缓释性粒子、其制造方法、成型材料以及成型品与流程
本发明涉及缓释性粒子、其制造方法、成型材料以及成型品,详细而言,涉及对抗生物活性化合物进行缓释的缓释性粒子、其制造方法、成型材料以及成型品。
背景技术:
已知通过使杀虫剂、防虫剂、防蚁剂、杀菌剂、防腐剂,除草剂、防藻剂、驱避剂等抗生物活性化合物微胶囊化,从而对抗生物活性化合物进行缓释,确保效力持续性的粒子。
例如提出了如下方法:通过对包含新烟碱系化合物、分散介质和聚异氰酸酯成分的浆料进行水分散,然后配合聚胺进行界面聚合,使新烟碱系化合物微胶囊化(例如,参照下述专利文献1)。
现有技术文献
专利文献
专利文献1:日本特开2000-247821号公报
技术实现要素:
发明所要解决的课题
然而,根据用途和目的,有时对粒子要求缓释性。但是,通过如专利文献1所记载的方法获得的微胶囊存在不能充分满足上述要求这样的不良。
此外,在将专利文献1中所记载的微胶囊与树脂、橡胶混炼时,存在微胶囊因混炼时的剪切而被破坏这样的不良。
本发明的目的在于,提供缓释性优异且坚固的缓释性粒子、其制造方法、使用该缓释性粒子的成型材料以及成型品。
用于解决课题的方法
本发明人等对上述目的的缓释性粒子、其制造方法、使用该缓释性粒子的成型材料以及成型品进行深入研究,结果发现,通过具备如下工序的制造方法能够得到缓释性优异且坚固的缓释性粒子、使用其的成型材料以及成型品,从而完成了第1发明组,上述制造方法具备如下工序:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。
即,第1发明组如下:
(1)一种缓释性粒子,其特征在于,通过具备如下工序的制造方法来获得:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于上述疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对上述油相成分进行水分散来调制水分散液的水分散工序;以及对上述聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序;
(2)根据上述(1)记载的缓释性粒子,其特征在于,在上述聚合工序中,在存在芳香族磺酸与甲醛的缩合物的盐的情况下,对上述聚合性乙烯基单体进行悬浮聚合;及/或上述聚合性乙烯基单体含有(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体;
(3)根据上述(1)或(2)记载的缓释性粒子,其特征在于,上述抗生物活性化合物为新烟碱系杀虫剂;
(4)根据上述(3)记载的缓释性粒子,其特征在于,上述新烟碱系杀虫剂含有选自由(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、以及1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺组成的组中的至少一种;
(5)一种缓释性粒子,其特征在于,具有由结构域和包含聚合物的基质(domain)形成的2相结构,上述结构域包含相对于用于生成上述聚合物的单体实质上为不溶性的抗生物活性化合物,且分散于上述基质中;
(6)根据上述(5)记载的缓释性粒子,其特征在于,在上述缓释性粒子的表面,露出上述基质和上述结构域这两者;
(7)根据上述(5)记载的缓释性粒子,其特征在于,上述结构域被上述基质所被覆;
(8)根据上述(7)记载的缓释性粒子,其特征在于,在上述基质的表面还附着有上述抗生物活性化合物;
(9)根据上述(5)~(8)中任一项记载的缓释性粒子,其特征在于,上述抗生物活性化合物为新烟碱系杀虫剂;
(10)根据上述(9)记载的缓释性粒子,其特征在于,上述新烟碱系杀虫剂含有选自由(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、以及1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺组成的组中的至少一种;
(11)根据上述(1)~(10)中任一项记载的缓释性粒子,其特征在于,调制成粒剂;
(12)一种成型材料,其特征在于,含有热塑性树脂和上述(1)~(11)中任一项记载的缓释性粒子;
(13)一种成型品,其特征在于,含有热塑性树脂和上述(1)~(11)中任一项记载的缓释性粒子;
(14)一种缓释性粒子的制造方法,其特征在于,具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于上述疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对上述油相成分进行水分散来调制水分散液的水分散工序;以及对上述聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序;
(15)根据上述(14)记载的缓释性粒子的制造方法,其特征在于,在上述聚合工序中,在存在芳香族磺酸与甲醛的缩合物的盐的情况下,对上述聚合性乙烯基单体进行悬浮聚合;及/或上述聚合性乙烯基单体含有(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体;
(16)根据上述(14)或(15)记载的缓释性粒子的制造方法,其特征在于,进一步具备将通过上述聚合工序获得的悬浮液与固体载体配合,使它们干燥,调制粒剂的工序;
(17)根据上述(14)~(16)中任一项记载的缓释性粒子的制造方法,其特征在于,上述抗生物活性化合物为新烟碱系杀虫剂;
(18)根据上述(17)记载的缓释性粒子的制造方法,其特征在于,上述新烟碱系杀虫剂含有选自由(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、以及1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺组成的组中的至少一种。
此外,本发明人等对上述第1发明组的缓释性粒子、其制造方法、使用该缓释性粒子的成型材料以及成型品进行深入研究,结果发现通过如下制造方法能够得到缓释性和耐碱性优异且坚固的缓释性粒子、使用其的成型材料以及成型品的见解,从而完成第2发明组,上述制造方法具备如下工序:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于上述疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对上述油相成分进行水分散来调制水分散液的水分散工序;以及对上述聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序,在聚合工序中,对聚合性乙烯基单体进行悬浮聚合,并且对疏水性壳层形成成分和亲水性壳层形成成分进行界面聚合,形成被覆悬浮聚合物的壳层。
即,第2发明组如下:
(1)一种缓释性粒子,其特征在于,具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于上述疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对上述油相成分进行水分散来调制水分散液的水分散工序;以及对上述聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序,在上述油相成分调制工序、上述水分散工序和上述聚合工序的至少任一个工序中,添加疏水性壳层形成成分和亲水性壳层形成成分,在上述聚合工序中,对上述聚合性乙烯基单体进行悬浮聚合,并且对上述疏水性壳层形成成分和上述亲水性壳层形成成分进行界面聚合,形成被覆悬浮聚合物的壳层;
(2)根据上述(1)记载的缓释性粒子,其特征在于,在开始悬浮聚合的同时开始界面聚合,或者在开始悬浮聚合之前开始界面聚合;
(3)根据上述(1)或(2)记载的缓释性粒子,其特征在于,上述疏水性壳层形成成分含有聚异氰酸酯,上述亲水性壳层形成成分含有聚胺;
(4)根据上述(1)~(3)中任一项记载的缓释性粒子,其特征在于,上述抗生物活性化合物为新烟碱系杀虫剂;
(5)根据上述(4)记载的缓释性粒子,其特征在于,上述新烟碱系杀虫剂含有选自由(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、以及1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺组成的组中的至少一种;
(6)一种缓释性粒子,其特征在于,含有包含聚合物的基质、结构域和被覆上述基质的壳层,上述结构域包含相对于用于生成上述聚合物的单体实质上为不溶性的抗生物活性化合物,且分散于上述基质中;
(7)根据上述(6)记载的缓释性粒子,其特征在于,上述壳层包含聚脲;
(8)根据上述(6)或(7)记载的缓释性粒子,其特征在于,在上述壳层的表面附着有抗生物活性化合物;
(9)根据上述(6)~(8)中任一项记载的缓释性粒子,其特征在于,上述抗生物活性化合物为新烟碱系杀虫剂;
(10)根据上述(9)记载的缓释性粒子,其特征在于,上述新烟碱系杀虫剂含有选自由(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、以及1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺组成的组中的至少一种;
(11)根据上述(1)~(10)中任一项记载的缓释性粒子,其特征在于,调制成粒剂;
(12)一种成型材料,其特征在于,含有热塑性树脂和上述(1)~(11)中任一项记载的缓释性粒子;
(13)一种成型品,其特征在于,含有热塑性树脂和上述(1)~(11)中任一项记载的缓释性粒子;
(14)一种缓释性粒子的制造方法,其特征在于,具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于上述疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对上述油相成分进行水分散来调制水分散液的水分散工序;以及对上述聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序,在上述油相成分调制工序、上述水分散工序和上述聚合工序的至少任一个工序中,添加疏水性壳层形成成分和亲水性壳层形成成分,在上述聚合工序中,对上述聚合性乙烯基单体进行悬浮聚合,并且对上述疏水性壳层形成成分和上述亲水性壳层形成成分进行界面聚合,形成被覆悬浮聚合物的壳层;
(15)根据上述(14)记载的缓释性粒子的制造方法,其特征在于,在聚合工序中,在开始悬浮聚合的同时开始界面聚合,或者在开始悬浮聚合之前开始界面聚合;
(16)根据上述(14)或(15)记载的缓释性粒子的制造方法,其特征在于,上述疏水性壳层形成成分为聚异氰酸酯,上述亲水性壳层形成成分为聚胺;
(17)根据上述(14)~(16)中任一项记载的缓释性粒子的制造方法,其特征在于,进一步具备将通过聚合工序获得的悬浮液与固体载体配合,使它们干燥,调制粒剂的工序;
(18)根据上述(14)~(17)中任一项记载的缓释性粒子的制造方法,其特征在于,上述抗生物活性化合物为新烟碱系杀虫剂;
(19)根据上述(18)记载的缓释性粒子的制造方法,其特征在于,上述新烟碱系杀虫剂含有选自由(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、以及1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺组成的组中的至少一种。
发明效果
第1发明组的缓释性粒子可以通过如下的制造方法获得,因此能够获得缓释性优异且坚固的缓释性粒子,上述制造方法具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。
根据第1发明组的缓释性粒子的制造方法,能够得到坚固且缓释性优异的缓释性粒子。
第1发明组的缓释性粒子具有由结构域和包含聚合物的基质形成的2相结构,上述结构域是包含抗生物活性化合物的结构域,且分散于基质中,因此抗生物活性化合物的缓释性优异,并且坚固性优异,由此与树脂的混炼性优异。
第1发明组的成型材料含有上述缓释性粒子,因此能够对第1发明组的成型品赋予优异的抗生物活性化合物的缓释性。
第2发明组的缓释性粒子可以通过如下的制造方法获得,因此能够获得缓释性和耐碱性优异、坚固的缓释性粒子,上述制造方法具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。
根据第2发明组的缓释性粒子的制造方法,能够得到坚固且缓释性和耐碱性优异的缓释性粒子。
进一步,关于第2发明组的缓释性粒子,在聚合工序中,对聚合性乙烯基单体进行悬浮聚合,并且对疏水性壳层形成成分和亲水性壳层形成成分进行界面聚合,形成被覆悬浮聚合物的壳层,因此能够提高抗生物活性化合物的内包率,并且抗生物活性化合物的耐碱性优异。
第2发明组的缓释性粒子含有结构域和包含聚合物的基质,上述结构域是包含抗生物活性化合物的结构域,且分散于基质中,从而抗生物活性化合物的缓释性优异,并且坚固性优异,因此与树脂的混炼性优异。
进一步,第2发明组的缓释性粒子包含被覆基质的壳层,因此抗生物活性化合物的缓释性和耐碱性优异。
]第2发明组的成型材料含有上述缓释性粒子,因此能够对第2发明组的成型品赋予优异的抗生物活性化合物的缓释性和耐碱性。
附图说明
图A1表示第1发明组的缓释性粒子的第1实施方式的截面示意图;
图A2表示第1发明组的缓释性粒子的第2实施方式(结构域被基质所被覆,附着物附着于基质表面的方式)的截面示意图;
图A3表示第2实施方式的变形例(基质的整个表面露出的方式)的截面示意图;
图A4表示实施例A1的缓释性粒子的SEM照片的图像处理图;
图A5表示实施例A2的缓释性粒子的SEM照片的图像处理图;
图A6表示实施例A3的缓释性粒子的SEM照片的图像处理图;
图A7表示实施例A4的缓释性粒子的SEM照片的图像处理图;
图A8表示实施例A9的缓释性粒子的SEM照片的图像处理图;
图A9表示实施例A19的缓释性粒子的SEM照片的图像处理图;
图A10表示实施例A20的料股的断裂面的SEM照片的图像处理图;
图A11表示实施例A21的料股的断裂面的SEM照片的图像处理图;
图A12表示实施例A1的缓释性粒子的TEM照片的图像处理图;
图A13表示实施例A2的缓释性粒子的TEM照片的图像处理图;
图A14表示实施例A3的缓释性粒子的TEM照片的图像处理图;
图B1表示第2发明组的缓释性粒子的第3实施方式的截面示意图;
图B2表示第2发明组的缓释性粒子的第4实施方式的截面示意图;
图B3表示实施例B1的缓释性粒子的SEM照片的图像处理图;
图B4表示实施例B2的缓释性粒子的SEM照片的图像处理图;
图B5表示实施例B6的缓释性粒子的SEM照片的图像处理图;
图B6表示实施例B30的缓释性粒子的SEM照片的图像处理图;
图B7表示实施例B35的缓释性粒子的SEM照片的图像处理图;
图B8表示实施例B2的缓释性粒子的TEM照片的图像处理图;
图B9表示参考例B1的缓释性粒子的TEM照片的图像处理图;
图B10表示参考例B2的缓释性粒子的TEM照片的图像处理图;
图B11表示参考例B3的缓释性粒子的TEM照片的图像处理图。
具体实施方式
以下,对包含于本发明且相互关联的第1发明组和第2发明组依次进行说明。
第1发明组
<缓释性粒子的制造方法的说明>
第1发明组的缓释性粒子的制造方法具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。
以下,对上述的各工序中所使用的原料进行说明。
(抗生物活性化合物)
作为抗生物活性化合物,选自具有杀虫(包括灭蚁)、防虫(包括防蚁)、杀菌、抗菌、防腐、除草、防藻、防霉等抗生物活性的杀虫剂(包括灭蚁剂)、防虫剂(包括防蚁剂)、杀菌剂、抗菌剂、防腐剂、除草剂、防藻剂、防霉剂、引诱剂、驱避剂和杀鼠剂等。
具体地说,关于抗生物活性化合物,例如作为杀虫剂,可以举出噻虫胺((E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍)、吡虫啉(1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺)、噻虫啉、噻虫嗪((EZ)-3-(2-氯-1,3-噻唑-5-基甲基)-5-甲基-1,3,5-二嗪-4-基叉(硝基)胺)、呋虫胺等新烟碱系杀虫剂,氟虫双酰胺、氯虫苯甲酰胺等二酰胺系,除虫脲、氟苯脲、氟啶脲、虫酰肼、甲氧虫酰肼、环丙氨嗪等昆虫生长调节剂,四螨嗪等杀螨剂,吡蚜酮、油酸钠等其它合成药剂等。例如,作为杀菌剂,可以举出碱性氯化铜、碱性硫酸铜、羟基喹啉铜等铜系杀菌剂,金属银等银系杀菌剂,福代锌等有机硫系杀菌剂,四氯苯酞、三环唑等黑色素生物合成抑制剂,甲基硫菌灵、多菌灵(MBC)、乙霉威等苯并咪唑系杀菌剂,异噻菌胺等酸酰胺系杀菌剂,嗪氨灵等甾醇生物合成抑制剂,1,2-苯并异噻唑-3-酮等异噻唑啉酮系杀菌剂,哒菌酮、氟酰亚胺、克菌丹、百菌清、灭螨猛、喹酸、苯噻菌胺、氰霜唑、吡啶硫酮锌等其它合成抑制剂等。例如,作为除草/防藻剂,可以举出3-(3,4-二氯苯基)-1,1-二甲基脲(DCMU)、苄草隆、卡灵草等脲系药剂,乙氧嘧磺隆、氯吡嘧磺隆、啶嘧磺隆、烟嘧磺隆、噻吩磺隆、唑吡嘧磺隆、环丙嘧磺隆、氟吡磺隆、三氟啶磺隆钠盐等磺脲系药剂,西玛津(CAT)、阿特拉津、三嗪氟草胺、环草啶、氟氯氰菊酯、特丁净等三嗪系药剂,草甘膦等氨基酸系,丙炔氟草胺等苯基邻苯二甲酰亚胺系,甲基磺草酮等三酮系,莫克草、环酯草醚等其它药剂等。作为抗生物活性化合物,优选从种选择性、安全性的观点考虑,可以举出新烟碱系杀虫剂,并且从通用性、效力的观点考虑,可以举出吡啶硫酮锌,更优选从难溶性的观点考虑,可以举出噻虫胺、吡虫啉、吡啶硫酮锌,进一步优选可以举出噻虫胺、吡虫啉。特别优选地,从对哺乳动物的安全性的观点考虑,可以举出噻虫胺。
抗生物活性化合物实质上为疏水性,具体地说,例如,室温(20~30℃,更具体地为25℃)时相对于水的溶解度极小,更具体地说,例如,室温的溶解度为1.5质量份/水100容量份(15g/L)以下、优选为0.5质量份/水100容量份(5g/L)以下、进一步优选为0.1质量份/水100容量份(1g/L)以下。
抗生物活性化合物相对于聚合性乙烯基单体实质上为不溶性,具体地说,例如,室温(20~30℃,更具体地为25℃)时相对于聚合性乙烯基单体的溶解度极小,具体地说,室温的溶解度例如为0.1质量份/(使用的)聚合性乙烯基单体(混合物)100容量份(1g/L)以下、优选为0.05质量份/(使用的)聚合性乙烯基单体(混合物)100容量份(0.5g/L)以下。
此外,抗生物活性化合物的熔点例如为80℃以上、优选为100℃以上,并且,如果抗生物活性化合物是不含有金属原子的化合物,则例如为300℃以下。
(聚合性乙烯基单体)
作为聚合性乙烯基单体,可以举出例如(甲基)丙烯酸酯系单体、芳香族乙烯基单体、乙烯基酯系单体、马来酸酯系单体、乙烯基卤、亚乙烯基卤、含氮乙烯基单体、交联性单体等。
作为(甲基)丙烯酸酯系单体,可以举出例如甲基丙烯酸酯和/或丙烯酸酯,具体可以举出(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸正丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸正丁酯、(甲基)丙烯酸异丁酯(i-BMA/i-BA)、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸正戊酯、(甲基)丙烯酸正己酯、(甲基)丙烯酸环己酯等具有烷基部分为直链状、支链状或环状的碳原子数1~6的烷基部分的(甲基)丙烯酸烷基酯;例如(甲基)丙烯酸2-甲氧基乙酯等(甲基)丙烯酸烷氧基烷基酯;例如(甲基)丙烯酸羟基乙酯等(甲基)丙烯酸羟基烷基酯;例如(甲基)丙烯酸缩水甘油酯等含有环氧基的(甲基)丙烯酸酯等。优选可以举出(甲基)丙烯酸烷基酯。
作为(甲基)丙烯酸烷基酯,更优选可以举出具有碳原子数1~6的烷基部分的(甲基)丙烯酸烷基酯,特别优选可以举出甲基丙烯酸异丁酯(i-BMA)。
作为芳香族乙烯基单体,可以举出例如苯乙烯(乙烯基苯)、对甲基苯乙烯、邻甲基苯乙烯、α-甲基苯乙烯、乙基乙烯基苯等苯乙烯系单体(单乙烯基苯)等。优选可以举出苯乙烯、乙基乙烯基苯。
作为乙烯基酯系单体,可以举出例如乙酸乙烯酯、丙酸乙烯酯等。
作为马来酸酯系单体,可以举出例如马来酸二甲酯、马来酸二乙酯、马来酸二丁酯等。
作为乙烯基卤,可以举出例如氯乙烯、氟乙烯等。
作为亚乙烯基卤,可以举出例如偏氯乙烯、偏氟乙烯等。
作为含氮乙烯基单体,可以举出例如(甲基)丙烯腈、N-苯基马来酰亚胺、乙烯基吡啶等。
作为交联性单体,可以举出例如乙二醇二(甲基)丙烯酸酯、二乙二醇二(甲基)丙烯酸酯等单或聚乙二醇二(甲基)丙烯酸酯,例如1,3-丙二醇二(甲基)丙烯酸酯、1,4-丁二醇二(甲基)丙烯酸酯、1,5-戊二醇二(甲基)丙烯酸酯等链烷二醇二(甲基)丙烯酸酯;例如三羟甲基丙烷三(甲基)丙烯酸酯、季戊四醇四(甲基)丙烯酸酯(PETA/PETM)等链烷多元醇聚(甲基)丙烯酸酯等(甲基)丙烯酸酯系交联性单体;例如烯丙基(甲基)甲基丙烯酸酯、三烯丙基(异)氰脲酸酯等烯丙基系单体;例如二乙烯基苯、三乙烯基苯等芳香族交联性单体。优选可以举出单或聚乙二醇二(甲基)丙烯酸酯、二乙烯基苯,更优选可以举出乙二醇二(甲基)丙烯酸酯、二乙烯基苯。
聚合性乙烯基单体可以单独使用或并用。
另外,为了使通过聚合性乙烯基单体的聚合而得到的聚合物在室温下具有坚固的表面,玻璃化转变温度例如为30℃以上、优选为50℃以上,以达到该玻璃化转变温度的方式选择聚合性乙烯基单体。
聚合性乙烯基单体例如实质上为疏水性,具体地说,例如室温下相对于水的溶解度极小,更具体地说,室温时的溶解度例如为10质量份/水100容量份(100g/L)以下、优选为8质量份/水100容量份(80g/L)以下。另外,在并用不同种类的聚合性乙烯基单体的情况下,作为聚合性乙烯基单体整体(即,不同种类的聚合性乙烯基单体的混合物)实质上为疏水性。
接下来,对缓释性粒子的制造方法中的各工序依次进行说明。
(油相成分调制工序)
在油相成分调制工序中,通过在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分。
具体地说,配合上述的聚合性乙烯基单体和抗生物活性化合物而不配合溶剂(己烷、甲苯、乙酸乙酯等疏水性有机溶剂),并搅拌。由此,调制疏水性浆料。疏水性浆料被包含于油相成分中。
为了将抗生物活性化合物分散于聚合性乙烯基单体中,可以使用例如涂料搅拌器、均质分散器(高速分散机)、珠磨机(包括分批式珠磨机)、球磨机、棒磨机等分散机。分散机可以单独使用或并用。作为分散机,优选从能够在广粘度区域中使用且能够在大规模工业生产中使用的观点考虑,使用分批式珠磨机。
通过上述分散,对抗生物活性化合物进行湿式粉碎。
另外,可以将聚合性乙烯基单体的全部配合于抗生物活性化合物,或者还可以将聚合性乙烯基单体分开配合于抗生物活性化合物。在将聚合性乙烯基单体分开配合的情况下,首先,将聚合性乙烯基单体的一部分与抗生物活性化合物配合,并将它们分散而调制疏水性浆料,然后,将聚合性乙烯基单体的剩余部分配合于疏水性浆料。
由此,调制包含疏水性浆料的油相成分。
关于抗生物活性化合物相对于聚合性乙烯基单体的配合比例,以质量比例(即,抗生物活性化合物的质量份/聚合性乙烯基单体的质量份)计,例如为1/99以上、优选为10/90以上、更优选为15/85以上,并且,例如为90/10以下、优选为75/25以下、更优选为70/30以下、进一步优选为65/35以下、特别优选为60/40以下。
此外,相对于聚合性乙烯基单体100质量份,抗生物活性化合物的配合比例例如为1质量份以上、优选为10质量份以上、更优选为20质量份以上,并且,例如为900质量份以下、优选为300质量份以下、更优选为200质量份以下、进一步优选为150质量份以下。
抗生物活性化合物在油相成分中的含有比例例如为1质量%以上、优选为10质量%以上,并且,例如为90质量%以下、优选为80质量%以下、更优选为70质量%以下、更优选为60质量%以下。
此外,在上述分散中,还可以根据需要配合分散剂(第1分散剂)。作为分散剂,可以举出两亲性高分子型分散剂、非离子系表面活性剂(第1表面活性剂)等。
作为两亲性高分子型分散剂,可以举出例如EFKA4008、EFKA4009(以上为汽巴精化制氨基甲酸酯系高分子分散剂),DISPERBYK-2164、DISPERBYK-164(以上为毕克化学公司制颜料分散用官能团改性共聚物),NUOSPERSE 2008、NUOSPERSE FA-196、NUOSPERSE657(以上为海名斯公司制),FLOWLEN D-90、POLYFLOW KL-100、POLYFLOW KL-700(以上为共荣社化学公司制),HOMOGENOL L-95(花王公司制)等非离子系两亲性高分子型分散剂。此外,作为两亲性高分子型分散剂,可以举出例如FLOWLEN G-900(共荣社化学公司制羧基改性高分子),DISPARLON DA-234、DISPARLON DA-325、DISPARLON DA-375、DISPARLON DA-550、DISPARLON AQ-330(以上为楠本化成公司制聚醚磷酸酯盐)等阴离子系两亲性高分子型分散剂。进而,作为两亲性高分子型分散剂,可以举出例如NOPCOSPERSE 092(圣诺普科公司制)等阳离子系两亲性高分子型分散剂。
作为非离子系表面活性剂,可以举出例如AMOGEN CBH(烷基甜菜碱)、AMOGEN SH(烷基酰胺甜菜碱)、NOIGEN 100E(聚氧乙烯油基醚)、NOIGEN EA73(聚氧乙烯十二烷基苯基醚)、NOIGEN ES99(单油酸聚乙二醇酯)、Dianol CME(椰子油脂肪酸单乙醇酰胺)、Dianol 300(椰子油脂肪酸单乙醇二酰胺)、SORGEN 30(失水山梨醇倍半油酸酯)、SORGEN 40(失水山梨醇单油酸酯)、SORGEN 50(失水山梨醇单硬脂酸酯)、EPAN 420(聚氧乙烯聚氧丙烯二醇)、EPAN 720(聚氧乙烯聚氧丙烯二醇)(以上为花王公司制)等。
作为分散剂,优选可以举出两亲性高分子型分散剂,更优选可以举出非离子系两亲性高分子型分散剂、阴离子系两亲性高分子型分散剂,进一步优选可以举出非离子系两亲性高分子型分散剂,特别优选可以举出颜料分散用官能团改性共聚物分散剂、氨基甲酸酯系高分子分散剂。
相对于抗生物活性化合物,分散剂的配合比例例如为0.1质量%以上、优选为1质量%以上,并且,例如为40质量%以下、优选为20质量%以下。
油相成分中的抗生物活性化合物的平均粒径例如为5μm以下、优选为2.5μm以下,并且,例如为0.05μm以上、优选为0.1μm以上。
在该方法中,例如在调制疏水性浆料的同时、或者在调制疏水性浆料后,配合聚合引发剂。优选在调制疏水性浆料后,将聚合引发剂配合于调制的疏水性浆料中。在该情况下,可以将聚合性乙烯基单体分开配合于抗生物活性化合物,具体地说,将聚合性乙烯基单体的一部分配合于抗生物活性化合物来调制疏水性浆料,接着使聚合引发剂溶解于聚合性乙烯基单体的剩余部分,将其配合于调制的疏水性浆料中。由此,调制含有聚合引发剂和疏水性浆料的油相成分。
聚合引发剂可以举出悬浮聚合中通常使用的自由基聚合引发剂,具体可以举出油溶性聚合引发剂等。
作为油溶性聚合引发剂,可以举出例如过氧化二月桂酰、过氧化2-乙基己酸1,1,3,3-四甲基丁酯、过氧化-2-乙基己酸叔己酯、过氧化二碳酸二异丙酯,过氧化苯甲酰等油溶性有机过氧化物,例如2,2′-偶氮二异丁腈、2,2'-偶氮双(2,4-二甲基戊腈)、2,2'-偶氮双(2-甲基丁腈)等油溶性偶氮化合物等。优选可以举出过氧化二月桂酰、过氧化-2-乙基己酸叔己酯、2,2′-偶氮二异丁腈。
聚合引发剂可以单独使用或并用两种以上。
相对于聚合性乙烯基单体100质量份,聚合引发剂的配合比例例如为0.01质量份以上、优选为0.1质量份以上、更优选为0.5质量份以上,例如为5质量份以下、优选为3质量份以下、更优选为1.0质量份以下。在聚合引发剂的配合比例超过上述上限的情况下,有时聚合物的分子量过度降低,在小于上述下限的情况下,转化率提高不充分,有时未反应的聚合性乙烯基单体残存数个百分点以上。
(水分散工序)
接着,对上述油相成分进行水分散(悬浮)。
即,将油相成分和水配合,并均匀地搅拌,从而对油相成分进行水分散(悬浮)。由此,得到油相成分的水分散(悬浮)液。
水分散的条件没有特别限制,例如可以在常温下实施,或者也可以加热来实施。
在油相成分的水分散中,优选配合分散剂(第2分散剂)、表面活性剂(第2表面活性剂)。
作为分散剂(第2分散剂),可以举出例如聚乙烯醇(PVA)、聚乙烯吡咯烷酮、明胶、阿拉伯胶、羟乙基纤维素、羟丙基纤维素、羧甲基纤维素、阳离子化淀粉、聚丙烯酸及其钠盐、苯乙烯马来酸共聚物及其钠盐等水溶性聚合物,例如磷酸三钙、胶体二氧化硅、蒙脱石、碳酸镁、氢氧化铝、氧化锌等无机系分散剂等。
分散剂中,优选可以举出聚乙烯醇(PVA)、磷酸三钙。进一步优选可以举出聚乙烯醇(PVA)。
相对于油相成分100质量份,分散剂的配合比例例如为0.01质量份以上、优选为0.1质量份以上、更优选为1质量份以上,并且,例如为10质量份以下、优选为5质量份以下。
关于表面活性剂(第2表面活性剂),为了有效防止自由基聚合中的粒子凝聚,优选与上述的分散剂(第2分散剂)并用,可以举出例如十二烷基苯磺酸钠、月桂基硫酸钠、双-2-乙基己基磺基琥珀酸钠、十二烷基二苯醚二磺酸钠、壬基二苯醚磺酸钠、芳香族磺酸与甲醛的缩合物的盐等阴离子系表面活性剂,例如聚氧乙烯月桂醚、聚氧乙烯壬基苯基醚、聚氧乙烯单硬脂酸酯、聚氧乙烯失水山梨醇单硬脂酸酯、聚氧乙烯聚氧丙烯嵌段共聚物等非离子系表面活性剂等。优选可以举出非离子系表面活性剂、阴离子系表面活性剂,更优选可以举出聚氧乙烯聚氧丙烯嵌段共聚物、芳香族磺酸与甲醛的缩合物的盐。
相对于油相成分100质量份,表面活性剂的配合比例例如为0.0001质量份以上、优选为0.001质量份以上,并且,例如为1.0质量份以下、优选为0.1质量份以下。
关于这些分散剂、或分散剂和表面活性剂,在例如将油相成分配合于水之前或配合后均可以进行配合,优选配合于将油相成分配合之前的水。由此,调制分散剂的水溶液、或分散剂和表面活性剂的水溶液。
在上述的油相成分的水分散(悬浮)中,可以使用例如均相混合机(homomixer)、超声波均质器、加压式均质器、乳化分散机(Milder)、多孔膜压入分散机等分散机,优选使用均相混合机。
水分散的条件可以适当地设定,在使用均相混合机的情况下,将其转数设定为例如100rpm以上、优选为1000rpm以上,并且,设定为例如10000rpm以下,例如8000rpm以下。
由此,调制水相中分散有油相成分的水分散液。
此外,在水分散液中配合有分散剂(第2分散剂)、或分散剂和表面活性剂的情况下,水分散液中的油相成分的液滴因分散剂、或分散剂和表面活性剂而更加稳定化。
将水(或水溶液)的配合比例调节成相对于油相成分100质量份例如为50质量份以上、优选为100质量份以上、更优选为150质量份以上,并且,例如为1900质量份以下、优选为900质量份以下、更优选为400质量份以下。
(聚合工序)
在聚合工序中,对聚合性乙烯基单体进行悬浮聚合,生成聚合物。为了对聚合性乙烯基单体进行悬浮聚合,将水分散液升温至预定温度。在悬浮聚合中,一边将水分散液搅拌以维持水分散液的水分散状态,一边使聚合性乙烯基单体进行反应(具体地为自由基聚合),生成聚合性乙烯基单体的聚合物。就悬浮聚合而言,由于成为聚合物的聚合性乙烯基单体全部仅存在于水分散粒子(疏水性液相)中,因此是原位(in-situ)聚合。
具体地说,悬浮聚合中,将水分散液边搅拌边加热,从而使聚合性乙烯基单体直接在水分散粒子中开始聚合,生成聚合物。
搅拌可以利用例如具有搅拌叶片的搅拌器来实施。关于搅拌速度,搅拌叶片的圆周速度例如为10m/分钟以上、优选为20m/分钟以上,并且为400m/分钟以下、优选为200m/分钟以下。
将水分散液加热成其温度达到例如为40℃以上、优选为50℃以上、更优选为60℃以上,并且,例如为100℃以下、优选为90℃以下、更优选为80℃以下。
并且,抗生物活性化合物以与聚合物不相溶的状态进行悬浮聚合。
加热时间例如为2小时以上、优选为3小时以上,并且,例如为12小时以下、优选为8小时以下。进而,也可以在加热至预定温度后,将其温度维持预定时间,然后反复进行加热以及温度维持,从而阶段性地进行加热。
在悬浮聚合中,抗生物活性化合物相对于聚合性乙烯基单体实质上为不溶性,抗生物活性化合物从聚合开始至聚合结束后,相对于聚合性乙烯基单体和/或聚合物维持不相溶状态。
然后,通过例如放冷等将聚合后的水分散液冷却,用100目(网眼)的滤布等进行过滤,从而得到缓释性粒子的水分散液(悬浮液)。
冷却温度例如为室温(20~30℃,更具体地为25℃)。
所得到的缓释性粒子中的抗生物活性化合物的浓度例如为1质量%以上、优选为5质量%以上、更优选为10质量%以上,并且,例如为50质量%以下、优选为40质量%以下、更优选为35质量%以下。
此外,缓释性粒子在悬浮液中的含有比例根据油相成分以及对其进行分散的水(或水溶液)的配合量而决定,具体地说,例如为10质量%以上、优选为20质量%以上,并且,例如为50质量%以下、优选为40质量%以下。
缓释性粒子的平均粒径例如为1μm以上、优选为2μm以上,并且,例如为20mm以下、优选为10mm以下。此外,平均粒径作为中位径进行计算。
通过上述缓释性粒子的制造方法来制造的缓释性粒子具有由后述的基质和分散于基质中的后述的结构域形成的2相结构。
另外,在通过上述制造方法得到的含有缓释性粒子的水分散液(悬浮液)中,可以根据需要适当地配合其它分散剂、增稠剂、防冻剂、防腐剂、微生物增殖抑制剂、比重调节剂等公知的添加剂。
这样得到的缓释性粒子可以以保持原样的状态(悬浮液),即以悬浮剂形态使用,此外还可以通过喷雾干燥而直接以粉剂形态使用。或者,也可以通过离心分离、压滤等进行固液分离,根据需要在清洗后例如通过流动干燥、架子干燥等进行干燥,根据需要利用雾化器、滑磨机(feather mill)等进行破碎,利用振动筛等进行分级,制成粉剂或粒剂等公知的剂型来使用。
此外,为了将缓释性粒子制成粒剂,例如在固体载体中配合缓释性粒子的悬浮液并混合,然后使它们干燥(粒剂化工序)。即,缓释性粒子的制造方法除了具有油相成分调制工序、水分散工序以及聚合工序之外,还可以具有粒剂化工序。
作为固体载体,可以举出例如浮石、膨润土、粘土、高岭土、滑石、酸性白土、沸石、蛭石、珠光体、碳酸钙、硅砂等。作为固体载体,优选可以举出浮石。固体载体可以使用市售品,具体可以使用卡卡(Kagalite)系列(天然浮石的细粒、卡卡工业公司制)等。固体载体的平均粒径例如为100μm以上、优选为300μm以上,并且,例如为5.00mm以下、优选为2.00mm以下。
在粒剂化工序中,将缓释性粒子的悬浮液的配合比例调节成抗生物活性化合物在所得到的粒剂(固体载体和缓释性粒子)中的浓度例如为0.01质量%以上、优选为0.05质量%以上,例如为2质量%以下、优选为1质量%以下。具体地说,相对于固体载体100质量份,缓释性粒子的悬浮液(包含水)的配合比例例如为0.01质量份以上、优选为0.05质量份以上、更优选为0.1质量份以上,进一步优选为0.2质量份以上,例如为10质量份以下、优选为5质量份以下。
<第1发明组的缓释性粒子的效果>
第1发明组的缓释性粒子通过如下制造方法获得,因此能够得到缓释性优异且坚固的缓释性粒子,该制造方法具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。
然而,根据如专利文献1中所记载的方法得到的微胶囊仅通过界面聚合来得到,因此在微胶囊中残存分散介质(溶剂),由此有时其表面硬度变得不充分。其结果是,微胶囊的分散液在经历施加高剪切力的工序的情况下、或长期保存的情况下,有时微胶囊发生凝聚而再分散变得困难。
进而,由于微胶囊的表面硬度不充分,因此有时微胶囊容易粘连(blocking)而难以以干燥粒子形态取出微胶囊。
另一方面,第1发明组的缓释性粒子通过如下制造方法获得,因此能够防止上述界面聚合中溶剂的存在所引起的缓释性粒子的表面硬度的降低,得到坚固的缓释性粒子,进而,所得到的缓释性粒子的再分散性和抗粘连性优异,该制造方法具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。
根据该缓释性粒子的制造方法,能够得到坚固并且再分散性和抗粘连性优异的缓释性粒子。
这样的缓释性粒子能够应用于各种工业制品,例如可以添加于屋内外的涂料、橡胶、纤维、树脂(包括塑料)、粘接剂、接缝剂、密封剂、建筑材料、填缝剂、木材处理剂、土壤处理剂、造纸工序中的白水、颜料、印刷版用处理液、冷却用水、油墨、切削油、化妆用品、非织造布、纺丝油、皮革等。另外,缓释性粒子中的抗生物活性化合物相对于这些工业制品的添加量例如为10mg/kg~100g/kg(制品质量)。
接下来,对将由缓释性粒子制成的粉剂与热塑性树脂配合的方式进行说明。
在该方法中,首先,使缓释性粒子的悬浮液干燥,制成粉剂。
接着,将粉剂和热塑性树脂进行熔融混炼,调制混炼物。
为了调制混炼物,例如具体而言可以使用挤出机、班伯里混合机。作为挤出机,可以使用例如双轴挤出机、单轴挤出机。混炼物是用于成型得到成型品的成型材料,具体地说,暂时进行冷却而调制成颗粒状成型材料(混炼物颗粒或母料)。另一方面,也可以将混炼物不以固体的成型材料的形态取出而直接连续地以熔融状态(熔融混炼物)供于后述的成型。
将粉剂配合于热塑性树脂,以使粉剂的抗生物活性化合物的含有比例相对于热塑性树脂为例如0.01质量%以上、优选为0.1质量%以上,并且,例如为10质量%以下、优选为3质量%以下。但是,在调制混炼物作为母料的情况下,不限于此,具体地说,将粉剂配合于热塑性树脂,以使抗生物活性化合物的含有比例相对于热塑性树脂为例如1质量%以上、优选为5质量%以上,并且,例如为50质量%以下、优选为30质量%以下,从而作为母料。
热塑性树脂没有特别限定,可以举出例如聚乙烯、聚丙烯等聚烯烃系树脂;聚苯乙烯或者聚甲基丙烯酸甲酯、丙烯腈-苯乙烯共聚树脂(AS树脂)、甲基丙烯酸甲酯-苯乙烯共聚物(MS树脂)、丙烯腈-苯乙烯-丁二烯共聚树脂(ABS树脂)等苯乙烯系和/或丙烯酸系树脂;聚对苯二甲酸乙二醇酯、聚乳酸等聚酯系树脂;6-尼龙等聚酰胺系树脂;氯乙烯树脂、偏氯乙烯树脂等乙烯基卤系树脂;聚碳酸酯、聚苯醚、聚甲醛、热塑性聚氨酯等。热塑性树脂可以单独使用或并用。优选可以举出聚烯烃系树脂、氯乙烯树脂、热塑性聚氨酯,更优选可以举出聚烯烃系树脂、氯乙烯树脂,进一步优选可以举出聚乙烯、聚丙烯。
接着,由混炼物颗粒或熔融混炼物成型得到成型品。
作为成型方法,可以采用例如注射成型、挤出成型、吹胀成型(inflation molding)、拉挤成型(Pultrusion molding)、压缩成型等。
由此,可以得到成型为预定形状的添加了粉剂(缓释性粒子)的成型品。
上述说明中,虽然将由缓释性粒子制成的粉剂添加于热塑性树脂,但只要是树脂就没有特别限定,也可以添加于例如热固化性树脂。
可以将粉剂适合地混合于特别是环氧树脂、硅树脂等液状树脂。
这样的成型品可用于各种用途,可以用作例如建筑材料,例如电线电缆材料及其电线电缆的被覆材料,例如气体等的导管及其导管的被覆材料,例如衣类、蚊帐等纤维制品。
<第1发明组的成型品的效果>
就这样的成型品而言,由于粉剂的缓释性粒子具有由基质和结构域形成的坚固的2相结构,因此粉剂在混炼、成型时不会被破坏,分散于成型品或局部存在于表面,从而添加了上述粉剂的成型品在抗生物活性化合物的缓释性方面优异。换言之,由于上述成型材料含有上述缓释性粒子,因此能够分散于上述成型品或局部存在于表面,对成型品赋予优异的抗生物活性化合物的缓释性。
此外,通过将缓释性粒子设为1mm~20mm直径的珠状,敷设、常备、固定于流体(气体、液体)的流动通道,从而能够对通过的流体稳定地赋予杀菌等抗生物活性效果。
并且,通过上述缓释性粒子的制造方法得到的缓释性粒子具体包括下面描述的缓释性粒子的第1实施方式以及第2实施方式。
[第1实施方式]
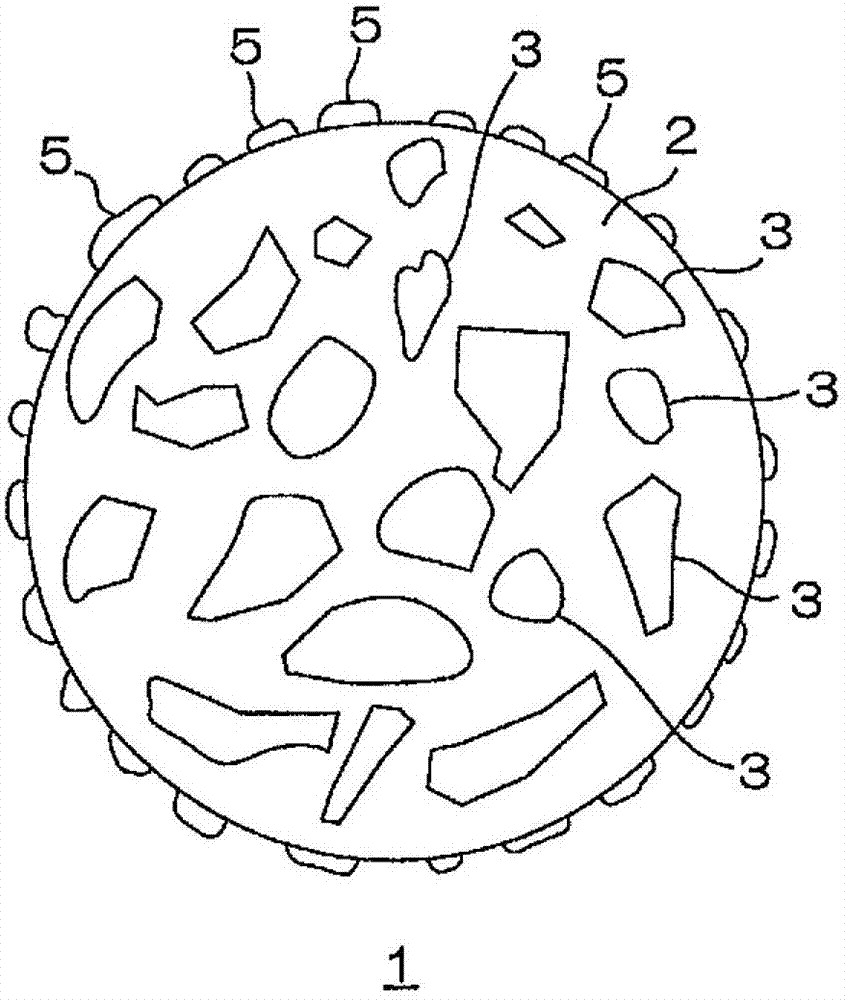
参照图A1对缓释性粒子的第1实施方式进行说明。
如图A1的截面图所示,缓释性粒子1形成为例如球状粒子。缓释性粒子1具有由基质2和分散于基质2中的结构域3形成的2相结构。基质2包含由上述聚合性乙烯基单体得到的聚合物。结构域3包含上述抗生物活性化合物。
具体地说,在缓释性粒子1中,基质2形成介质或连续相,形成有多个结构域3以孤立状分散的多结构域(multidomain)结构或海岛结构(或多核结构)。此外,在该缓释性粒子1中,基质2和结构域3彼此不相溶,形成彼此分离的相分离结构。
基质2在缓释性粒子1中存在于结构域3以外的区域,形成为除去结构域3的形状。
多个结构域3在基质2中形成分散相。结构域3的形状没有特别限定,可以形成为例如不定形状、球状、块状、板状等适宜形状。结构域3的最大长度的平均值例如为0.05μm以上、优选为0.1μm以上,并且,例如为20μm以下、优选为10μm以下。
此外,结构域3包含从基质2的内部向外侧突出的突出物4。突出物4从基质2的表面露出。由此,在缓释性粒子1的表面,露出基质2和结构域3这两者。突出物4具有埋设于基质2的表层部的埋设部8。突出物4起到加快缓释性粒子1中的抗生物活性化合物的初期缓释速度的作用、以及显著提高缓释性粒子1的抗粘连性的作用。相对于缓释性粒子1的整个表面,突出物4在基质2的整个表面的露出率(即,结构域3的露出率)例如为0.1%以上、优选为1%以上,并且,例如为50%以下、优选为30%以下。基质2的露出率是从缓释性粒子1的整个表面扣除突出物4的露出率的比例。
另外,通过结构域3的一部分从基质2脱离(脱落),从而在该缓释性粒子1的表面形成有孔6。孔6形成为与构成结构域3的抗生物活性化合物的形状对应。
为了得到该缓释性粒子1,在上述缓释性粒子1的制造方法中,尤其是在油相成分调制工序中,作为聚合性乙烯基单体,不使用(甲基)丙烯酸酯系单体以及(甲基)丙烯酸酯系交联性单体,并且,在水分散工序中,优选地,作为表面活性剂(第2表面活性剂),不配合芳香族磺酸与甲醛的缩合物的盐。
在油相成分调制工序中,作为聚合性乙烯基单体,优选使用芳香族乙烯基单体与芳香族交联性单体的组合。
在聚合性乙烯基单体为芳香族乙烯基单体与芳香族交联性单体的组合的情况下,相对于芳香族乙烯基单体与芳香族交联性单体的总量100质量份,芳香族乙烯基单体的含有比例例如为10质量份以上、优选为20质量份以上、更优选为30质量份以上,并且,例如为90质量份以下、优选为80质量份以下、更优选为70质量份以下。
在水分散工序中,作为表面活性剂(第2表面活性剂),不配合芳香族磺酸与甲醛的缩合物的盐,而优选配合分散剂(第2分散剂)。
<第1实施方式的效果>
就用于制造缓释性粒子的第1实施方式的缓释性粒子的制造方法而言,在油相成分调制工序中,作为聚合性乙烯基单体,不使用(甲基)丙烯酸酯系单体以及(甲基)丙烯酸酯系交联性单体,并且在水分散工序中,不配合芳香族磺酸与甲醛的缩合物的盐,接着在聚合工序中,对上述聚合性乙烯基单体进行悬浮聚合,因此能够可靠地形成突出物4。
在缓释性粒子1的表面,露出基质2和结构域3这两者。尤其在该缓释性粒子1的表面,以抗生物活性化合物向外侧突出的方式露出,从而构成突出物4。此外,缓释性粒子1具有由基质2和结构域3形成的2相结构,而不具有壳层。
此外,在缓释性粒子1中,突出物4从基质2露出,因此能够借助于突出物4进一步提高抗粘连性。
此外,在缓释性粒子1中,如果形成露出的突出物4的抗生物活性化合物能够从初期开始缓释,并且突出物4脱落,则进一步加速抗生物活性化合物的初期缓释速度,因此能够加快抗生物活性化合物的初期缓释速度,调节抗生物活性化合物的缓释速度。
如图A1所示,该缓释性粒子1具有由包含聚合物的基质2和结构域3形成的2相结构,上述结构域3是包含抗生物活性化合物的结构域3,且分散于基质2中,因此抗生物活性化合物的缓释性优异,并且坚固性优异。由此,缓释性粒子在与上述树脂的混炼性方面优异。
[第2实施方式]
参照图A2对缓释性粒子的第2实施方式进行说明。
如图A2的截面图所示,在缓释性粒子1的表面,没有露出结构域3,全部结构域3内包于基质2中。即,在缓释性粒子1中,形成结构域3的抗生物活性化合物被基质2所被覆而得到保护。
在缓释性粒子1中的基质2的表面,附着有例如抗生物活性化合物。具体地说,包含抗生物活性化合物的附着物5以被覆基质2的整个表面的全部或一部分的方式附着。附着物5与第1实施方式的缓释性粒子1(参照图A1)的突出物4不同,不具有埋设部8,与基质2的表面接触。附着物5的形状没有特别限定,形成为例如不定形状、球状、块状、板状等适宜形状。尤其,附着物5的内面(与基质2的表面接触的接触面)形成有与基质2的表面(球面)对应的凹面,具体地说,形成有向外侧凹陷的弯曲面。附着物5的大小与结构域3相同或小于结构域3,相对于结构域3的最大长度的平均值,例如为100%以下、优选为50%以下,并且,例如为0.01%以上,具体地说,附着物5的最大长度的平均值例如为10μm以下、优选为5μm以下,例如为0.05μm以上、优选为0.1μm以上。附着物5相对于基质2的整个表面的被覆率例如为10%以上、优选为20%以上,并且,例如为100%以下、优选为90%以下。
此外,为了得到该缓释性粒子1,在上述缓释性粒子1的制造方法的水分散工序中,配合芳香族磺酸与甲醛的缩合物的盐作为表面活性剂(第2表面活性剂),和/或在油相成分调制工序中,配合(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体作为聚合性乙烯基单体。
第2表面活性剂优选与上述第2分散剂并用。
作为芳香族磺酸,可以举出例如苯磺酸、甲苯磺酸、枯烯磺酸、萘磺酸等。优选可以举出α-萘磺酸、β-萘磺酸等萘磺酸。
作为用于形成盐的阳离子,可以举出例如1价阳离子。作为1价阳离子,可以举出钠阳离子、钾阳离子等碱金属阳离子,例如铵阳离子等。优选可以举出碱金属阳离子。
作为芳香族磺酸与甲醛的缩合物的盐,具体可以举出萘磺酸与甲醛的缩合物的盐(萘磺酸甲醛缩合物钠盐)。作为芳香族磺酸与甲醛的缩合物的盐,可以使用市售品,具体可以举出DEMOL NL(β-萘磺酸甲醛缩合物钠盐、41%水溶液、花王公司制)等。
相对于疏水性浆料100质量份,芳香族磺酸与甲醛的缩合物的盐的配合比例例如为0.0001质量份以上、优选为0.001质量份以上,并且,例如为1.0质量份以下、优选为0.2质量份以下、更优选为0.1质量份以下。
(甲基)丙烯酸酯系交联性单体在聚合性乙烯基单体中的含有比例例如为10质量%以上、优选为30质量%以上,并且,例如为100质量%以下。
关于聚合物,由于聚合性乙烯基单体含有(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体,因此(甲基)丙烯酸酯系单体的聚合物具有由(甲基)丙烯酸酯系交联性单体或其聚合物交联而成的交联结构。
<第2实施方式的效果>
就用于制造缓释性粒子的第2实施方式的缓释性粒子的制造方法而言,在水分散工序中,配合芳香族磺酸与甲醛的缩合物的盐,和/或在油相成分调制工序中,使用(甲基)丙烯酸酯系单体以及(甲基)丙烯酸酯系交联性单体作为聚合性乙烯基单体,在聚合工序中,对(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体进行悬浮聚合。因此,能够抑制包含抗生物活性化合物的结构域3露出于缓释性粒子1的表面(参照图A1)。即,如图A2所示,能够由基质2被覆结构域3而进行保护。
此外,在聚合工序中,在存在萘磺酸与甲醛的缩合物的盐(优选为萘磺酸甲醛缩合物钠盐)的情况下,对聚合性乙烯基单体进行悬浮聚合时,使聚合工序中的悬浮聚合物与水连续相之间的界面更加稳定化,因此能够抑制抗生物活性化合物向缓释性粒子之外漏出。其结果是,利用萘磺酸与甲醛的缩合物的盐的配合比例,能够调节缓释性粒子中的抗生物活性化合物的缓释性。
此外,在聚合工序中,对(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体进行悬浮聚合时,使抗生物活性化合物粒子在油相中的分散稳定化,因此能够抑制抗生物活性化合物向缓释性粒子之外漏出。其结果是,通过使用(甲基)丙烯酸酯系单体和(甲基)丙烯酸酯系交联性单体作为聚合性乙烯基单体,能够调节缓释性粒子中的抗生物活性化合物的缓释性。
具体地说,就通过包括上述水分散工序的缓释性粒子的制造方法得到的缓释性粒子而言,如图A2所示,能够由基质2被覆结构域3,能够使附着物5附着于基质2的表面。因此,第2实施方式的缓释性粒子1因附着物5而抗粘连性优异。此外,能够借助于附着物5,加快抗生物活性化合物的初期缓释速度,调节抗生物活性化合物的缓释速度。
并且,如图A2所示,在第2实施方式中,虽然附着物5附着于基质2的表面,但结构域3被基质2所被覆,因此与具备突出物4的第1实施方式的缓释性粒子1(参照图A1)相比,耐碱性优异。即,第2实施方式的缓释性粒子1与第1实施方式的缓释性粒子1相比,即使保存于碱水溶液中,也能够抑制缓释性粒子中的突出物4所引起的抗生物活性化合物的浓度减少。
<第2实施方式的变形例>
如图A3所示,不使附着物5附着于基质2的表面,也能够使基质2的表面全部露出。
[第2发明组]
<缓释性粒子的制造方法的说明>
对第2发明组的缓释性粒子的制造方法进行说明。
缓释性粒子的制造方法具备:油相成分调制工序,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制含有疏水性浆料的油相成分;对油相成分进行水分散来调制水分散液的水分散工序;以及对聚合性乙烯基单体进行悬浮聚合来生成聚合物的聚合工序。此外,关于缓释性粒子的制造方法,在油相成分调制工序、水分散工序和聚合工序的至少任一个工序中,添加疏水性壳层形成成分和亲水性壳层形成成分。
关于该缓释性粒子的制造方法,优选在油相成分调制工序中,将抗生物活性化合物分散于聚合性乙烯基单体中来调制疏水性浆料,接着,将疏水性浆料与疏水性壳层形成成分配合,调制包含疏水性浆料和疏水性壳层形成成分的油相成分。此外,关于缓释性粒子的制造方法,优选在水分散工序和聚合工序的至少任一个工序中,添加亲水性壳层形成成分,更优选在聚合工序中,配合亲水性壳层形成成分。
以下,对上述抗生物活性化合物、聚合性乙烯基单体、疏水性壳层形成成分以及亲水性壳层形成成分依次进行说明。
(抗生物活性化合物)
作为抗生物活性化合物,选自具有杀虫(包括灭蚁)、防虫(包括防蚁)、杀菌、抗菌、防腐、除草、防藻、防霉等抗生物活性的杀虫剂(包括灭蚁剂)、防虫剂(包括防蚁剂)、杀菌剂、抗菌剂、防腐剂、除草剂、防藻剂、防霉剂、引诱剂、驱避剂和杀鼠剂等。
具体地说,关于抗生物活性化合物,例如作为杀虫剂,可以举出噻虫胺((E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍)、吡虫啉(1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺)、噻虫啉、噻虫嗪((EZ)-3-(2-氯-1,3-噻唑-5-基甲基)-5-甲基-1,3,5-二嗪-4-基叉(硝基)胺)、呋虫胺等新烟碱系杀虫剂,氟虫双酰胺、氯虫苯甲酰胺等二酰胺系,除虫脲、氟苯脲、氟啶脲、虫酰肼、甲氧虫酰肼、环丙氨嗪等昆虫生长调节剂,四螨嗪等杀螨剂,吡蚜酮、油酸钠等其它合成药剂等。例如,作为杀菌剂,可以举出碱性氯化铜、碱性硫酸铜、羟基喹啉铜等铜系杀菌剂,金属银等银系杀菌剂,福代锌等有机硫系杀菌剂,四氯苯酞、三环唑等黑色素生物合成抑制剂,甲基硫菌灵、多菌灵(MBC)、乙霉威等苯并咪唑系杀菌剂,异噻菌胺等酸酰胺系杀菌剂,嗪氨灵等甾醇生物合成抑制剂,1,2-苯并异噻唑-3-酮等异噻唑啉酮系杀菌剂,哒菌酮、氟酰亚胺、克菌丹、百菌清、灭螨猛、喹酸、苯噻菌胺、氰霜唑、吡啶硫酮锌等其它合成抑制剂等。例如,作为除草/防藻剂,可以举出3-(3,4-二氯苯基)-1,1-二甲基脲(DCMU)、苄草隆、卡灵草等脲系药剂,乙氧嘧磺隆、氯吡嘧磺隆、啶嘧磺隆、烟嘧磺隆、噻吩磺隆、唑吡嘧磺隆、环丙嘧磺隆、氟吡磺隆、三氟啶磺隆钠盐等磺脲系药剂,西玛津(CAT)、阿特拉津、三嗪氟草胺、环草啶、氟氯氰菊酯、特丁净等三嗪系药剂,草甘膦等氨基酸系,丙炔氟草胺等苯基邻苯二甲酰亚胺系,甲基磺草酮等三酮系,莫克草、环酯草醚等其它药剂等。作为抗生物活性化合物,优选从种选择性、安全性的观点考虑,可以举出新烟碱系杀虫剂,并且从通用性、效力的观点考虑,可以举出吡啶硫酮锌,更优选从难溶性的观点考虑,可以举出噻虫胺、吡虫啉、吡啶硫酮锌,进一步优选可以举出噻虫胺、吡虫啉。特别优选从对哺乳动物的安全性的观点考虑,可以举出噻虫胺。
抗生物活性化合物实质上为疏水性,具体地说,例如,室温(20~30℃,更具体地为25℃)时相对于水的溶解度极小,更具体地说,例如,室温时的溶解度为1.5质量份/水100容量份(15g/L)以下、优选为0.5质量份/水100容量份(5g/L)以下、进一步优选为0.1质量份/水100容量份(1g/L)以下。
抗生物活性化合物相对于聚合性乙烯基单体实质上为不溶性的,具体地说,例如,室温(20~30℃,更具体地为25℃)时相对于聚合性乙烯基单体的溶解度极小,具体地说,室温时的溶解度例如为0.1质量份/(使用的)聚合性乙烯基单体(混合物)100容量份(1g/L)以下、优选为0.05质量份/(使用的)聚合性乙烯基单体(混合物)100容量份(0.5g/L)以下。
此外,抗生物活性化合物的熔点例如为80℃以上、优选为100℃以上,此外,如果抗生物活性化合物是不含有金属原子的化合物,则例如为300℃以下。
(聚合性乙烯基单体)
作为聚合性乙烯基单体,可以举出例如(甲基)丙烯酸酯系单体、芳香族乙烯基单体、乙烯基酯系单体、马来酸酯系单体、乙烯基卤、亚乙烯基卤、含氮乙烯基单体、交联性单体等。
作为(甲基)丙烯酸酯系单体,可以举出例如甲基丙烯酸酯和/或丙烯酸酯,具体可以举出(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸正丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸正丁酯、(甲基)丙烯酸异丁酯(i-BMA/i-BA)、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸正戊酯、(甲基)丙烯酸正己酯、(甲基)丙烯酸环己酯等具有烷基部分为直链状、支链状或环状的碳原子数1~6的烷基部分的(甲基)丙烯酸烷基酯;例如(甲基)丙烯酸2-甲氧基乙酯等(甲基)丙烯酸烷氧基烷基酯;例如(甲基)丙烯酸羟基乙酯等(甲基)丙烯酸羟基烷基酯;例如(甲基)丙烯酸缩水甘油酯等含有环氧基的(甲基)丙烯酸酯等。优选可以举出(甲基)丙烯酸烷基酯。
作为(甲基)丙烯酸烷基酯,更优选可以举出具有碳原子数1~6的烷基部分的(甲基)丙烯酸烷基酯,特别优选可以举出甲基丙烯酸异丁酯(i-BMA)。
作为芳香族乙烯基单体,可以举出例如苯乙烯(乙烯基苯)、对甲基苯乙烯、邻甲基苯乙烯、α-甲基苯乙烯、乙基乙烯基苯等苯乙烯系单体(单乙烯基苯)等。
作为乙烯基酯系单体,可以举出例如乙酸乙烯酯、丙酸乙烯酯等。
作为马来酸酯系单体,可以举出例如马来酸二甲酯、马来酸二乙酯、马来酸二丁酯等。
作为乙烯基卤,可以举出例如氯乙烯、氟乙烯等。
作为亚乙烯基卤,可以举出例如偏氯乙烯、偏氟乙烯等。
作为含氮乙烯基单体,可以举出例如(甲基)丙烯腈、N-苯基马来酰亚胺、乙烯基吡啶等。
作为交联性单体,可以举出例如乙二醇二(甲基)丙烯酸酯、二乙二醇二(甲基)丙烯酸酯等单或聚乙二醇二(甲基)丙烯酸酯,例如1,3-丙二醇二(甲基)丙烯酸酯、1,4-丁二醇二(甲基)丙烯酸酯、1,5-戊二醇二(甲基)丙烯酸酯等链烷二醇二(甲基)丙烯酸酯;例如三羟甲基丙烷三(甲基)丙烯酸酯、季戊四醇四(甲基)丙烯酸酯(PETA/PETM)等链烷多元醇聚(甲基)丙烯酸酯等(甲基)丙烯酸酯系交联性单体;例如烯丙基(甲基)甲基丙烯酸酯、三烯丙基(异)氰脲酸酯等烯丙基系单体;例如二乙烯基苯、三乙烯基苯等芳香族交联性单体。优选可以举出单或聚乙二醇二(甲基)丙烯酸酯、二乙烯基苯,更优选可以举出乙二醇二(甲基)丙烯酸酯、二乙烯基苯。
聚合性乙烯基单体可以单独使用或并用。
作为聚合性乙烯基单体,优选可以举出(甲基)丙烯酸酯系单体与交联性单体的组合、芳香族乙烯基单体与交联性单体的组合。
在聚合性乙烯基单体为(甲基)丙烯酸酯系单体与交联性单体的组合的情况下,相对于(甲基)丙烯酸酯系单体与交联性单体的总量100质量份,(甲基)丙烯酸酯系单体的含有比例例如为10质量份以上、优选为20质量份以上、更优选为30质量份以上,并且,例如为90质量份以下、优选为80质量份以下、更优选为70质量份以下。在聚合性乙烯基单体为芳香族乙烯基单体与交联性单体的组合的情况下,相对于芳香族乙烯基单体与交联性单体的总量100质量份,芳香族乙烯基单体的配合比例例如为10质量份以上、优选为20质量份以上、更优选为30质量份以上,并且,例如为90质量份以下、优选为80质量份以下、更优选为70质量份以下。
为了在室温时具有坚固的表面,聚合物的玻璃化转变温度例如为30℃以上、优选为50℃以上,以达到该玻璃化转变温度的方式选择聚合性乙烯基单体。
聚合性乙烯基单体例如实质上为疏水性,具体地说,例如室温时相对于水的溶解度极小,更具体地说,室温时的溶解度例如为10质量份/水100容量份(100g/L)以下、优选为8质量份/水100容量份(80g/L)以下。另外,在并用不同种类的聚合性乙烯基单体的情况下,作为聚合性乙烯基单体整体(即,不同种类的聚合性乙烯基单体的混合物),实质上为疏水性。
(疏水性壳层形成成分和亲水性壳层形成成分)
疏水性壳层形成成分和亲水性壳层形成成分是通过加聚或缩聚(缩合聚合)等进行反应的彼此不同的2种成分。
疏水性壳层形成成分例如实质上为疏水性,具体地说,室温时相对于水的溶解度极小,更具体地说,例如,室温时的溶解度为1质量份/水100容量份(10g/L)以下、优选为0.5质量份/水100容量份(5g/L)以下、更优选为0.1质量份/水100容量份(1g/L)以下。
疏水性壳层形成成分是通过与亲水性壳层形成成分进行加聚或缩聚来形成壳层的油溶性化合物,可以举出例如聚异氰酸酯、聚羧酸氯化物、聚磺酰氯等。
作为聚异氰酸酯,可以举出例如二苯基甲烷二异氰酸酯、甲苯二异氰酸酯等芳香族聚异氰酸酯(芳香族二异氰酸酯),例如六亚甲基二异氰酸酯等脂肪族聚异氰酸酯(脂肪族二异氰酸酯),例如异佛尔酮二异氰酸酯(IPDI)、氢化二甲苯二异氰酸酯、氢化二苯基甲烷二异氰酸酯等脂环族聚异氰酸酯(脂环族二异氰酸酯),例如二甲苯二异氰酸酯、四甲基二甲苯二异氰酸酯等芳香脂肪族聚异氰酸酯(芳香脂肪族二异氰酸酯)等。
此外,还可以举出上述聚异氰酸酯的多聚物,具体可以举出二聚物、三聚物(含有异氰脲酸酯基的聚异氰酸酯、环状三聚物)、五聚物、七聚物等。优选可以举出三聚物,具体地为IPDI的三聚物。
进而,还可以举出上述聚异氰酸酯的改性物(多聚物除外),可以举出例如三羟甲基丙烷的IPDI加合物等多元醇改性聚异氰酸酯等。
作为聚羧酸氯化物,可以举出例如癸二酰氯、己二酰氯、壬二酰氯、对苯二甲酰氯、苯三甲酰氯等。
作为聚磺酰氯,可以举出例如苯磺酰氯等。
疏水性壳层形成成分可以单独使用或并用。
作为疏水性壳层形成成分,优选可以举出聚异氰酸酯,更优选可以举出二异氰酸酯的环状三聚物、三羟甲基丙烷的加合物。
亲水性壳层形成成分是在界面聚合之前存在于水相的水溶性化合物。亲水性壳层形成成分是含有活性烃基的化合物,这样的含有活性烃基的化合物是具有例如氨基、羟基等活性烃基的化合物,具体可以举出例如聚胺、多元醇、水等。
作为聚胺,可以举出例如乙二胺、丙二胺、己二胺、二氨基甲苯、苯二胺、哌嗪等二胺,例如二亚乙基三胺、三亚乙基四胺、四亚乙基五胺、五亚乙基六胺等3价以上的聚胺等。优选可以举出3价以上的聚胺,更优选可以举出二亚乙基三胺。
作为多元醇,可以举出例如乙二醇、丙二醇、1,4-丁二醇、1,6-己二醇、新戊二醇、二乙二醇、三乙二醇、二丙二醇、环己烷二甲醇、聚乙二醇、聚丙二醇等二醇,例如甘油、三羟甲基丙烷等三醇,例如季戊四醇等四醇等。
亲水性壳层形成成分可以单独使用或并用。
作为亲水性壳层形成成分,优选可以举出聚胺、多元醇,更优选为聚胺。
接下来,对油相成分调制工序、水分散工序以及聚合工序依次进行说明。
(油相成分调制工序)
在油相成分调制工序中,在不存在溶剂的情况下,将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体中,从而调制疏水性浆料,接着,将疏水性浆料与疏水性壳层形成成分配合,调制包含疏水性浆料和疏水性壳层形成成分的油相成分。
具体地说,首先,配合上述聚合性乙烯基单体和抗生物活性化合物,而不配合溶剂(己烷、甲苯、乙酸乙酯等疏水性有机溶剂),并搅拌。由此,调制疏水性浆料。疏水性浆料被包含于油相成分中。
为了将抗生物活性化合物分散于聚合性乙烯基单体中,可以使用例如涂料搅拌器、均质分散器(高速分散机)、珠磨机(包括分批式珠磨机)、球磨机、棒磨机等分散机。分散机可以单独使用或并用。作为分散机,优选从能够在广粘度区域中使用且能够在大规模工业生产中使用的观点考虑,可以使用分批式珠磨机。
通过上述分散,对抗生物活性化合物进行湿式粉碎。
关于抗生物活性化合物相对于聚合性乙烯基单体的配合比例,以质量比例(即,抗生物活性化合物的质量份/聚合性乙烯基单体的质量份)计,例如为1/99以上、优选为10/90以上、更优选为15/85以上,并且,例如为90/10以下、优选为75/25以下、更优选为70/30以下、进一步优选为65/35以下、特别优选为60/40以下。
此外,相对于聚合性乙烯基单体100质量份,抗生物活性化合物的配合比例例如为1质量份以上、优选为10质量份以上、更优选为20质量份以上,并且,例如为900质量份以下、优选为300质量份以下、更优选为200质量份以下、进一步优选为150质量份以下。
在上述分散中,还可以根据需要配合分散剂(第1分散剂)。作为分散剂,可以举出两亲性高分子型分散剂、非离子系表面活性剂(第1表面活性剂)等。
作为两亲性高分子型分散剂,可以举出例如EFKA4008、EFKA4009(以上为汽巴精化制氨基甲酸酯系高分子分散剂),DISPERBYK-2164、DISPERBYK-164(以上为毕克化学公司制颜料分散用官能团改性共聚物),NUOSPERSE 2008、NUOSPERSE FA-196、NUOSPERSE 657(以上为海名斯公司制),FLOWLEN D-90、POLYFLOW KL-100、POLYFLOW KL-700(以上为共荣社化学公司制),HOMOGENOL L-95(花王公司制)等非离子系两亲性高分子型分散剂。此外,作为两亲性高分子型分散剂,可以举出例如FLOWLEN G-900(共荣社化学公司制羧基改性高分子),DISPARLON DA-234、DISPARLON DA-325、DISPARLON DA-375、DISPARLON DA-550、DISPARLON AQ-330(以上为楠本化成公司制聚醚磷酸酯盐)等阴离子系两亲性高分子型分散剂。进而,作为两亲性高分子型分散剂,可以举出例如NOPCOSPERSE 092(圣诺普科公司制)等阳离子系两亲性高分子型分散剂。
作为非离子系表面活性剂,可以举出例如AMOGEN CBH(烷基甜菜碱)、AMOGEN SH(烷基酰胺甜菜碱)、NOIGEN 100E(聚氧乙烯油基醚)、NOIGEN EA73(聚氧乙烯十二烷基苯基醚)、NOIGEN ES99(单油酸聚乙二醇酯)、Dianol CME(椰子油脂肪酸单乙醇酰胺)、Dianol 300(椰子油脂肪酸单乙醇二酰胺)、SORGEN 30(失水山梨醇倍半油酸酯)、SORGEN 40(失水山梨醇单油酸酯)、SORGEN 50(失水山梨醇单硬脂酸酯)、EPAN 420(聚氧乙烯聚氧丙烯二醇)、EPAN 720(聚氧乙烯聚氧丙烯二醇)(以上为花王公司制)等。
作为分散剂,优选可以举出两亲性高分子型分散剂,更优选可以举出非离子系两亲性高分子型分散剂、阴离子系两亲性高分子型分散剂,进一步优选可以举出非离子系两亲性高分子型分散剂,特别优选可以举出颜料分散用官能团改性共聚物分散剂、氨基甲酸酯系高分子分散剂。
相对于抗生物活性化合物,分散剂的配合比例例如为0.1质量%以上、优选为1质量%以上,并且,例如为40质量%以下、优选为20质量%以下。
调制疏水性浆料后,将疏水性浆料与疏水性壳层形成成分配合。
具体地说,将疏水性壳层形成成分配合于疏水性浆料。
优选将疏水性壳层形成成分与聚合引发剂一起配合于疏水性浆料。
聚合引发剂可以举出悬浮聚合中通常使用的自由基聚合引发剂,具体可以举出油溶性聚合引发剂等。
作为油溶性聚合引发剂,可以举出例如过氧化二月桂酰(10小时半衰期温度T1/2:61.6℃)、过氧化2-乙基己酸1,1,3,3-四甲基丁酯(10小时半衰期温度T1/2:65.3℃)、过氧化-2-乙基己酸叔己酯(10小时半衰期温度T1/2:69.9℃)、过氧化二碳酸二异丙酯(10小时半衰期温度T1/2:40.5℃)、过氧化苯甲酰(10小时半衰期温度T1/2:73.6℃)等油溶性有机过氧化物,例如2,2′-偶氮二异丁腈(10小时半衰期温度T1/2:60℃)、2,2'-偶氮双(2,4-二甲基戊腈)(10小时半衰期温度T1/2:51℃)、2,2'-偶氮双(2-甲基丁腈)(10小时半衰期温度T1/2:67℃)等油溶性偶氮化合物等。优选可以举出过氧化二月桂酰、过氧化-2-乙基己酸叔己酯、2,2′-偶氮二异丁腈。
此外,聚合引发剂的10小时半衰期温度T1/2例如为40℃以上、优选为50℃以上,并且,例如为90℃以下、优选为80℃以下。将聚合引发剂的10小时半衰期温度T1/2设为对任意几个温度值时的浓度半衰期时间进行作图而得到的图的10小时值的温度。
聚合引发剂可以单独使用或并用两种以上。
相对于聚合性乙烯基单体100质量份,聚合引发剂的配合比例例如为0.01质量份以上、优选为0.1质量份以上、更优选为0.5质量份以上,例如为5质量份以下、优选为3质量份以下、更优选为2.0质量份以下。在聚合引发剂的配合比例超过上述上限的情况下,有时聚合物的分子量过度降低,在小于上述下限的情况下,转化率提高不充分,有时未反应的聚合性乙烯基单体残存数个百分点以上。
另外,可以将聚合性乙烯基单体分开配合,该情况下,首先,将聚合性乙烯基单体的一部分与抗生物活性化合物配合,并将它们分散而调制疏水性浆料,然后,使聚合引发剂和疏水性壳层形成成分溶解于聚合性乙烯基单体的剩余部分,并将其配合于疏水性浆料。
由此,调制含有聚合引发剂、疏水性壳层形成成分和疏水性浆料的油相成分。
相对于聚合性乙烯基单体100质量份,疏水性壳层形成成分的配合比例例如为2质量份以上、优选为5质量份以上、更优选为10质量份以上,进一步优选为20质量份以上,并且,例如为100质量份以下、优选为80质量份以下、更优选为70质量份以下、进一步优选为60质量份以下。
此外,相对于油相成分,疏水性壳层形成成分的配合比例例如为1质量%以上、优选为2质量%以上,并且,例如为60质量%以下、优选为40质量%以下。
另一方面,抗生物活性化合物在油相成分中的含有比例例如为1质量%以上、优选为10质量%以上,并且,例如为90质量%以下、优选为80质量%以下、更优选为70质量%以下、更优选为60质量%以下。
聚合性乙烯基单体在油相成分中的含有比例例如为10质量%以上、优选为30质量%以上、优选为50质量%以上,并且,例如为90质量%以下、优选为80质量%以下、更优选为70质量%以下。
油相成分中的抗生物活性化合物的平均粒径例如为5μm以下、优选为2.5μm以下,并且,例如为0.05μm以上、优选为0.1μm以上。
需要说明的是,在上文中,虽然将疏水性壳层形成成分和聚合引发剂配合于疏水性浆料,但是也可以例如将疏水性壳层形成成分和聚合引发剂配合于被调制为疏水性浆料之前的抗生物活性化合物和聚合性乙烯基单体。具体地说,首先,将疏水性壳层形成成分配合于抗生物活性化合物和聚合性乙烯基单体中,接着,将它们分散而调制疏水性浆料。由此,一次性调制含有抗生物活性化合物、聚合性乙烯基单体、疏水性壳层形成成分和聚合引发剂的油相成分。
(水分散工序)
接着,对上述油相成分进行水分散(悬浮)。
即,将油相成分和水配合,并均匀地搅拌,从而对油相成分进行水分散(悬浮)。由此,得到油相成分的水分散(悬浮)液。
水分散的条件没有特别限制,例如可以在常温下实施,或者也可以加热来实施。
在油相成分的水分散中,优选配合分散剂(第2分散剂)、表面活性剂(第2表面活性剂)。
作为分散剂(第2分散剂),可以举出例如聚乙烯醇(PVA)、聚乙烯吡咯烷酮、明胶、阿拉伯胶、羟乙基纤维素、羟丙基纤维素、羧甲基纤维素、阳离子化淀粉、聚丙烯酸及其钠盐、苯乙烯马来酸共聚物及其钠盐等水溶性聚合物,例如磷酸三钙、胶体二氧化硅、蒙脱石、碳酸镁、氢氧化铝、氧化锌等无机系分散剂等。
分散剂中,优选可以举出聚乙烯醇(PVA)、磷酸三钙。进一步优选可以举出聚乙烯醇(PVA)。
相对于油相成分100质量份,分散剂的配合比例例如为0.01质量份以上、优选为0.1质量份以上、更优选为1质量份以上,并且,例如为10质量份以下、优选为5质量份以下。
关于表面活性剂(第2表面活性剂),为了有效防止自由基聚合中的粒子凝聚,优选与上述的分散剂(第2分散剂)并用,具体可以举出十二烷基苯磺酸钠、月桂基硫酸钠、二-2-乙基己基磺基琥珀酸钠、十二烷基二苯醚二磺酸钠、壬基二苯醚磺酸钠、芳香族磺酸与甲醛的缩合物的盐等阴离子系表面活性剂,例如聚氧乙烯月桂醚、聚氧乙烯壬基苯基醚、聚氧乙烯单硬脂酸酯、聚氧乙烯失水山梨醇单硬脂酸酯、聚氧乙烯聚氧丙烯嵌段共聚物等非离子系表面活性剂等。优选可以举出非离子系表面活性剂、阴离子系表面活性剂,更优选可以举出聚氧乙烯聚氧丙烯嵌段共聚物、芳香族磺酸与甲醛的缩合物的盐。
表面活性剂可以单独使用或并用。优选可以举出非离子系表面活性剂与阴离子系表面活性剂的组合,更优选可以举出聚氧乙烯聚氧丙烯嵌段共聚物、与芳香族磺酸与甲醛的缩合物的盐的组合。
作为芳香族磺酸,可以举出例如苯磺酸、甲苯磺酸、枯烯磺酸、萘磺酸等。优选可以举出α-萘磺酸、β-萘磺酸等萘磺酸。
作为用于形成盐的阳离子,可以举出例如钠阳离子、钾阳离子等1价碱金属阳离子,例如铵阳离子等。优选可以举出1价碱金属阳离子。
作为芳香族磺酸与甲醛的缩合物的盐,具体可以举出萘磺酸与甲醛的缩合物的盐(萘磺酸甲醛缩合物钠盐)。作为芳香族磺酸与甲醛的缩合物的盐,可以使用市售品,具体可以举出DEMOL NL(β-萘磺酸甲醛缩合物钠盐、41%水溶液、花王公司制)等。
相对于油相成分100质量份,表面活性剂的配合比例例如为0.0001质量份以上、优选为0.001质量份以上,并且,例如为1.0质量份以下、优选为0.1质量份以下。在表面活性剂为非离子系表面活性剂与阴离子系表面活性剂的组合的情况下,相对于油相成分100质量份,非离子系表面活性剂和阴离子系表面活性剂各自的配合比例例如为0.0001质量份以上、优选为0.001质量份以上,并且,例如为1.0质量份以下、优选为0.1质量份以下。
关于分散剂、或分散剂和表面活性剂,在例如将油相成分配合于水之前或配合后均可以进行配合,优选配合于与油相成分配合之前的水。由此,调制分散剂的水溶液、或分散剂和表面活性剂的水溶液。
在上述的油相成分的水分散(悬浮)中,可以使用例如均相混合机(homomixer)、超声波均质器、加压式均质器、乳化分散机(Milder)、多孔膜压入分散机等分散机,优选使用均相混合机。
水分散的条件可以适当地设定,在使用均相混合机的情况下,将其转数设定为例如100rpm以上、优选为1000rpm以上,并且,设定为例如10000rpm以下,例如8000rpm以下。
由此,调制水相中分散有油相成分的水分散液。
此外,在水分散液中配合有分散剂(第2分散剂)、或分散剂和表面活性剂的情况下,水分散液中的油相成分的液滴因分散剂、或分散剂和表面活性剂而更加稳定化。
将水(或水溶液)的配合比例调节成相对于油相成分100质量份例如为50质量份以上、优选为100质量份以上、更优选为150质量份以上,并且,例如为1900质量份以下、优选为900质量份以下、更优选为400质量份以下。
(聚合工序)
在聚合工序中,对聚合性乙烯基单体进行悬浮聚合,并且对疏水性壳层形成成分和亲水性壳层形成成分进行界面聚合,形成被覆悬浮聚合物的壳层。即,将壳层形成为被覆通过悬浮聚合得到的聚合物、即悬浮聚合物。
<悬浮聚合>
在聚合工序中,对聚合性乙烯基单体进行悬浮聚合,生成聚合物。为了对聚合性乙烯基单体进行悬浮聚合,将水分散液升温至预定温度。在悬浮聚合中,一边将水分散液搅拌以维持水分散液的水分散状态,一边使聚合性乙烯基单体进行反应(具体地为自由基聚合),生成聚合性乙烯基单体的聚合物。就悬浮聚合而言,由于成为聚合物的聚合性乙烯基单体全部仅存在于水分散粒子(疏水性液相),因此是原位(in-situ)聚合。
具体地说,悬浮聚合中,将水分散液边搅拌边加热,从而使聚合性乙烯基单体直接在水分散粒子中开始聚合,生成聚合物。
搅拌可以利用例如具有搅拌叶片的搅拌器来实施。关于搅拌速度,搅拌叶片的圆周速度例如为10m/分钟以上、优选为20m/分钟以上,并且400m/分钟以下、优选为200m/分钟以下。
将水分散液加热以使温度达到例如为40℃以上、优选为50℃以上、更优选为60℃以上,并且,例如为100℃以下、优选为90℃以下、更优选为80℃以下。
并且,抗生物活性化合物以与聚合物不相溶的状态进行悬浮聚合。
加热时间例如为2小时以上、优选为3小时以上,并且,例如为12小时以下、优选为8小时以下。进而,也可以在加热至预定温度后,将其温度维持预定时间,然后反复进行加热以及温度维持,从而阶段性地进行加热。
在悬浮聚合中,抗生物活性化合物相对于聚合性乙烯基单体实质上为不溶性,抗生物活性化合物从聚合开始至聚合结束后,相对于聚合性乙烯基单体和/或聚合物维持不相溶状态。
通过悬浮聚合,以悬浮聚合物形态生成由聚合性乙烯基单体调制的聚合物。
<界面聚合>
为了与上述悬浮聚合一起实施界面聚合,使例如亲水性壳层形成成分包含于含有疏水性壳层形成成分的水分散液,同时使水分散液升温。具体地说,将亲水性壳层形成成分配合于含有疏水性壳层形成成分的水分散液,同时使水分散液升温至开始悬浮聚合的温度(具体地为聚合引发剂的分解温度以上的温度)。
开始界面聚合的温度(开始温度)Tip没有特别限定,例如为0℃以上、优选为10℃以上,并且,例如为100℃以下、优选为80℃以下。另外,在加热至温度例如为25℃以上、优选为40℃以上,并且,例如为100℃以下、优选为80℃以下时,促进反应。
此外,开始悬浮聚合的温度(开始温度)Ti例如与上述聚合引发剂的10小时半衰期温度T1/2存在下述式(1)的关系。
T1/2-10≤Ti≤T1/2+10 (1)
(式中,Ti表示开始悬浮聚合的温度,T1/2表示聚合引发剂的10小时半衰期温度。)
具体地说,开始悬浮聚合的温度例如为55℃以上、优选为60℃以上,并且,例如为100℃以下、优选为80℃以下。
因此,将悬浮聚合的开始温度Ti设定为例如比界面聚合的开始温度Tip高。具体地说,将悬浮聚合的开始温度Ti设定为比界面聚合的开始温度Tip例如高5℃以上、优选高10℃以上、更优选高20℃以上,并且,设定为例如高100℃以下。
<界面聚合的时机>
作为开始界面聚合和悬浮聚合的方法,可以举出例如(1)在开始悬浮聚合的同时开始界面聚合的方法、(2)在开始悬浮聚合之前开始界面聚合的方法、(3)在开始悬浮聚合之后开始界面聚合的方法。
(1)在开始悬浮聚合的同时开始界面聚合的方法中,例如,使包含含有疏水性壳层形成成分的油相成分的水分散液升温至开始悬浮聚合的温度以上,此时,在水分散液的温度达到开始悬浮聚合的温度时,将亲水性壳层形成成分配合于水分散液。
(2)在开始悬浮聚合之前开始界面聚合的方法中,在升温至开始悬浮聚合的温度之前,将亲水性壳层形成成分配合于包含含有疏水性壳层形成成分的油相成分的水分散液。即,首先,将亲水性壳层形成成分配合于例如室温(20~30℃)的水分散液,然后,将常温的水分散液升温至开始悬浮聚合的温度。
另外,也可以在将亲水性壳层形成成分配合于包含含有疏水性壳层形成成分的油相成分的水分散液后,立即将水分散液升温至小于开始悬浮聚合的温度的温度,然后,将水分散液升温至开始悬浮聚合的温度。在将水分散液升温至小于开始悬浮聚合的温度的温度的情况下,将水分散液加热至其温度例如小于55℃、优选小于50℃。由此,可以在开始悬浮聚合之前,充分促进界面聚合。
(3)在开始悬浮聚合之后开始界面聚合的方法中,首先,将水分散液升温至开始悬浮聚合的温度以上,然后,将亲水性壳层形成成分配合于水分散液。具体地说,从将水分散液升温至开始悬浮聚合的温度以上到将亲水性壳层形成成分配合于水分散液为止的时间例如为0.5小时以上、优选为1小时以上,并且,例如为8小时以下、优选为5小时以下。
与(3)的方法相比,(1)或(2)的方法能够抑制抗生物活性化合物从基质(后述)脱落,因此,可以在形成壳层的同时,维持抗生物活性化合物分散在基质中的状态。即,就缓释性粒子而言,壳层可以将抗生物活性化合物可靠地内包于基质内。因此,能够提高缓释性粒子中的抗生物活性化合物的耐碱性。
(2)在开始悬浮聚合之前开始界面聚合的方法中,可以将壳层形成为被覆油相成分的液滴,因此,能够控制在悬浮聚合中内包的抗生物活性化合物从悬浮聚合物向水相界面(即,悬浮聚合物与水连续相的界面)移动。
在疏水性壳层形成成分为聚异氰酸酯的情况下,亲水性壳层形成成分的配合比例是疏水性壳层形成成分的异氰酸酯基相对于亲水性壳层形成成分的活性烃基(在亲水性壳层形成成分为聚胺的情况下,为氨基)的当量比(异氰酸酯基/氨基)达到例如为0.4以上、优选为0.6以上的比例,并且是达到例如为1.2以下、优选为1.0以下的比例。
另外,在上文中,虽然将亲水性壳层形成成分配合于含有疏水性壳层形成成分的水分散液,但例如在亲水性壳层形成成分为水的情况下,也可以不另行将亲水性壳层形成成分配于水分散液,而利用水分散液中所含有的水作为亲水性壳层形成成分,对该亲水性壳层形成成分与疏水性壳层形成成分进行界面聚合。在亲水性壳层形成成分为水的情况下,可以使用二月桂酸二丁基锡等加聚催化剂。
在界面聚合中,油相成分(油相)中的疏水性壳层形成成分与水相中的亲水性壳层形成成分在水分散粒子的表面进行界面聚合。
界面聚合的聚合时间取决于悬浮聚合的温度,可以通过聚合反应液的pH的降低(达到中和点)来确认。如果聚合温度为60~70℃,则结束界面聚合的时间例如为2小时~4小时。
通过开始界面聚合,优选可以在开始悬浮聚合之前或同时,形成被覆油相成分液滴的壳层。其结果是,能够控制在悬浮聚合中内包的抗生物活性化合物从悬浮聚合物向水相界面(悬浮聚合物与水连续相的界面)移动。
进而,在通过疏水性壳层形成成分和亲水性壳层形成成分的界面聚合得到的悬浮聚合物的表面,形成有包含疏水性壳层形成成分和亲水性壳层形成成分的聚合物的壳层。因此,缓释性粒子中的抗生物活性化合物的缓释速度变慢,能够长期持续缓释性。
在界面聚合和悬浮聚合后,通过例如放冷等将反应后的水分散液冷却,用100目(网眼)的滤布等进行过滤,从而得到缓释性粒子的水分散液(悬浮液)。
冷却温度例如为室温(20~30℃,更具体地为25℃)。
所得到的缓释性粒子中的抗生物活性化合物的浓度例如为1质量%以上、优选为5质量%以上、更优选为10质量%以上,并且,例如为50质量%以下、优选为40质量%以下、更优选为35质量%以下。
此外,缓释性粒子在水分散液(悬浮液)中的含有比例根据油相成分以及对其进行分散的水(或水溶液)的配合量而决定,具体地说,例如为10质量%以上、优选为20质量%以上,并且,例如为50质量%以下、优选为40质量%以下。
壳层在缓释性粒子中的浓度例如为1质量%以上、优选为2质量%以上,并且,例如为50质量%以下、优选为40质量%以下。
缓释性粒子的平均粒径例如为1μm以上、优选为2μm以上,并且,例如为20mm以下、优选为10mm以下。此外,平均粒径作为中位径进行计算。
<第2发明组的缓释性粒子的效果>
第2发明组的缓释性粒子通过如下制造方法获得,因此能够得到缓释性和耐碱性优异且坚固的缓释性粒子,该制造方法具备:油相成分调制工序,在不存在溶剂的情况下,调制含有将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体而成的疏水性浆料、以及疏水性壳层形成成分的油相成分;水分散工序,对油相成分进行水分散来调制水分散液;以及聚合工序,对疏水性壳层形成成分与亲水性壳层形成成分进行界面聚合而形成成为壳层的聚合物,对聚合性乙烯基单体进行悬浮聚合,生成成为芯层的聚合物。
然而,利用如专利文献1所记载的方法得到的微胶囊由于仅通过界面聚合而得到,因此在微胶囊中残存分散介质(溶剂),由此有时其表面硬度不充分。其结果是,在微胶囊的分散液经历施加高剪切力的工序的情况、或长期保存的情况下,有时微胶囊发生凝聚而再分散变得困难。
进而,由于微胶囊的表面硬度不充分,因此微胶囊容易粘连,有时难以以干燥粒子形态取出微胶囊。
另一方面,第2发明组的缓释性粒子通过如下制造方法获得,因此能够防止界面聚合中溶剂的存在所引起的缓释性粒子的表面硬度的降低,得到坚固的缓释性粒子,进而,所得到的缓释性粒子的再分散性和抗粘连性优异,该制造方法具备:油相成分调制工序,在不存在溶剂的情况下,调制含有将疏水性且相对于疏水性的聚合性乙烯基单体实质上为不溶性的抗生物活性化合物分散于疏水性的聚合性乙烯基单体而成的疏水性浆料、以及疏水性壳层形成成分的油相成分;水分散工序,对油相成分进行水分散来调制水分散液;以及聚合工序,对疏水性壳层形成成分与亲水性壳层形成成分进行界面聚合而形成成为壳层的聚合物,对聚合性乙烯基单体进行悬浮聚合,生成成为芯层的聚合物。
根据该缓释性粒子的制造方法,能够得到坚固并且再分散性和抗粘连性优异的缓释性粒子。
并且,根据该缓释性粒子的制造方法,形成被覆悬浮聚合而成的悬浮聚合物的壳层,因此能够提高抗生物活性化合物的内包率(抗生物活性化合物在缓释性粒子中的浓度),并且抗生物活性化合物的缓释性和耐碱性优异。另外,缓释性粒子的缓释性与缓释性粒子中的抗生物活性化合物的耐碱性相互关联,具体地说,如果缓释性粒子中的抗生物活性化合物的耐碱性提高,则缓释性粒子的缓释性会提高。
特别是,在疏水性壳层形成成分含有聚异氰酸酯、亲水性壳层形成成分含有聚胺的情况下,由于壳层含有聚脲,因此形成与热塑性氨基甲酸酯树脂的熔融混合性优异的缓释性粒子。
这样的缓释性粒子能够应用于各种工业制品,例如可以添加于屋内外的涂料、橡胶、纤维、树脂(包括塑料)、粘接剂、接缝剂、密封剂、建筑材料、填缝剂、木材处理剂、土壤处理剂、造纸工序中的白水、颜料、印刷版用处理液、冷却用水、油墨、切削油、化妆用品、非织造布、纺丝油、皮革等。另外,缓释性粒子中的抗生物活性化合物相对于这些工业制品的添加量例如为10mg/kg~100g/kg(制品质量)。
接着,对将由缓释性粒子制成的粉剂与热塑性树脂配合的方式进行说明。
在该方法中,首先,使缓释性粒子的悬浮液干燥,制成粉剂。
接着,将粉剂和热塑性树脂进行熔融混炼,调制混炼物。
为了调制混炼物,例如具体可以使用挤出机、班伯里混合机。作为挤出机,可以使用例如双轴挤出机、单轴挤出机。混炼物是用于成型得到成型品的成型材料,具体地说,暂时进行冷却而调制成颗粒状成型材料(混炼物颗粒或母料)。另一方面,也可以将混炼物不以固体的成型材料的形态取出,而直接连续地以熔融状态(熔融混炼物)供于后述的成型。
将粉剂配合于热塑性树脂,以使粉剂的抗生物活性化合物的含有比例相对于热塑性树脂为例如0.01质量%以上、优选为0.1质量%以上,并且,例如为10质量%以下、优选为3质量%以下。但是,在调制混炼物作为母料的情况下,不限于此,具体地说,将粉剂配合于热塑性树脂,以使抗生物活性化合物的含有比例相对于热塑性树脂为例如1质量%以上、优选为5质量%以上,并且,例如为50质量%以下、优选为30质量%以下,从而作为母料。
热塑性树脂没有特别限定,可以举出例如聚乙烯、聚丙烯等聚烯烃系树脂;聚苯乙烯或者聚甲基丙烯酸甲酯、丙烯腈-苯乙烯共聚树脂(AS树脂)、甲基丙烯酸甲酯-苯乙烯共聚物(MS树脂)、丙烯腈-苯乙烯-丁二烯共聚树脂(ABS树脂)等苯乙烯系、和/或丙烯酸系树脂;聚对苯二甲酸乙二醇酯、聚乳酸等聚酯系树脂;6-尼龙等聚酰胺系树脂;氯乙烯树脂、偏氯乙烯树脂等乙烯基卤系树脂;聚碳酸酯、聚苯醚、聚甲醛、热塑性聚氨酯等。优选可以举出聚烯烃系树脂、氯乙烯树脂、热塑性聚氨酯。
接着,由混炼物颗粒或熔融混炼物成型为成型品。
作为成型方法,可以采用例如注射成型、挤出成型、吹胀成型(inflation molding)、拉挤成型(Pultrusion molding)、压缩成型等。
由此,可以得到成型为预定形状的添加了粉剂(缓释性粒子)的成型品。
上述说明中,虽然将由缓释性粒子制成的粉剂添加于热塑性树脂,但只要是树脂就没有特别限定,也可以添加于例如热固化性树脂。
可以将粉剂适合地混合于特别是环氧树脂、硅树脂等液状树脂。
这样的成型品用于各种用途,可以用作例如建筑材料,例如电线电缆材料及其电线电缆的被覆材料,例如气体等的导管及其导管的被覆材料,例如衣类、蚊帐等纤维制品。
<第2发明组的成型品的效果>
并且,由于这样的成型品由含有上述缓释性粒子的成型材料成型,因此具有优异的抗生物活性化合物的缓释性和耐碱性。
并且,通过上述第2发明组的缓释性粒子的制造方法得到的缓释性粒子具体包括下面描述的缓释性粒子的第3实施方式以及第4实施方式。
[第3实施方式]
参照图B1对缓释性粒子的第3实施方式进行说明。
如图B1的截面图所示,该缓释性粒子1形成为例如球状粒子。缓释性粒子1包含基质2、分散于基质2中的结构域3和被覆基质2的壳层7。
基质2包含由上述聚合性乙烯基单体调制的聚合物。结构域3包含上述抗生物活性化合物。壳层7包含由上述疏水性壳层形成成分和亲水性壳层形成成分调制的聚合物。
具体地说,在缓释性粒子1中,基质2形成介质或连续相,形成多个结构域3在基质2中以孤立状分散的多结构域结构或海岛结构(或多核结构)。此外,在该缓释性粒子1中,基质2和结构域3彼此不相溶,形成彼此分离的相分离结构或2相结构。此外,基质2和结构域3形成相对于后述壳层7的芯层。
具体地说,多个结构域3在基质2中形成分散相。结构域3的形状没有特别限定,可以形成为例如不定形状、球状、块状、板状等适宜形状。结构域3的最大长度的平均值例如为0.05μm以上、优选为0.1μm以上,并且,例如为20μm以下、优选为10μm以下。
壳层7形成在基质2(通过对上述聚合性乙烯基单体进行悬浮聚合而得到的聚合物)的表面。具体地说,壳层7被覆着例如基质2表面的至少一部分,优选为基质2表面的全部。即,壳层7与由基质2和结构域3构成的芯层一起形成芯壳结构。
另外,在图B1中,在基质2与壳层7之间,清晰地形成有截面为圆形的界面,如图B8的TEM照片所示,也可以不清晰地形成基质2与壳层7的界面。如图B8的TEM照片所示,壳层7由以疏水性壳层形成成分和亲水性壳层形成成分调制的聚合物构成,详细而言,最外层(最表面)实质上仅由界面聚合的聚合物构成,并且构成为,随着从最外层(最表面)朝向内侧,由疏水性壳层形成成分和亲水性壳层形成成分调制的聚合物相对于基质2(聚合物)的浓度变淡。由此,壳层7以包围结构域3的方式位(偏在)于基质2的表层。
并且,为了得到该缓释性粒子1,在上述缓释性粒子1的制造方法的油相成分调制工序中,配合抗生物活性化合物,以使缓释性粒子中的抗生物活性化合物的浓度达到例如30质量%以下。
<第3实施方式的效果>
第3实施方式的缓释性粒子1包括包含聚合性乙烯基单体的聚合物的基质2,以及结构域3,该结构域3是包含抗生物活性化合物的结构域3,且分散于基质2中,因此抗生物活性化合物的缓释性优异,并且坚固性优异,由此与树脂的混炼性优异。
举出作为第2发明组的参考的参考方式,进一步详细说明第3实施方式的效果。
在参考方式中,如图B9所示,结构域3包含从基质2的内部向外侧突出的突出物4。突出物4从基质2的表面露出。由此,在缓释性粒子1的表面,露出基质2和结构域3这两者。突出物4具有埋设于基质2的表层部的埋设部8。此外,缓释性粒子1具有由基质2和结构域3形成的2相结构,而不具有壳层7。
除了不配合疏水性壳层形成成分和亲水性壳层形成成分,并且不实施界面聚合以外,利用上述制造方法,制造图B9所示的缓释性粒子1。
与图B9的参考方式的缓释性粒子1不同,上述图B1所示的第3实施方式的缓释性粒子1没有突出物4,悬浮聚合物被壳层7所被覆,因此长期缓释持续性优异。具体地说,根据第3实施方式,能够如图B1所示那样借助于壳层7保护缓释性粒子1的结构域3(抗生物活性化合物)。因此,第3实施方式的缓释性粒子1与参考方式的缓释性粒子1相比,抗生物活性化合物的缓释性和耐碱性优异。
(第4实施方式)
参照图B2对缓释性粒子的第4实施方式进行说明。
如图B2的截面图所示,在第4实施方式中,包含抗生物活性化合物的附着物5附着于壳层7的表面。
附着物5的形状没有特别限定,形成为例如不定形状、球状、块状、板状等适宜形状。尤其,附着物5的内面(与壳层7的表面接触的接触面)形成有与壳层7的表面(球面)对应的凹面,具体地说,形成有向外侧凹陷的弯曲面。附着物5的大小与结构域3相同或小于结构域3,相对于结构域3的最大长度的平均值,例如为100%以下、优选为50%以下,并且,例如为0.01%以上,具体地说,附着物5的最大长度的平均值例如为10μm以下、优选为5μm以下,例如为0.05μm以上、优选为0.1μm以上。附着物5相对于壳层7的整个表面的被覆率例如为10%以上、优选为20%以上,并且,例如为100%以下、优选为90%以下。
为了得到上述缓释性粒子1,在上述缓释性粒子1的制造方法的油相成分调制工序中,配合抗生物活性化合物,以使缓释性粒子中的抗生物活性化合物的浓度例如超过28质量%,优选为30质量%以上、更优选为35质量%。
<第4实施方式的效果>
根据第4实施方式的缓释性粒子,能够借助于附着物5进一步提高抗粘连性。
此外,第2发明组可以混合包含第3实施方式的缓释性粒子和第4实施方式的缓释性粒子这二者,该情况下,以质量基准计,它们的配合比例(第3实施方式的缓释性粒子/第4实施方式的缓释性粒子)例如为1/99以上、进一步为10/90以上,并且,例如为99/1以下、进一步为90/10以下。
实施例
[1]对应于第1发明组的实施例A等
以下所示的调制例A以及实施例A的数值可以替代为上述“具体实施方式”栏中所记载的数值(即,上限值或下限值)。此外,在调制例A、实施例A以及比较例A中,关于%等单位,只要没有特殊记载,就是指质量%。
首先,将各调制例A、各实施例A以及各比较例A中使用的简称的详细内容记载于下文中。
噻虫胺:(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、分子量250、熔点177℃、在水中的溶解度:0.33g/L、住友化学公司制
吡虫啉:1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺、分子量256、熔点144℃、在水中的溶解度:0.48g/L、丸善公司制
EGDMA:乙二醇二甲基丙烯酸酯、商品名“LIGHT ESTER EG”、不溶于水、共荣社化学公司制
i-BMA:甲基丙烯酸异丁酯、在水中的溶解度:0.6g/L、日本触媒公司制
DVB-570:商品名、不溶于水、组成:二乙烯基苯(上限60%)、乙基乙烯基苯(上限40%)、新日铁住金化学公司制
苯乙烯:在水中的溶解度:0.3g/L、和光特级试剂、和光纯药公司制
DISPERBYK-164:商品名、颜料分散用官能团改性共聚物(含有叔胺的聚酯改性聚氨酯系聚合物、分子量10000~50000)的乙酸丁酯溶液、固体成分浓度60%、毕克化学公司制
PEROYL L:商品名、过氧化二月桂酰、日油公司制
PERHEXYL O:商品名、过氧化-2-乙基己酸叔己酯、日油公司制
PRONON 208:商品名、聚氧乙烯聚氧丙烯嵌段共聚物、日油公司制
PVA-217:商品名“KURARAY POVAL 217”、局部皂化聚乙烯醇、KURARAY公司制
DEMOL NL:商品名、β-萘磺酸甲醛缩合物钠盐的41%水溶液、花王公司制
(疏水性浆料的调制)
调制例A1(噻虫胺浆料(浆料A)的调制)
将90g的EGDMA、90g的i-BMA、20g的DISPERBYK-164和100g的噻虫胺投入玻璃瓶中,进而投入玻璃瓶的1/3容量的直径1.0mm的氧化锆珠,用涂料调节器(涂料搅拌器、商品名“THECLASSIC型号1400”、Red Devil公司制)进行2小时的湿式粉碎,得到含有噻虫胺33.3%的浆料(疏水性浆料,以下称为“浆料A”)。
用高浓度粒径分析仪FPAR-1000(大塚电子公司制)测定的结果,浆料A中的噻虫胺的平均粒径为1.38μm。
调制例A2(噻虫胺浆料(浆料B)的调制)
利用分批式介质型分散机(分批式珠磨机、商品名“AD MILL(AD-5)、氧化锆珠径1.5mm”、浅田铁工公司制),将7200g的DVB-570、804g的DISPERBYK-164搅拌分散至均匀后,进而投入噻虫胺3996g,进行150分钟的湿式粉碎,得到含有噻虫胺33.3%的浆料(疏水性浆料,以下称为“浆料B”。)。
用高浓度粒径分析仪FPAR-1000(大塚电子公司制)测定的结果,浆料B中的噻虫胺的平均粒径为0.45μm。
调制例A3~调制例A8
(噻虫胺浆料(浆料C~H)的调制)
将配合处方变更为表A1中所记载的处方,除此以外,与调制例A1同样地处理,得到噻虫胺浆料(疏水性浆料,以下称为“浆料C~H”。)。
将各浆料C~H中的噻虫胺的平均粒径记载于表A1。
调制例A9
(吡虫啉浆料(浆料I)的调制)
将配合处方变更为表A1中所记载的处方,除此以外,与调制例A1同样地处理,得到吡虫啉浆料(疏水性浆料,以下称为“浆料I”。)。
将浆料I中的吡虫啉的平均粒径记载于表A1。
[表A1]
(水分散工序以及聚合工序)
实施例A1(含有噻虫胺的缓释性粒子的合成:对应于第1实施方式)
在200mL的烧杯(1)中,放入100g调制例A2中调制的浆料B、以及0.5g的PEROYL L,在室温下搅拌,从而使PEROYL L溶解于浆料B。由此,调制包含PEROYL L和浆料B的油相成分。
另外,在500mL的烧杯(2)中,放入离子交换水258.50g、PVA-217的10%水溶液40g以及PRONON 208的1%水溶液1g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数6000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,将悬浮液(水分散液)转移至装备了搅拌器、回流冷却器、温度计及氮气导入管的500mL四口烧瓶中,在氮气气流下,边搅拌边升温,实施悬浮聚合。
关于悬浮聚合,将达到55℃的时刻设为聚合开始,然后,连续地在70±1℃实施5小时、在80±1℃实施2小时。
然后,将反应后的悬浮液冷却至30℃以下,从而得到含有噻虫胺的中位径28.2μm的缓释性粒子的悬浮液(悬浮剂)。
需要说明的是,利用激光衍射散射式粒径分布测定装置LA-920(堀场制作所公司制)来测定缓释性粒子的中位径。关于中位径的测定,以下的实施例以及比较例中也同样。
实施例A2(含有噻虫胺的缓释性粒子的合成:对应于第2实施方式)
在200mL的烧杯(1)中,投入100g调制例A2中调制的浆料B、以及0.5g的PEROYL L,在室温下搅拌,从而使PEROYL L溶解于浆料B。由此,调制包含PEROYL L和浆料B的油相成分。
另外,在500mL的烧杯(2)中,放入离子交换水258.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数6000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,在与实施例A1同样的条件下实施悬浮聚合,得到含有噻虫胺的中位径24.5μm的缓释性粒子的悬浮液(悬浮剂)。
实施例A3(含有噻虫胺的缓释性粒子的合成:对应于第2实施方式)
在200mL的烧杯(1)中,投入100g调制例A1中调制的浆料A,以及0.5g的PEROYL L,在室温下搅拌,从而使PEROYL L溶解于浆料A。由此调制包含PEROYL L和浆料A的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水258.50g、PVA-217的10%水溶液40g以及PRONON 208的1%水溶液1g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数3000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,在与实施例A1同样的条件下实施悬浮聚合,得到含有噻虫胺的中位径43.5μm的缓释性粒子的悬浮液(悬浮剂)。
实施例A4(含有噻虫胺的缓释性粒子的合成:对应于第2实施方式)
在200mL的烧杯(1)中,投入50g调制例A1中调制的浆料A、25g的i-BMA、25g的EGDMA以及0.5g的PEROYL L,在室温下搅拌,从而使i-BMA、EGDMA以及PEROYL L溶解于浆料A。由此,调制包含i-BMA、EGDMA、PEROYL L以及浆料A的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水258.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数5000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,在与实施例A1同样的条件下实施悬浮聚合,得到含有噻虫胺的中位径9.3μm的缓释性粒子的悬浮液(悬浮剂)。
实施例A5~实施例A8、实施例A13、实施例A15、实施例A16(含有噻虫胺的缓释性粒子的合成:对应于第2实施方式)
根据表A2以及表A3的记载变更配合处方,除此以外,与实施例A4同样地处理,得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。将悬浮液中的缓释性粒子各自的平均粒径示于表A2及表A3。
实施例A9(含有噻虫胺的缓释性粒子的合成:对应于第2实施方式)
在200mL的烧杯(1)中,投入50g调制例A1中调制的浆料C、25g苯乙烯、25g的EGDMA、以及0.5g的PEROYL L,在室温下搅拌,从而使苯乙烯、EDGMA以及PEROYL L溶解于浆料C。由此,调制包含苯乙烯、EDGMA、PEROYL L以及浆料C的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水258.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数5000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,在与实施例A1同样的条件下实施悬浮聚合,得到含有噻虫胺的中位径14.5μm的缓释性粒子的悬浮液(悬浮剂)。
实施例A10~实施例A12、实施例A14、实施例A17、实施例A18(含有噻虫胺的缓释性粒子的合成:对应于第2实施方式)
根据表A3的记载变更配合处方,除此以外,与实施例A9同样地处理,得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。将悬浮液中的缓释性粒子各自的平均粒径示于表A3。
实施例A19(含有吡虫啉的缓释性粒子的合成:对应于第2实施方式)
根据表A3的记载变更配合处方,除此以外,与实施例A4同样地处理,得到含有吡虫啉的缓释性粒子的悬浮液(悬浮剂)。将悬浮液中的缓释性粒子的平均粒径示于表A3。
[表A2]
[表A3]
在表A2和表A3中,聚合条件栏中的“1”是指将聚合工序中的悬浮液的温度调整为“在70±1℃5小时、在80±1℃2小时”,“2”是指将聚合工序中的悬浮液的温度调整为“在80±1℃3小时、在85±1℃3小时”。
此外,缓释性粒子的方式栏中的“1”表示具有图A1所示的第1实施方式的结构,“2”表示具有图A2所示的第2实施方式的结构。
(缓释性粒子的粉剂与热塑性树脂的混炼以及成型)
实施例A20(实施例A1的粉剂与聚乙烯的混炼以及成型)
利用100目滤布,将实施例A1中制作的缓释性粒子的悬浮液过滤后,在室温下干燥1天,得到缓释性粒子的粉末(粉剂)。将所得到的缓释性粒子的粉末(粉剂)与高密度聚乙烯(HDPE)HI-ZEX 6300M(Prime Polymer公司制、熔体流动速率0.11g/10分钟)进行干式混合,以使噻虫胺相对于HDPE为0.25%,投入双轴挤出-注射成型并设机DSMX plore MC15M(DSM公司制),进行220℃×5分钟的熔融混炼而得到料股,接着,以熔融状态直接通过注射成型而得到长条型成型品(10mm×76mm×4mm)。
实施例A21(实施例A3的粉剂与聚乙烯的混炼以及成型)
使用实施例A3中制作的缓释性粒子的悬浮液,来代替实施例A1中制作的缓释性粒子的悬浮液,除此以外,与实施例A20同样地处理,得到长条型成型品。
(缓释性粒子的粒剂的制剂化)
实施例A22
相对于卡卡(Kagalite)2号(卡卡工业公司制、浮石的细粒、粒径425~1400μm)100质量份,配合实施例A1中制作的缓释性粒子的悬浮液(噻虫胺浓度8.3质量%)1.2质量份,接着,将它们干燥,得到噻虫胺的粒剂。粒剂中的噻虫胺浓度为约0.1质量%。
实施例A23
配合实施例A3中制作的缓释性粒子的悬浮液(噻虫胺浓度8.3质量%)1.2质量份,来代替实施例A1中制作的缓释性粒子的悬浮液,除此以外,与实施例A22同样地处理,得到噻虫胺的粒剂。粒剂中的噻虫胺浓度为约0.1质量%。
比较例A1(噻虫胺微胶囊悬浮液的干燥品与聚乙烯的混炼)
使用将通过界面聚合制造的日本环境化学公司制噻虫胺微胶囊悬浮液“Xylamon MC”在室温下干燥1天并破碎而成的样品,来代替由实施例A1调制的缓释性粒子的粉末(粉剂),除此以外,试图与实施例A6同样地处理,但在熔融混炼中胶囊被破坏而溶剂雾化,无法进行混炼。
(评价)
1.SEM(扫描式电子显微镜、Scanning Electron Microscope)观察
将实施例A1~实施例A4、实施例A9以及实施例A19各自的悬浮液(悬浮剂)滴加于试样台,然后将水蒸馏除去,然后对所得到的缓释性粒子利用扫描式电子显微镜日立TM-3000(日立高新技术公司制)进行SEM观察。将实施例A1~实施例A4、实施例A9以及实施例A19各自所得到的缓释性粒子的SEM图像分别示于图A4~图A9。
此外,将实施例A20以及实施例A21的料股浸渍于液态氮,利用扫描式电子显微镜日立TM-3000(日立高新技术公司制)对脆性断裂的断裂面进行SEM观察。将实施例A20以及实施例A21的截面SEM图像分别示于图A10以及图A11。
2.TEM(透射式电子显微镜、Transmission Electron Microscope)观察
将实施例A1~实施例A3的悬浮液(悬浮剂)冷冻干燥,分散于双酚型液状环氧树脂,用胺进行固化。利用超薄切片机将其切断从而使截面露出,用四氧化锇进行染色,根据需要进一步用四氧化钌进行染色,利用超薄切片机将其切成超薄切片而调制样品。利用透射式电子显微镜(型号“H-7100”、日立制作所社制),对调制的样品进行TEM观察。
将实施例A1~实施例A3的TEM照片的图像处理图分别示于图A12~图A14。
需要说明的是,在图A12~图A14中,符号3所示的空白是在使切出的超薄切片浮于水而回收的过程中,噻虫胺溶解脱落的痕迹,表示噻虫胺的结构域的形状。
3.耐碱性试验
3-1.缓释性粒子的悬浮剂
按照下面的顺序实施缓释性粒子的悬浮剂的耐碱性试验。
利用100目滤布,将实施例A1~实施例A3中制作的缓释性粒子的悬浮液过滤后,在室温下干燥1天,得到缓释性粒子的粉末(粉剂)。用去离子水将这些粉末稀释成1000倍,量取其中的6.3mL至玻璃瓶,添加饱和氢氧化钙溶液2mL而制成试验溶液。在40℃的恒温条件下静置该试验溶液。
在开始试验1天后及7天后,向试验溶液添加乙腈10mL,提取噻虫胺,利用HPLC对噻虫胺的量进行定量,计算出残存率。
作为对照,使用噻虫胺原药的水溶液,同样地实施试验。
将结果示于表A4。
[表A4]
由表A4可知,就含有实施例A1~实施例A3的缓释性粒子的悬浮剂而言,在开始试验后第1天,噻虫胺的残存率高,为91~93%,在开始试验后第7天,均为12~16%,虽然与开始试验后第1天相比降低,但如果考虑对照为7%,则可知仍然达到实用化水平。进而还可知,与相当于第1实施方式的实施例A1的缓释性粒子相比,相当于第2实施方式的实施例A2以及实施例A3的缓释性粒子在试验开始后第1天及第7天耐碱性均优异。
3-2.缓释性粒子的粒剂
量取1.0g的实施例A22以及实施例A23中得到的粒剂,添加去离子水3.6mL和饱和氢氧化钙水溶液2mL,调制试验溶液。在40℃恒温的条件下静置该试验溶液。
在开始试验1天后及7天后,向试验溶液添加乙腈10mL,提取噻虫胺,用HPLC对噻虫胺的量进行定量,计算出残存率。
作为对照,使用噻虫胺原药的水溶液,同样地实施试验。
将结果示于表A5。
[表A5]
由表A5可知,就含有实施例A1以及实施例A3的缓释性粒子的实施例A22以及实施例A23的粒剂而言,在试验开始后第1天,噻虫胺的残存率高,为90~92%,在开始试验后第7天,均为11~15%,虽然与开始试验后第1天相比降低,但如果考虑对照为7%,则可知仍然达到实用化水平。进而还可知,与含有相当于第1实施方式的实施例A1的缓释性粒子的实施例A22的粒剂相比,含有相当于第2实施方式的实施例A3的缓释性粒子的实施例A23的粒剂在试验开始后第1天以及第7天抗生物活性化合物的耐碱性均优异。
4.成型品的防蚁试验
将以含水率为8%(白蚁活动的最适含水率)的方式注水的硅砂填充于塑料容器,接着,在硅砂的表面设置实施例A20以及实施例A21的长条型成型品。
作为比较对照,实施了设置未混炼缓释性粒子的、仅包含HDPE的长条型成型品的试验。
在上述塑料容器内投入家白蚁工蚁50只,对白蚁的死亡只数(=死亡率)以及行动观察了7天(以n=2实施试验)。关于实施例A20以及实施例A21的长条型成型品,在试验开始第2天以及第3天白蚁全部死亡。
另一方面,关于作为比较对照的仅包含HDPE的长条型成型品,在7天后白蚁也没有死亡,并且没有发现白蚁的行动有变化。
即,对于实施例A20以及实施例A21,证实了显著的灭蚁效果。
[2]对应于第2发明组的实施例B等
以下所示的调制例B、实施例B以及参考例B的数值可以替代为上述“具体实施方式”栏中所记载的数值(即,上限值或下限值)。此外,在调制例B、实施例B以及参考例B中,关于%等单位,只要没有特殊记载,就是指质量%。
首先,将各调制例B、各实施例B、各参考例B以及各比较例B中使用的简称的详细记载于下文中。
噻虫胺:(E)-1-(2-氯噻唑-5-基甲基)-3-甲基-2-硝基胍、分子量250、熔点177℃、在水中的溶解度:0.33g/L、住友化学公司制
吡虫啉:1-(6-氯-3-吡啶基甲基)-N-硝基亚咪唑烷-2-基胺、分子量256、熔点144℃、在水中的溶解度:0.48g/L、丸善公司制
EGDMA:乙二醇二甲基丙烯酸酯、商品名“LIGHT ESTER EG”、不溶于水、共荣社化学公司制
i-BMA:甲基丙烯酸异丁酯、在水中的溶解度:0.6g/L、日本触媒公司制
DVB-570:商品名、不溶于水、组成:二乙烯基苯(上限60%)、乙基乙烯基苯(上限40%)、新日铁住金化学公司制
苯乙烯:在水中的溶解度:0.3g/L、和光特级试剂、和光纯药公司制
DISPERBYK-164:商品名、颜料分散用官能团改性共聚物(含有叔胺的聚酯改性聚氨酯系聚合物、分子量10000~50000)的乙酸丁酯溶液、固体成分浓度60%、毕克化学公司制
PEROYL L:商品名、过氧化二月桂酰、10小时半衰期温度T1/2:61.6℃、日油公司制
PRONON 208:商品名、聚氧乙烯聚氧丙烯嵌段共聚物、日油公司制
PVA-217:商品名“KURARAY POVAL 217”、局部皂化聚乙烯醇、KURARAY公司制
DEMOL NL:商品名、β-萘磺酸甲醛缩合物钠盐的41%水溶液、花王公司制
T-1890:商品名“VESTANAT T 1890/100”、异佛尔酮二异氰酸酯(IPDI)的环状三聚物、在水中的溶解度:0.02g/L、赢创工业公司制
DETA:二亚乙基三胺、和光一级试剂、和光纯药工业公司制
(疏水性浆料的调制)
调制例B1(噻虫胺浆料(浆料A)的调制)
将90g的EGDMA、90g的i-BMA、20g的DISPERBYK-164和100g的噻虫胺投入玻璃瓶中,进而投入玻璃瓶的1/3容量的直径1.0mm的氧化锆珠,用涂料调节器(涂料搅拌器、商品名“THECLASSIC型号1400”、Red Devil公司制)进行2小时的湿式粉碎,得到含有噻虫胺33.3%的浆料(疏水性浆料,以下称为“浆料A”。)。
用高浓度粒径分析仪FPAR-1000(大塚电子公司制)测定的结果,浆料A中的噻虫胺的平均粒径为1.38μm。
调制例B2(噻虫胺浆料(浆料B)的调制)
利用分批式介质型分散机(分批式珠磨机、商品名“AD MILL(AD-5)、氧化锆珠径1.5mm”、浅田铁工公司制),将7200g的DVB-570、804g的DISPERBYK-164搅拌分散至均匀后,进而投入噻虫胺3996g,进行150分钟的湿式粉碎,得到含有噻虫胺33.3%的浆料(疏水性浆料,以下称为“浆料B”。)。
用高浓度粒径分析仪FPAR-1000(大塚电子公司制)测定的结果,浆料B中的噻虫胺的平均粒径为0.45μm。
调制例B3~调制例B8
(噻虫胺浆料(浆料C~H)的调制)
将配合处方变更为表B1中所记载的处方,除此以外,与调制例B1同样地处理,得到噻虫胺浆料(疏水性浆料,以下称为“浆料C~H”。)。
将各浆料C~H中的噻虫胺的平均粒径记载于表B1。
调制例B9
(吡虫啉浆料(浆料I)的调制)
将配合处方变更为表B1中所记载的处方,除此以外,与调制例B1同样地处理,得到吡虫啉浆料(疏水性浆料,以下称为“浆料I”。)。
将浆料I中的吡虫啉的平均粒径记载于表B1。
[表B1]
(水分散工序以及聚合工序)
实施例B1(聚脲被覆/含有噻虫胺的缓释性粒子的合成:对应于第3实施方式)
在200mL的烧杯(1)中,向85g调制例B2中调制的浆料B中放入15g的T-1890、以及0.5g的PEROYL L,在室温下搅拌,从而使T-1890以及PEROYL L溶解于浆料B。由此,调制包含T-1890、PEROYL L以及浆料B的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水240.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数6000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,将悬浮液(水分散液)转移至装备了搅拌器、回流冷却器、温度计及氮气导入管的500mL四口烧瓶中,在氮气气流下进行搅拌。
接着,在悬浮液中添加二亚乙基三胺的10质量%水溶液18g,开始界面聚合,然后使悬浮液升温,开始悬浮聚合。
具体地说,首先,在室温的悬浮液中,将二亚乙基三胺的10质量%水溶液18g添加于悬浮液,之后立即将悬浮液的温度升温至70℃,在该温度下维持5小时。然后,将悬浮液升温至80℃,在该温度下维持2小时。
在投入二亚乙基三胺的10质量%水溶液的时刻,开始界面聚合,在达到将悬浮液升温至70℃的中途温度、即55℃的时刻,开始悬浮聚合。
由此,得到噻虫胺被分散于通过悬浮聚合形成的基质中、基质被聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
然后,将反应后的悬浮液冷却至30℃以下,从而得到噻虫胺被分散于基质中、基质被通过界面聚合形成的聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
悬浮液中的缓释性粒子的中位径利用激光衍射散射式粒径分布测定装置LA-920(堀场制作所公司制)来测定。将其结果记载于表B2。需要说明的是,关于中位径的测定,以下的参考例以及比较例中也同样,将它们的结果记载于表B2~表B6。
实施例B2(聚脲被覆/含有噻虫胺的缓释性粒子的合成:对应于第3实施方式)
在200mL的烧杯(1)中,向50g调制例B1中调制的浆料A中投入17.5g的i-BMA、17.5g的EGDMA、15g的T-1890、以及0.5g的PEROYL L,在室温下搅拌,从而使i-BMA、EGDMA、T-1890以及PEROYL L溶解于浆料A。由此,调制含有i-BMA、EGDMA、T-1890、PEROYL L以及浆料A的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水240.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数5000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,将悬浮液(水分散液)转移至装备了搅拌器、回流冷却器、温度计及氮气导入管的500mL四口烧瓶中,在氮气气流下进行搅拌。
接着,在悬浮液中添加二亚乙基三胺的10质量%水溶液18g,开始界面聚合,然后使悬浮液升温,开始悬浮聚合。
具体地说,首先,在室温的悬浮液中,将二亚乙基三胺的10质量%水溶液18g添加于悬浮液,之后立即将悬浮液的温度升温至70℃,在该温度下维持5小时。然后,将悬浮液升温至80℃,在该温度下维持2小时。
在投入二亚乙基三胺的10质量%水溶液的时刻,开始界面聚合,在达到将悬浮液升温至70℃的中途温度、即55℃的时刻,开始悬浮聚合。
由此,得到噻虫胺被分散于通过悬浮聚合形成的基质中、基质被通过界面聚合形成的聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
然后,将反应后的悬浮液冷却至30℃以下,从而得到噻虫胺被分散于基质中、基质被聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
实施例B3(聚脲被覆/含有噻虫胺的缓释性粒子的合成:对应于第3实施方式)
将聚合条件变更为如下所述,除此以外,与实施例B2同样地处理,从而得到噻虫胺被分散于基质中、基质被聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
即,在添加二亚乙基三胺的水溶液之后,立即将悬浮液升温至60℃,在该温度下维持1小时,接着,将悬浮液升温至70℃,在该温度下维持2小时,然后,将悬浮液升温至80℃,在该温度下维持1小时。
在投入二亚乙基三胺的10质量%水溶液的时刻,开始界面聚合,在达到将悬浮液升温至60℃的中途温度、即55℃的时刻,开始悬浮聚合。
实施例B4(聚脲被覆/含有噻虫胺的缓释性粒子的合成:对应于第3实施方式)
将聚合条件变更为如下所述,除此以外,与实施例B2同样地处理,从而得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
即,在添加二亚乙基三胺的水溶液之后,将悬浮液升温至50℃,在该温度下维持2小时,接着,将悬浮液升温至60℃,在该温度下维持1小时,接着,将悬浮液升温至70℃,在该温度下维持2小时,然后,将悬浮液升温至80℃,在该温度下维持1小时。
在投入二亚乙基三胺的10质量%水溶液的时刻,开始界面聚合,在开始界面聚合后的、达到将悬浮液升温至60℃的中途温度、即55℃的时刻,开始悬浮聚合。
实施例B5(聚脲被覆/含有噻虫胺的缓释性粒子的合成:对应于第3实施方式)
将聚合条件变更为如下所述,除此以外,与实施例B2同样地处理,从而得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
即,首先,将悬浮液升温至60℃,在该温度下维持1小时,然后,添加二亚乙基三胺的水溶液,之后立即将悬浮液的温度升温至70℃,在该温度下维持2小时,然后,将悬浮液升温至80℃,在该温度下维持1小时。
即,在达到将悬浮液升温至60℃的中途温度、即55℃的时刻,开始悬浮聚合,在开始悬浮聚合后的、投入二亚乙基三胺的10质量%水溶液的时刻,开始界面聚合。
实施例B10~实施例B13、实施例B19~实施例B23、实施例B27、实施例B28、实施例B31以及实施例B32
(聚脲被覆/含有噻虫胺的缓释性粒子的合成)
(实施例B10~实施例B13、实施例B19~实施例B23、实施例B27、实施例B31以及实施例B32:对应于第3实施方式)
(实施例B28:对应于第4实施方式)
根据表B3~表B5的记载变更配合处方,除此以外,与实施例B2同样地处理,得到被聚脲所被覆的、含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
实施例B6(聚脲被覆/含有噻虫胺的缓释性粒子的合成:对应于第3实施方式)
在200mL的烧杯(1)中,向50g调制例B3中调制的浆料C中投入17.5g苯乙烯、17.5g的EGDMA、15g的T-1890、以及0.5g的PEROYL L,在室温下搅拌,从而使苯乙烯、EGDMA、T-1890以及PEROYL L溶解于浆料C。由此,调制含有苯乙烯、EGDMA、T-1890、PEROYL L以及浆料C的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水240.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入溶解有苯乙烯、EGDMA、T-1890以及PEROYL L的油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数5000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,将悬浮液(水分散液)转移至装备了搅拌器、回流冷却器、温度计及氮气导入管的500mL四口烧瓶中,在氮气气流下进行搅拌。
接着,在悬浮液中添加二亚乙基三胺的10质量%水溶液18g,开始界面聚合,然后使悬浮液升温,开始悬浮聚合。
具体地说,首先,在室温的悬浮液中,将二亚乙基三胺的10质量%水溶液18g添加于悬浮液,之后立即将悬浮液的温度升温至70℃,在该温度下维持5小时。然后,将悬浮液升温至80℃,在该温度下维持2小时。
在投入二亚乙基三胺的10质量%水溶液的时刻,开始界面聚合,在达到将悬浮液升温至70℃的中途温度、即55℃的时刻,开始悬浮聚合。
由此,得到噻虫胺被分散于通过悬浮聚合形成的基质中、基质被通过界面聚合形成的聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
然后,将反应后的悬浮液冷却至30℃以下,从而得到噻虫胺被分散于基质中、基质被聚脲所被覆的缓释性粒子的悬浮液(悬浮剂)。
实施例B7~实施例B9、实施例B14~实施例B18、实施例B24~实施例B26、实施例B29、实施例B30、实施例B33、实施例B34
(聚脲被覆/含有噻虫胺的缓释性粒子的合成)
(实施例B7~实施例B9、实施例B14~实施例B18、实施例B24~实施例B26、实施例B33以及实施例B34:对应于第3实施方式)
(实施例B29以及实施例B30:对应于第4实施方式)
根据表B2~表B5的记载变更配合处方以及聚合条件,除此以外,与实施例B6同样地处理,得到被聚脲所被覆的、含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
实施例B35(含有吡虫啉的缓释性粒子的合成:对应于第3实施方式)
根据表B5的记载变更配合处方,除此以外,与实施例B2同样地处理,得到含有吡虫啉的缓释性粒子的悬浮液(悬浮剂)。
参考例B1(含有噻虫胺的缓释性粒子的合成:对应于参考方式)
在200mL的烧杯(1)中,投入100g调制例B2中调制的浆料B、以及0.5g的PEROYL L,在室温下搅拌,从而使PEROYL L溶解于浆料B。由此,调制含有PEROYL L以及浆料B的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水258.50g、PVA-217的10%水溶液40g以及PRONON 208的1%水溶液1g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入配合了PEROYL L的油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数6000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,将悬浮液(水分散液)转移至装备了搅拌器、回流冷却器、温度计及氮气导入管的500mL四口烧瓶中,在氮气气流的条件下边搅拌边升温,实施悬浮聚合。
关于悬浮聚合,将达到55℃的时刻设为聚合开始,然后,连续地在70±1℃实施5小时、在80±1℃实施2小时。
然后,将反应后的悬浮液冷却至30℃以下,从而得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
参考例B2(含有噻虫胺的缓释性粒子的合成:对应于参考方式)
在200mL的烧杯(1)中,投入100g调制例B2中调制的浆料B、以及0.5g的PEROYL L,在室温下搅拌,从而使PEROYL L溶解于浆料B。由此,调制含有PEROYL L以及浆料B的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水258.26g、PVA-217的10%水溶液40g、PRONON 208的1%水溶液1g以及DEMOL NL 0.24g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数6000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,在与参考例B1同样的条件下实施悬浮聚合,得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
参考例B3(含有噻虫胺的缓释性粒子的合成:对应于参考方式)
在200mL的烧杯(1)中,投入100g调制例B1中调制的浆料A、以及0.5g的PEROYL L,在室温下搅拌,从而使PEROYL L溶解于浆料A。由此,调制含有PEROYL L以及浆料A的油相成分。
另外,在500mL的烧杯(2)中,投入离子交换水258.50g、PVA-217的10%水溶液40g以及PRONON 208的1%水溶液1g,在室温下搅拌,从而得到均匀的水溶液。
接着,在500mL的烧杯(2)中,加入油相成分,利用T.K.均相混合机MARK 2.5型(PRIMIX公司制),以转数6000rpm搅拌5分钟,从而对油相成分进行水分散,调制悬浮液(水分散液)。
然后,在与参考例B1同样的条件下实施悬浮聚合,得到含有噻虫胺的缓释性粒子的悬浮液(悬浮剂)。
[表B2]
[表B3]
[表B4]
[表B5]
[表B6]
在表B2~表B5中,聚合条件栏中的“1”表示在悬浮液中添加二亚乙基三胺水溶液后,立即将悬浮液升温至70℃,在该温度下维持5小时,然后,将悬浮液升温至80℃,在该温度下维持2小时的情况。
表B2~表B6中,聚合条件栏中的“2”表示在悬浮液中添加二亚乙基三胺的水溶液后,立即将悬浮液升温至60℃,在该温度下维持1小时,接着,将悬浮液升温至70℃,在该温度下维持2小时,然后,将悬浮液升温至80℃,在该温度下维持1小时的情况。
表B2~表B5中,聚合条件栏中的“3”表示在悬浮液中添加二亚乙基三胺水溶液后,立即将悬浮液升温至50℃,在该温度下维持2小时,接着,将悬浮液升温至60℃,在该温度下维持1小时,接下来,将悬浮液升温至70℃,在该温度下维持2小时,然后,将悬浮液升温至80℃,在该温度下维持1小时的情况。
表B2~表B5中,聚合条件栏中的“4”表示在将悬浮液升温至60℃,在该温度下维持1小时后,添加二亚乙基三胺的水溶液,之后立即将悬浮液升温至70℃,在该温度下维持2小时,然后,将悬浮液升温至80℃,在该温度下维持1小时的情况。
此外,表B2~表B5中,缓释性粒子的方式栏中的“1”表示具有图B1中所示的第3实施方式的结构,“2”表示具有图B2中所示的第4实施方式的结构。
(缓释性粒子的粉剂与热塑性树脂的混炼以及成型)
实施例B36(实施例B1的粉剂与聚乙烯的混炼以及成型)
利用100目滤布将实施例B1中制作的缓释性粒子的悬浮液过滤后,在室温下干燥1天,得到缓释性粒子的粉末(粉剂)。将所得到的缓释性粒子的粉末(粉剂)与高密度聚乙烯(HDPE)HI-ZEX 6300M(Prime Polymer公司制、熔体流动速率0.11g/10分钟)进行干式混合,以使噻虫胺相对于HDPE为0.25%,并投入双轴挤出-注射成型并设机DSMX plore MC15M(DSM公司制),进行220℃×5分钟的熔融混炼而得到料股,接着,以熔融状态直接通过注射成型而得到长条型成型品(10mm×76mm×4mm)。
实施例B37(实施例B27的粉剂与聚乙烯的混炼以及成型)
使用实施例B27中制作的缓释性粒子的悬浮液,来代替实施例B1中制作的缓释性粒子的悬浮液,除此以外,与实施例B36同样地处理,得到长条型成型品。
参考例B4(参考例B1的粉剂与聚乙烯的混炼以及成型)
使用参考例B1中制作的缓释性粒子的悬浮液,来代替实施例B1中制作的缓释性粒子的悬浮液,除此以外,与实施例B36同样地处理,得到长条型成型品。
参考例B5(参考例B3的粉剂与聚乙烯的混炼以及成型)
使用实施例B3中制作的缓释性粒子的悬浮液,来代替实施例B1中制作的缓释性粒子的悬浮液,除此以外,与实施例B36同样地处理,得到长条型成型品。
(缓释性粒子的粒剂的制剂化)
实施例B38
相对于卡卡(Kagalite)2号(卡卡工业公司制、浮石的细粒、粒径425~1400μm)100质量份,配合实施例B1中制作的缓释性粒子的悬浮液(噻虫胺浓度7.0质量%)1.4质量份,接着,将它们干燥,得到噻虫胺粒剂。粒剂中的噻虫胺浓度为约0.1质量%。
实施例B39
配合实施例B27中制作的缓释性粒子的悬浮液(噻虫胺浓度7.0质量%)1.4质量份,来代替实施例B1中制作的缓释性粒子的悬浮液,与实施例B38同样地处理,得到噻虫胺粒剂。粒剂中的噻虫胺浓度为约0.1质量%。
参考例B6
配合参考例B1中制作的缓释性粒子的悬浮液(噻虫胺浓度8.3质量%)1.2质量份,来代替实施例B1中制作的悬浮液,除此以外,与实施例B38同样地处理,得到噻虫胺粒剂。粒剂中的噻虫胺浓度为约0.1质量%。
参考例B7
配合参考例B3中制作的缓释性粒子的悬浮液(噻虫胺浓度8.3质量%)1.2质量份,来代替实施例B1中制作的悬浮液,除此以外,与实施例B38同样地处理,得到噻虫胺粒剂。粒剂中的噻虫胺浓度为约0.1质量%。
1.SEM(扫描式电子显微镜,Scanning Electron Microscope)观察
将实施例B1、实施例B2、实施例B6、实施例B30以及实施例B35各自的悬浮液(悬浮剂)滴加于试样台,然后将水蒸除去,然后对所得到的缓释性粒子利用扫描式电子显微镜日立TM-3000(日立高新技术公司制)进行SEM观察。将实施例B1、实施例B2、实施例B6、实施例B30以及实施例B35中所得到的缓释性粒子的SEM图像分别示于图B3~图B7。
2.TEM(透射式电子显微镜,Transmission Electron Microscope)观察
将实施例B2以及参考例B1~参考例B3各自的悬浮液(悬浮剂)冷冻干燥,分散于双酚型液状环氧树脂,用胺进行固化。利用超薄切片机将其切断从而使截面露出,用四氧化锇进行染色,根据需要进一步用四氧化钌进行染色,利用超薄切片机将其切成超薄切片而调制样品。利用透射式电子显微镜(型号“H-7100”、日立制作所社制)对调制的样品进行TEM观察。
将实施例B2以及参考例B1~参考例B3的TEM照片的图像处理图分别示于图B8~图B11。
需要说明的是,在图B8~图B11中,由符号3表示的空白是在使切出的超薄切片浮于水而回收的过程中,噻虫胺溶解脱落的痕迹,表示由噻虫胺形成的结构域的形状。
此外,在图B8中,壳层7由聚脲构成,具体构成为:随着从最外层(最表面)朝向内侧,聚脲相对于基质2的浓度变淡。此外,壳层7以包围结构域3的方式位(偏在)于基质2的表层部。
另一方面,由图B9~图B11可知,在参考例B1~参考例B3的缓释性粒子1中,没有形成壳层7(参照图B8)。
3.耐碱性试验
3-1.缓释性粒子的悬浮剂
按照下面的顺序实施缓释性粒子的悬浮剂的耐碱性试验(试验A和B)。
(试验A)
利用去离子水将实施例B1~实施例B35各自的悬浮剂稀释成抗生物活性化合物的浓度(对于实施例B1~实施例B34而言是噻虫胺的浓度,对于实施例B35而言是吡虫啉的浓度)为0.25%。称取悬浮剂1mL至玻璃瓶,添加饱和氢氧化钙溶液4mL,调制试验溶液。在40℃恒温的条件下静置该试验溶液。
在开始静置第7天后,向试验溶液添加乙腈5mL,提取抗生物活性化合物,利用HPLC对抗生物活性化合物进行定量,计算出抗生物活性化合物的残存率。
将这些结果示于表B2~表B6。
另外,作为对照,使用噻虫胺的0.25%水溶液以及吡虫啉的0.25%水溶液,同样地实施试验。其结果是,噻虫胺的0.25%水溶液的试验A的残存率为2.5%,吡虫啉的0.25%水溶液的试验A的残存率为0%。
由表B2~表B6至少可知以下的点。
就实施例B2~实施例B4而言,由于在开始悬浮聚合之前开始界面聚合,因此能够良好地进行含有噻虫胺的基质与壳层的相分离。另一方面,就实施例B5而言,由于在开始悬浮聚合之后开始界面聚合,因此无法良好地进行基质与壳层的相分离,与实施例B5相比,实施例B2~实施例B4的耐碱性更优异。
就实施例B6~实施例B8而言,由于在开始悬浮聚合之前开始界面聚合,因此能够良好地进行含有噻虫胺的基质与壳层的相分离。另一方面,就实施例B9而言,由于在开始悬浮聚合之后开始界面聚合,因此无法良好地进行基质与壳层的相分离,与实施例B9相比,实施例B6~实施例B8的耐碱性更优异。
实施例B13、实施例B12、实施例B11、实施例B2以及实施例B10中,T-1890相对于i-BMA和EGDMA的配合比例依次增大。因此,实施例B13、实施例B12、实施例B11、实施例B2以及实施例B10的壳层的厚度(缓释性粒子中的壳层的浓度)依次增大。因此,实施例B13、实施例B12、实施例B11、实施例B2以及实施例B10的耐碱性依次提高。
实施例B18、实施例B17、实施例B16、实施例B15、实施例B6以及实施例B14中,T-1890相对于苯乙烯和EGDMA的配合比例依次增大。因此,实施例B18、实施例B17、实施例B16、实施例B15、实施例B6以及实施例B1的壳层的厚度(缓释性粒子中的壳层的浓度)依次增大。因此,实施例B18、实施例B17、实施例B16、实施例B15、实施例B6以及实施例B14的耐碱性依次提高。
实施例B28、实施例B27以及实施例B2的缓释性粒子中的噻虫胺的配合比例依次降低,实施例B28、实施例B27以及实施例B2的耐碱性依次提高。
实施例B30、实施例B29以及实施例B2的缓释性粒子中的噻虫胺的配合比例依次降低,耐碱性提高。
疏水性更高的悬浮聚合物更良好地进行与聚脲的相分离。因此,在实施例B2以及实施例B19~实施例B23中,与i-BMA的配合比例极低的实施例B22及实施例B23相比,i-BMA的配合比例比较高的实施例B2以及实施例B19~实施例B21更良好地进行壳层与包含噻虫胺的基质的相分离。因此,与实施例B22以及实施例B23相比,实施例B2、实施例B20以及实施例B21的耐碱性更优异。
在实施例B6以及实施例B23~实施例B26中,与苯乙烯的配合比例极低的实施例B23以及实施例B26相比,苯乙烯的配合比例比较高的实施例B6、实施例B24以及实施例B25更良好地进行壳层与包含噻虫胺的基质的相分离。因此,与实施例B23以及实施例B26相比,实施例B6、实施例B24以及实施例B25的耐碱性更优异。
实施例B6含有苯乙烯作为聚合性乙烯基单体,实施例B2含有i-BMA作为聚合性乙烯基单体,与实施例B2的i-BMA相比,实施例B6的苯乙烯的疏水性更高,因此良好地进行壳层与聚合物的相分离。由此,与实施例B2相比,实施例B6的耐碱性更优异。
(试验B)
利用100目滤布将实施例B1、实施例B2以及参考例B1~实施例B3中制作的缓释性粒子的悬浮液过滤后,在室温下干燥1天,得到缓释性粒子的粉末(粉剂)。用去离子水将这些粉末稀释成1000倍,称取其中的6.3mL至玻璃瓶,添加饱和氢氧化钙溶液2mL,制成试验溶液。在40℃恒温的条件下静置该试验溶液。
在试验开始1天后及7天后,向试验溶液添加乙腈10mL,提取噻虫胺,利用HPLC对噻虫胺量进行定量,计算出残存率。
作为对照,使用噻虫胺原药的水溶液,同样地实施试验。
将结果示于表B7。
[表B7]
由表B7可知,与含有不具有壳层的参考例B1~参考例B3的缓释性粒子的悬浮剂相比,含有具有壳层(参照图B1中的符号7)的实施例B1和B2的缓释性粒子的悬浮剂中,噻虫胺的残存率在试验开始后第1天及第7天均更高。
3-2.缓释性粒子的粒剂
称取1.0g的实施例B38、实施例B39以及参考例B6、参考例B7中得到的粒剂,添加去离子水3.6mL和饱和氢氧化钙水溶液2mL,调制试验溶液。在40℃恒温的条件下静置该试验溶液。
在开始试验1天后及7天后,向试验溶液添加乙腈10mL,提取噻虫胺,用HPLC对噻虫胺量进行定量,计算出残存率。
作为对照,使用噻虫胺原药的水溶液,同样地实施试验。
将结果示于表8。
[表B8]
由表8可知,与含有不具有壳层的参考例B1及参考例B3的缓释性粒子的参考例B6及参考例B7的粒剂相比,含有具有壳层(参照图B1中的符号7)的实施例B1及实施例B2的缓释性粒子的实施例B38及实施例B39的粒剂中,噻虫胺的残存率在试验开始1天后及7天后均更高。
4.成型品的防蚁试验
将以含水率为8%(白蚁活动的最适含水率)的方式注水的硅砂填充于塑料容器,接着,在硅砂的表面设置实施例B36以及实施例B37的长条型成型品。
作为比较对照,实施了设置未混炼缓释性粒子的仅包含HDPE的长条型成型品的试验。
在上述塑料容器内投入家白蚁工蚁50只,对白蚁的死亡只数(=死亡率)以及行动观察了7天(以n=2实施试验)。关于实施例B36以及实施例B37的长条型成型品,在试验开始第2天以及第3天白蚁全部死亡。
另一方面,关于作为比较对照的仅包含HDPE的长条型成型品,在7天后白蚁也没有死亡,并且没有发现白蚁的行动有变化。
即,对于实施例B36以及实施例B37,证实了显著的灭蚁效果。
需要说明的是,上述发明虽然以本发明例示的实施方式提供,但其只不过是例示而已,不应解释为限定性的。对于本技术领域的技术人员而言显而易见的本发明的变形例均包含于所附权利要求的范围内。
符号的说明
1:缓释性粒子,2:基质,3:结构域,5:附着物,7:壳层
工业实用性
通过缓释性粒子的制造方法得到的缓释性粒子可用于各种用途,可以用作例如建筑材料,例如电线电缆材料及其电线电缆的被覆材料,例如气体等的导管及其导管的被覆材料,例如衣类、蚊帐等纤维制品。
- 还没有人留言评论。精彩留言会获得点赞!