-cGAMP)、环状[A(3’,5’)pA(3’,5’)p](CDA)、Rp,Rp二硫代环状[A(3’,5’)pA(3’,5’)p](RR-CDA)、环状[A(2’,5’)pA(3’,5’)p](ML-CDA)、Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)、环状[G(3’,5’)pG(3’,5’)p](CDG)、Rp,Rp二硫代环状[G(3’,5’)pG(3’,5’)p](RR-CDG)、环状[G(2’,5’)pG(3’,5’)p](ML-CDG)或Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)对每个STING细胞系进行刺激。20分钟后,除去刺激混合物并添加200ul标准RPMI培养基。刺激6小时后,制备细胞裂解物并使用Dual双荧光素酶测定系统(Promega)在Spectramax M3光度计上测量报道基因基因活性。
图14描通过测量IFNβ-LUC报道基因(在y-轴上进行RLU绘图)的诱导倍数而得到的编码人STING变体等位基因的HEK293细胞系的刺激。如图14中所示,Rp,Rp二硫代混合键化合物、Rp,Rp二硫代环状[G(2’,5’)pA(3’,5’)p](ML RR-cGAMP)、Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)通过所有人STING等位基因强力诱导IFNβ报道基因活性。抗性人STING等位基因hSTING(REF)和hSTING(Q)对具有典型核苷酸间磷酸桥键的天然分子:环状[G(3’,5’)pA(3’,5’)p](cGAMP)、环状[A(3’,5’)pA(3’,5’)p](CDA);和环状[G(3’,5’)pG(3’,5’)p](CDG)的刺激应答不良。强烈的相比之下,表达抗性人STING等位基因的细胞系对合成Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA):ML RR-CDA;ML RR-CDG;和ML RR-cGAMP的刺激有应答。表达小鼠STING的细胞对所有测试的分子均有应答,这证实经修饰的合成CDN衍生物分子与人STING信号传导途径的活化有关。这些结果证实,Rp,Rp二硫代混合键化合物Rp,Rp二硫代环状[G(2’,5’)pA(3’,5’)p](ML RR-cGAMP)、Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)可强效地活化所有测试的人STING等位基因,这表明这些分子将在整个宽范围的人群体中有效地诱导先天免疫。
为了证实合成CDN衍生物分子诱导人树突细胞(DC)的成熟,将来自人PBMC的CD14+单核细胞用50ng/ml GM-CSF和25ng/ml IL-4测试6天。七天后,用直接加入培养基的LPS(1μg/ml)或CDN(50μM)刺激单核细胞衍生的DC。48小时后,通过对CD11c+DC群门控的FACS测定MHC I类(HLA-ABC)、CD80、CD83和CD86的表面表达。图15A描绘了条形图,其指示在用图中所示的CDN分子刺激后的平均荧光强度(MFI)的平均值。还在图15B中示出人DC中CD80、CD86、CD83和MHC I类(HLA-ABC)的代表性直方图。填充的直方图对应于未经刺激的细胞,虚线代表LPS刺激,实线代表Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)刺激。这些结果证实,具有包含核苷酸间磷酸桥键的非桥接氧原子的Rp,Rp二硫代取代的结构以及2’-5,3’-5’非典型或混合键(ML)磷酸桥键结构的合成CDN分子活化所有人STING等位基因中的信号传导,并且强效活化人DC的成熟。
实施例8.CDN-诱导的抗原-特异性T细胞应答
为了测定由不同环状二核苷酸分子诱导的OVA-特异性CD8T细胞应答,用在含有10μg卵清蛋白质的2%角鲨烯-水溶液(squalene-and-water)中配制的0μg(无CDN)或5μg或25μg[G(2’,5’)pG(3’,5’)p](混合键或ML-CDG)对C57BL/6小鼠(n=5)进行皮下免疫。疫苗接种后七天,收集来自每个动物的血液并准备PBMC。在IFNγELISpot测定法中用单独的培养基(未经刺激的)或用1μM OVA257-264肽在1×105个作为喂养细胞的初次接受实验的脾细胞的存在下对5×104个PBMC刺激过夜。使用CTL读板仪和ImmunoSpot软件显现和定量IFNγELISpots。
如图16中所示,环状[G(2’,5’)pG(3’,5’)p](ML-CDG)的两种剂量均诱导C57BL/6小鼠中的OVA-特异性CD8免疫应答。这些应答显著高于由未经刺激的对照和无CDN的对照组诱导的应答。这些结果证实含有抗原的环状[G(2’,5’)pG(3’,5’)p](ML-CDG)的制剂可刺激体内抗原特异性CD8T细胞应答。
为了测定STING信号传导是否为c[G(2’,5’)pG(3’,5’)p](ML-CDG)诱导OVA-特异性CD8T细胞应答所需,用在含有10μg卵清蛋白质的2%角鲨烯-水溶液中配制的0μg(无CDN)或25μg c[G(2’,5’)pG(3’,5’)p](ML-CDG)对C57BL/6小鼠(n=3或5)和goldenticket小鼠(n=3)进行皮下免疫。疫苗接种后七天,收集来自每个动物的血液并准备PBMC。在IFNγELISpot测定法中用单独的培养基(未经刺激的)或用1μM OVA257-264肽在1×105个作为喂养细胞的初次接受实验的脾细胞的存在下对5×104个PBMC刺激过夜。使用CTL读板仪和ImmunoSpot软件显现和定量IFNγELISpots。
图17示出c[G(2’,5’)pG(3’,5’)p](ML-CDG)诱导依赖于功能性STING分子的存在的OVA-特异性CD8T细胞应答。在具有功能性STING分子的野生型C57BL/6小鼠中,c[G(2’,5’)pG(3’,5’)p](ML-CDG)和卵清蛋白质的制剂与未经刺激的对照和无CDN的对照相比诱导显著的OVA257-264免疫应答。在不表达功能性STING分子的goldenticket小鼠中(Sauer,Infection and Immunity 2011),c[G(2’,5’)pG(3’,5’)p](ML-CDG)诱导的OVA-特异性应答与不含CDN(无CDN)的对照制剂诱导的OVA-特异性应答相比显著不同。这些结果指示c[G(2’,5’)pG(3’,5’)p](ML-CDG)诱导的免疫应答需要功能性STING分子。
实施例9.多种CDN衍生物的比较数据
为了评估衍生物分子促进抗肿瘤免疫的能力,将B16黑素瘤细胞(100μL PBS中的5×104个细胞)皮下植入到6-8周龄雌性C57BL/6小鼠(每组8只小鼠)的下背部。在肿瘤植入后14天,当肿瘤的体积达到大约75mm3时开始治疗。通过用27规格针皮下注入肿瘤中央区域来施用CDN化合物(25μg,在总体积为40μL的HBSS中)。每三天重复注射,总共三次瘤内注射。测试的CDN为环状[G(3’,5’)pG(3’,5’)p](CDG);环状[G(2’,5’)pG(3’,5’)p](混合键或ML CDG);Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG);环状[A(3’,5’)pA(3’,5’)p](CDA);环状[A(2’,5’)pA(3’,5’)p](混合键或ML CDA);和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)。
如图18中所示,ML RR-CDG和ML RR-CDA衍生物与环状MLCDG和环状ML CDA环状二核苷酸分子相比诱导强效的抗肿瘤功效。ML RR-CDA分子比ML CDA衍生物诱导更多的肿瘤排斥(P=0.0004,斯氏t检验),并且截止肿瘤植入后44天,ML RR-CDG肿瘤组中的小鼠保持几乎不存在肿瘤。这些数据证实ML RR-CDN衍生物与ML CDN衍生物分子相比具有增强的效力,并且ML RR-CDN分子在B16黑素瘤小鼠模型中具有显著的抗肿瘤功效。
为了进一步评估衍生物分子促进抗肿瘤免疫的能力,将CT26结肠癌细胞(100μL PBS中的2×105个细胞)静脉注入6-8周龄雌性BALB/c小鼠并且评估整体存活率。在肿瘤植入后一天,通过皮下注入尾根部来施用CDN化合物(25μg,在总体积为100μL的HBSS中)。一周后,通过额外注射进行总共两次疫苗接种来加强小鼠的免疫。
如图19A中所示,Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)与环状[G(2’,5’)pG(3’,5’)p](ML CDG)分子(P=0.0018,时序检验)相比诱导显著更高的存活率,并且Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)与环状[A(2’,5’)pA(3’,5’)p](ML CDA)分子(P=0.0005,时序检验)相比诱导显著更高的存活率。这证实在CT26肝转移存活模型中,ML RR-CDN衍生物与ML CDN衍生物分子相比具有显著的抗肿瘤功效。这些结果证实CDN衍生物分子可成功地进行皮下施用。
为了证实肿瘤引发的T细胞触发的活化和CDN衍生物分子诱导的抗肿瘤功效并不限于单一肿瘤模型和小鼠遗传背景,测试合成CDN在其它肿瘤模型中促进抗肿瘤免疫的能力。将CT26结肠癌细胞(100μL PBS中的1×105个细胞)或4T1乳腺癌细胞(100μL PBS中的1×105个细胞)皮下植入到6-8周龄雌性BALB/c小鼠(每组8只小鼠)的胁腹。在肿瘤植入后大约14天,当肿瘤的体积达到大约75mm3时开始治疗。将化合物Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)或Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)化合物(25μg,在总体积为40μL的HBSS中)或HBSS媒介物对照和Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)(50μg,在总体积为40μL的HBSS中)或HBSS媒介物对照使用27规格针通过皮下注入到肿瘤的中央区域。每三天重复注射,总共三次瘤内注射。
如图19B中所示,ML RR-CDG完全抑制8只小鼠中7只的肿瘤生长,而ML RR-CDA完全抑制所有确立的CT26肿瘤的肿瘤生长。如图19C中所示,ML RR-CDA衍生物完全抑制所有确立的4T1乳房肿瘤的肿瘤生长。这些数据证实合成混合键RpRp二硫代环状二核苷酸(ML RR-CDN)衍生物在多种肿瘤模型中的显著效力和持久的抗肿瘤功效。
实施例10.CDN诱导的抗肿瘤功效为STING-依赖性的
为了测定衍生物分子的效应是否为STING-依赖性的,将B16黑素瘤细胞(100μL PBS中的5×104个细胞)植入到6-8周龄雌性goldenticket STING-/-小鼠或野生型C57BL/6对照小鼠(每组5只小鼠)的右胁腹。在肿瘤植入后14天,当肿瘤的体积达到大约75mm3时开始治疗。所施用的化合物为Rp,Rp二硫代环状[G(2’,5’)pG(3’,5’)p](ML RR-CDG)(25μg,在总体积为40μL的HBSS中)、Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)(50μg,在总体积为40μL的HBSS中)、TLR9激动剂CpG 1668(50μg,在总体积为40μL的HBSS中)或HBSS媒介物对照。使用27规格针仅皮下注入肿瘤中央区域来治疗小鼠。每三天重复注射,总共三次瘤内注射。
如图20A中所示,衍生物ML RR-CDN与HBSS对照相比在野生型C57BL/6小鼠中诱导强烈的肿瘤抑制,并且诱导比TLR9激动剂CpG 1668显著更多的肿瘤抑制(P=0.03,斯氏t-检验)。在图20B中,肿瘤生长未被ML RR-CDG或ML RR-CDA所抑制,这证实在野生型C57BL/6小鼠中观察到的抗肿瘤功效(图20A)完全依赖于功能性STING信号传导途径。相比之下,CpG 1668的肿瘤生长与HBSS对照在野生型和STING-/-小鼠中均相似(P=0.03,斯氏t-检验),这证实此化合物的作用是非STING依赖性的。
实施例11.CDN衍生物诱导持久有效的抗肿瘤特异性T细胞免疫
为了测定合成衍生物CDN分子是否引发持久有效的抗肿瘤T细胞免疫,向6-8周龄雌性BALB/c小鼠(每组8只小鼠)植入CT26结肠癌细胞(100μL PBS中的1×105个细胞)。将小鼠用Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)化合物(50μg,在总体积为40μL的HBSS中)或HBSS媒介物对照治疗,并且如前述实施例监测肿瘤生长。在肿瘤植入后18天对小鼠放血,并通过Ficoll梯度(Miltenyi Biotech)来分离PBMC。在IFNγELISpot测定法中用单独的培养基(未经刺激的)或用1μM内原性H-2Ld-限制性肿瘤排斥抗原AH1肽在1×105个作为喂养细胞的初次接受实验的脾细胞的存在下对5×104PBMC刺激过夜。使用CTL读板仪和ImmunoSpot软件显现和定量IFN-γELISpot板。在肿瘤植入55天,向存活小鼠和年龄匹配的初次接受实验的对照小鼠的对侧胁腹植入CT26或4T1(均为100μL PBS中的1×105个细胞)肿瘤细胞(每组4只小鼠),并且监测肿瘤生长。
如图21A中所示,所有用ML RR-CDA治疗的小鼠均排斥所确立的CT26结肠癌的生长。为了证实所述效应由CDN介导的适应性T细胞免疫应答的诱导所介导,在肿瘤诱导后18天通过ELISpot测定法评估PBMC应答于内源性肿瘤抗原AH1的刺激的IFN-γ产生。如图21B中所示,与HBSS治疗的对照组相比,分离自用ML RR-CDA治疗的小鼠的PBMC应答于AH1肽刺激产生显著更高的IFN-γ(P=0.003,斯氏t-检验)。另外,在图21C中,用对侧肿瘤再激发的存活小鼠表现出对同一CT26肿瘤类型的完全防护,而不抑制4T1肿瘤类型的生长。这些数据证实ML RR-CDA能够引发持续有效的肿瘤特异性T细胞介导的抗肿瘤免疫,其导致对经治疗肿瘤的排斥以及可排斥肿瘤激发的稳定肿瘤抗原特异性记忆T细胞群。
为了测定CDN衍生物分子是否在替代肿瘤模型中诱导有效且持久的抗肿瘤免疫,向6-8周龄雌性BALB/c小鼠(每组8只小鼠)植入4T1乳腺癌细胞(100μL PBS中的1×105个细胞)。将小鼠用Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)化合物(50μg,在总体积为40μL的HBSS中)或HBSS媒介物对照治疗,按照前述实验进行。在肿瘤植入35天,向存活小鼠和年龄匹配的初次接受实验的对照小鼠的对侧胁腹植入CT26或4T1(均为100μL PBS中的1×105个细胞)肿瘤细胞(每组4只小鼠),并且监测肿瘤生长。
如图22A中所示以及如前所证实,用ML RR-CDA治疗完全抑制所确立的4T1乳腺癌的肿瘤生长。另外,在图22B中,用4T1肿瘤细胞在对侧胁腹上再激发诱导完全的保护。用更具免疫原性的CT26肿瘤再激发也引发完全的保护,这表明这些肿瘤共有类似的肿瘤抗原,从而提供了合成CDN衍生物分子效力的进一步证据。
实施例12.通过瘤内注射CDN合成衍生物活化肿瘤引发的T细胞触发会诱导伴随远隔肿瘤抑制。
本文示出的实施例证实,合成CDN衍生物的瘤内(IT)注射由于促炎细胞因子的STING-依赖性活化导致显著持久的肿瘤破坏,从而有利于形成有效的肿瘤-特异性T细胞免疫。肿瘤-特异性T细胞免疫的STING-依赖性诱导保护动物免于自体同源肿瘤的后续激发。对本领域的技术人员而言显而易见的是进行性癌为转移性的,治疗要有效的话必须抑制远端肿瘤团块向外生长。对一种或选定病变的治疗抑制未治疗的远端肿瘤团块的肿瘤向外生长被称为伴随远隔效应。为了测试用合成CDN衍生物分子对选定肿瘤进行瘤内注射是否抑制未治疗的远端肿瘤的肿瘤向外生长,将(A)CT26结肠癌细胞(100μL PBS中的1×105个细胞)和(B)4T1乳腺癌细胞(100μL PBS中的1×105个细胞)皮下植入到6-8周龄雌性BALB/c小鼠(每组8只小鼠)的对侧胁腹。在肿瘤植入后13天,当肿瘤的体积达到大约75mm3时开始治疗。使用27规格针仅皮下注入原发(右侧)肿瘤的中央区域来施用Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)化合物(50μg,在总体积为40μL的HBSS中)或HBSS媒介物对照。每三天重复注射,总共三次瘤内注射。
如图23中所示,Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)化合物与HBSS媒介物对照相比在CT26(图23A)和4T1(图23B)荷瘤动物中诱导经治疗的原发性肿瘤的完全抑制。另外,与HBSS对照相比,两个肿瘤模型中的对侧(未经治疗的)肿瘤的向外生长也受到显著抑制(图23A P=0.0011,图23B P=0.0019,斯氏t-检验)。这些数据证实,ML RR-CDA衍生物在注入原发肿瘤中时具有显著的抗肿瘤功效以及显著的伴随远隔抗肿瘤免疫效应。
为了测定合成CDN衍生物分子是否在替代肿瘤模型和小鼠遗传背景促进伴随远隔抗肿瘤免疫,向6-8周龄雌性C57BL/6小鼠(每组8只小鼠)的右胁腹植入B16黑素瘤细胞(100μL PBS中的5×104个细胞)。一周后,向小鼠静脉内植入1×105个B16黑素瘤细胞以定殖于肺部,同时提供一组初次接受实验的年龄匹配的对照小鼠。当在第13天皮下胁腹肿瘤达到大约75mm3时,用Rp,Rp二硫代环状[A(2’,5’)pA(3’,5’)p](ML RR-CDA)(50μg,在总体积为40μL的HBSS中)或HBSS媒介物对照对小鼠进行瘤内治疗,如前述方案进行三次注射。在皮下肿瘤植入后28天(静脉内植入后21天),对小鼠实施安乐死,将肺收集并固定(10%中性缓冲福尔马林),并使用解剖显微镜对肺肿瘤结节的数量进行计数。
如图24A以及前述实验中所示,与HBSS对照组相比,用ML RR-CDA治疗显著抑制了原发胁腹肿瘤的肿瘤生长(P<0.001,斯氏t-检验)。另外,图24B和图24C中示出,与HBSS和初次接受实验的(仅静脉内)肿瘤组相比,用CDN衍生物治疗显著抑制了远端肺肿瘤结节的生长。此处示出的结果证实,合成CDN衍生物的瘤内(IT)注射导致伴随远隔抗肿瘤效应,如经治疗的肿瘤由于促炎细胞因子的STING-依赖性活化和有效肿瘤-特异性T细胞免疫的形成而被破坏所证实,所述伴随远隔抗肿瘤效应随后抑制未经治疗的远端肿瘤向外生长。
本领域技术人员易于了解,本发明充分适于执行目标并且获得所提到的以及其中固有的结果和优点。本文提供的实例代表优选的实施方案,是示例性的,并且无意限制本发明的范围。
应当理解,本发明在其申请中不限于在以下描述中所阐述的或在附图中示出的组件的构造和布置的细节。本发明除了所述的那些外还能够具有其它实施方案并能够以多种方式实践和执行。另外,应当理解,本文以及说明书摘要采用的词组和术语是为了进行说明而不应视为进行限制。
同样,本领域的技术人员将会了解,本公开所依据的概念可容易地作为设计其它结构、方法和系统的基础,以便实现本发明的若干目的。因此,重要的是,将权利要求书视为包括此类等同结构,只要它们不脱离本发明的精神和范围即可。
虽然为了使本领域的技术人员完成和利用本发明已经足够详细地描述和示例性地示出了本发明,但是在不脱离本发明精神和范围的前提下各种替代形式、修改形式和改进形式应是显而易见的。本文提供的实例代表优选的实施方案,是示例性的,并且无意限制本发明的范围。其中的修改形式和其它用途将是本领域的技术人员能够想到的。这些修改形式包括在本发明的精神内并由权利要求书的范围限定。
对于本领域技术人员将显而易见的是,在不脱离本发明精神和范围的前提下可对本文公开的发明进行各种替换和修改。
本说明书中提及的所有专利和出版物指示本发明所属领域的普通技术人员的水平。所有专利和出版物均以引用方式并入本文,达到如同各个出版物具体且单独地表明以引用方式并入的相同程度。
可将本文示例性描述的发明适宜地在不存在本文未具体公开的任何一个或多个要素、任何一个或多个限制的情况下实施。因此,例如,在本文各种情况下,术语“包含”、“基本上由...组成”和“由...组成”的任何一个均可使用另外两个术语中的任一个代替。已采用的术语和措辞是用作描述性而非限制性的术语,并且在此类术语和措辞的使用中并不意图排除所示的和所描述的特征或其部分的任何等同物,而是应认识到在受权利要求书保护的本发明的范围之内可以作出各种修改形式。因此,应当理解,虽然本发明已通过优选的实施方案和任选的特征具体地进行公开,但本领域技术人员可能采用本文所公开的构思的修改形式和变型,并且此类修改形式和变型被视为在本发明的范围内,本发明的范围由附随的权利要求书限定。
其它实施方案在以下权利要求书中示出。
- 还没有人留言评论。精彩留言会获得点赞!