用于局部给药的药用组合物的制作方法
本发明涉及一种用于局部给药的药用组合物,其包含式I化合物,3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸或其药学上可接受的盐;以及涉及治疗炎症的方法,其包括局部给予包含式I化合物的组合物。
背景
胞质磷脂酶A2 (cPLA2),磷脂酶A2 (PLA2)家族的一个成员,从磷脂膜中释放花生四烯酸并且是前列腺素(PGs)、血栓烷(Txs)和白三烯(LTs)的生物合成中的限速酶,所有这些都与身体的炎症反应有关。因此,这种酶和PLA2家族的其它成员的抑制是改善炎性疾病和病症的潜在途径(Tibes & Friebe, 开发中的磷脂酶A2抑制剂(Phospholipase A2 inhibitors in development). Expert Opin Investig Drugs. 1997 Mar; 6(3): 279-98)。
化合物3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸被鉴定为识别新的、高效力的cPLA2α抑制剂的工作的一部分(McKew等 J Med Chem. 2008 Jun 26; 51(12): 3388-413)。它可如在WO2006/128142中所述制备,其内容通过引用结合到本文中。
发明概述
皮肤炎症是许多皮肤疾病和病症的一种症状。这样的病症包括,例如,特应性皮炎、大疱性疾病、胶原性疾病、银屑病、银屑病性损伤、脂溢性皮炎或接触性皮炎、湿疹、荨麻疹、瘙痒症、酒渣鼻、结节性痒疹、增生性瘢痕、瘢痕疙瘩形成、硬皮病、项部瘢痕瘤性毛囊炎、川崎病、Sjögren-Larsson综合征、Grover病、一度、二度、三度和四度烧伤、皮肤粘蛋白沉积症、日光性角化病、鳞状细胞癌和黑色素瘤。用靶向PLA2家族的药物治疗这些病症中的炎症是可取的。
先前的研究者已经研究了经过口服和肺部途径靶向PLA2家族的炎症治疗。一般来说,这些治疗已集中在优化用于口服途径的治疗药物,当这样的药物靶向PLA2以治疗炎症时,其有利于良好的口服生物利用度和长的血清半衰期。虽然仍然是一个设计要求,当靶向口服途径时,药物的效力不太关键,因为它是通过将更多的药物加入剂型中或通过增加给药频率来直接增加剂量。事实上,现有技术的PLA2抑制剂的一个共同特征是它们的相对低的效力,例如,已显示SB 203347在酸提取的完整的人中性粒细胞匀浆中完全抑制PLA2活性,其IC50为4.7 µM (Marshall等J Pharmacol Exp Ther. 1995 Sep; 274(3): 1254-62)。
一般来说,口服途径是治疗皮肤疾病的次优方法。特别是,全身给药(经由口服途径或其它方式)带来其与所述病症无关的组织副作用的风险,例如胃肠道刺激和/或毒性。此外,口服给予的化合物通过肝经历首过代谢。相反,局部给药往往是选择的途径,如果它能够实现的话。局部给予药物在局部病症的治疗中有许多优势。由于该药物可在需要其的部位提供,系统循环的药物的等效浓度得以避免。这可减轻或消除上面提到的副作用。
因此,虽然已经开发出适合于口服给予的cPLA2α抑制剂,仍需要可局部给药的有效的cPLA2α抑制剂。
不幸的是,最适于口服给予的化合物一般不适合局部应用,往往是由于所讨论的低效力导致。对于皮肤给予,非常高的效力是必需的。当化合物不与其靶标结合时,从身体中快速地清除该化合物也是优选的。这组性质允许药物仅对皮肤施加有力的作用,并避免不要的系统作用。
虽然理想,但用于皮肤给药的制剂可能是复杂的,由于药物应用于皮肤,通常要考虑外源物进入的屏障。局部药物对应用于皮肤的早先的研究讲述了具有低分子量(<500道尔顿)和适度log P (0-3)的化合物对于这种应用是理想的。
令人吃惊地,本发明人已发现本发明的化合物,一种为口服给予而开发的cPLA2α抑制剂,当作为用于局部给予的组合物的部分传递时,是有效的和良好耐受的。到现在为止,尚未证实这种类型的酶的抑制剂经由局部途径是有用的。鉴于3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸的相对高的分子量(823.35道尔顿)及其高的log P,这一发现是特别令人惊讶的。
本发明的化合物当正确地配制时,令人惊讶和意外地显示在动物皮肤炎症模型中具有抗-炎作用并渗透进入人皮肤的较深层。
在导致本发明的研究过程中,已发现式I化合物可作为局部用组合物的活性组分应用于皮肤并可用于渗透到皮肤内。此外,所述化合物具有抗-炎作用。
因此,本发明提供一种用于局部给药的药用组合物,其包含式I化合物:
或其药学上可接受的盐和药学上可接受的稀释剂或载体。
贯穿本申请,式(I)化合物也称为化合物I。应该理解,无论何处提及化合物I时,也涵盖所述化合物的药学上可接受的盐的用途。
具有酸性部分的式(I)化合物的药学上可接受的盐可从有机和无机碱形成。与碱形成的合适的盐是,例如,金属盐,如碱金属或碱土金属盐,例如钠、钾或镁盐;或与氨或有机胺所成的盐,如吗啉、硫吗啉、哌啶、吡咯烷、单-、二-或三低级烷基胺,例如乙基-叔丁基-、二乙基-、二异丙基-、三乙基-、三丁基-或5二甲基丙胺,或单-、二-,或三羟基低级烷基胺,例如单-、二-或三乙醇胺。
术语“局部给药”涉及将物质直接应用于身体表面,如皮肤。在特别优选的实施方案中,局部给药是经表皮的,即直接给予皮肤表面。这有时可被称为皮肤给药。所述组合物可使用熟知的技术和赋形剂,以适合于局部给药的任何形式提供,包括但不限于软膏剂、凝胶剂、霜剂、洗剂、水包油(o/w)乳剂、油包水(w/o)乳剂、油/水/油(o/w/o)乳剂、水/油/水(w/o/w)乳剂、微乳剂、泡沫剂、喷雾剂、摩丝、贴剂、散剂、糊剂、药用膏药等。优选地,所述组合物以霜剂、软膏剂、洗剂或凝胶剂的形式提供。最优选地,所述组合物以霜剂或软膏剂的形式提供。
特别优选的组合物(其在本文也可称为制剂)在表3-7中描述。最优选的制剂包括,但不限于:
软膏剂O2 (表3)
软膏剂O2X (表3)
软膏剂O4v4 (表3)
软膏剂O4v4E (表3)
软膏剂O5v2 (表3)
软膏剂O7 (表3)
霜剂7PE (表5)
霜剂9 (表5)
凝胶剂1V3 (表7)
凝胶剂4V3 (表7)
乳化凝胶剂1V2 (表7)
在优选的实施方案中,式I化合物或其药学上可接受的盐以从约0.01%至约10% (w/w)存在。更优选地,式I化合物或其药学上可接受的盐以从约0.3%至约3%存在。在其它优选的实施方案中,式I化合物或其药学上可接受的盐以约0.01、0.02、0.03、0.04、0.05、0.06、0.07、0.08、0.09、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、2、3、4、5、6、7、8、9或10% (w/w)存在。
所述载体可以是活性组分可溶于其中的溶剂。
合适的溶剂包括,但不限于水(约0-约25% w/w);醇(约0-约20% w/w),选自具有从1至18个碳的一元醇,如甲醇、乙醇、丙醇、异丙醇、丁醇、己醇、十六烷醇、十八烷醇等;二元和多元醇,如丙二醇、丙三醇;己三醇,如1,2,6-己三醇、山梨醇、1,3-丁二醇、2,3-丁二醇等;具有从1至6个碳的二醇的低级烷基醚如乙二醇或二乙二醇或丙二醇或二丙二醇的单甲基或单乙基醚、丙二醇或二丙二醇单甲基醚、二乙二醇单乙基醚、乙二醇单甲基醚等;具有从100至8,000,优选地在4,000范围内的分子量的聚乙二醇;具有从2至22个碳的脂族一元和二元酸和具有从1至20个碳的一元醇、具有从2至20个碳的二-和多元醇,和糖醇的酯如肉豆蔻酸异丙酯、棕榈酸异丙酯、肉豆蔻酸肉豆蔻酯、硬脂酸鲸蜡酯、硬脂酸甲酯、癸二酸异丙酯、癸二酸甲酯、单月桂酸蔗糖、单硬脂酸蔗糖等。其它的合适的溶剂包括在0-20% (w/w)范围内的甘露醇、木糖醇、山梨醇和具有从1至6个碳的一元醇,如甲醇、乙醇、丙醇、异丙醇、丁醇,或己醇。
更特别地,溶剂可包含选自丙二醇、丁二醇、二乙二醇和二乙二醇醚的非-毒性二醇或二醇醚。其它优选的溶剂包括,但不限于,乙醇、去离子水、苄醇、PEG 400、二甲基异山梨醇酯(Arlasolve dmi)、二乙二醇单乙基醚(Transcutol P)、甘油(丙三醇)、肉豆蔻酸异丙酯(IPM)、异丙醇(异丙基醇)、辛基十二烷醇、苯氧基乙醇、油醇、矿物油(液体石蜡)、Crodamol GTCC (辛酸/癸酸甘油三酯或中链甘油三酯)、蓖麻油、棕榈酸异丙酯(IPP)、丙二醇二辛酸酯/二癸酸酯和杏仁油PEG- 6酯(Labrafil M1944CS)。
所述组合物还可包含共溶剂,其可选自以上提供的溶剂列表。特别是,共溶剂选自在0-20% (w/w)范围内的水、甘露醇、木糖醇、山梨醇和具有从1至6个碳的一元醇,如甲醇、乙醇、丙醇、异丙醇、丁醇,或己醇;和另一种共溶剂选自以下的一种或多种:丙二醇(在0-35% w/w范围内)、丙三醇(在0-10% w/w范围内)、聚乙二醇如PEG400 (在0-80% w/w范围内)或PEG4000 (或在该类型中的类似分子量的物质) (在0-30% w/w范围内)。
此外,两种或更多种不同的溶剂可合并成具有所需特性的溶剂系统。合适的溶剂系统的实例在表1中提供:
当组合物呈现为乳剂(水包油(o/w)、油包水(w/o)、油/水/油(o/w/o)和水/油/水(w/o/w))的形式时,所述组合物还可包含乳化剂。这可包括在内以调节油相中可具有至多200 µm的直径的油滴大小至较小的尺寸,通常在1-50 µm范围内,从而改进所述组合物的化妆用外观。乳化剂可适当地为油包水乳化剂,如选自油包水乳化剂,例如,聚氧化烯C12-20烷基醚,如聚氧乙烯-2-十六烷基醚、聚氧乙烯-2-月桂基醚、聚氧乙烯-2-油醚或聚氧乙烯-2-硬脂醚、聚氧化烯烷基酯、脱水山梨醇油酸酯、脱水山梨醇异硬脂酸酯、脱水山梨醇倍半油酸酯、异硬脂酸和己二酸的丙三醇酯、聚甘油-3-二异硬脂酸酯和聚甘油-6-六蓖麻醇酸酯。乳化剂可适当地为一种水包油乳化剂,如选自水包油乳化剂,例如,聚氧化烯C12-20烷基醚和酯,如聚氧乙烯-20-十六烷基醚、聚氧乙烯-20-月桂基醚、聚氧乙烯-20-油醚或聚氧乙烯-20-硬脂醚、聚山梨醇酯20、聚山梨醇酯60、聚山梨醇酯80等。
本发明的组合物可根据药物制剂领域的技术人员熟知的方法制备。所述组合物中各组分的量将在某种程度上取决于其中加入的活性组分的浓度。所述组合物中的活性组分的量可根据待治疗的病症的严重性,患者的年龄和情况以及临床医师的判断力而广泛变化。
除了上述成分,本组合物可包含一种或多种另外的成分如:
1. 应用于治疗皮肤炎性病症的其它治疗活性物质,包括:
a. 皮质类固醇,如氢化可的松;
b. 非-甾体抗炎药如水杨酸、水杨酸酯、维生素D类似物如卡泊三醇(Dovonex)、亲免素、p38激酶抑制剂、钙调磷酸酶抑制剂如他克莫司和吡美莫司;和大麻素类;
c. 血管调节因子(vasomodulators)如α-肾上腺素能受体配体;和
d. 局部麻醉剂如布比卡因、氯普鲁卡因、狄步卡因、氯胺酮和普莫卡因(pramoxine)。
2. 抗-感染药如:
a. 局部抗生素如克林霉素
b. 抗真菌剂
c. 抗病毒剂
其它的合适的共同给予的药物(co-administrants)是本领域技术人员熟知的,包括但不限于:
1. 组胺H1受体拮抗剂
2. 组胺H2受体拮抗剂
3. 组胺H3受体拮抗剂
4. 白三烯拮抗剂,包括LTB4、LTC4、LTD4和LTE4的拮抗剂,例如孟鲁司特
5. 磷酸二酯酶抑制剂,包括PDE3抑制剂、PDE4抑制剂、PDE5抑制剂、PDE7抑制剂和两种或更多种磷酸二酯酶的抑制剂,如双重PDE3/PDE4抑制剂
6. 神经递质重摄取抑制剂,特别是氟西汀、舍曲林、帕罗西汀、齐拉西酮
7. 5-脂氧化酶(5-LO)抑制剂或5-脂氧化酶激活蛋白(FLAP)抑制剂
8. α1-和α2-肾上腺素受体激动剂的血管收缩拟交感神经药物
9. 毒蕈碱M3受体拮抗剂或抗胆碱能药物
10. β2-肾上腺素受体激动药
11. 双重作用β2/M3剂
12. 黄嘌呤类,如茶碱、氨茶碱
13. 非-甾体类抗炎药,如色甘酸钠和奈多罗米钠
14. 酮替芬
15. COX-1抑制剂(NSAIDs)和COX-2选择性抑制剂
16. 口服、吸入、鼻内和局部糖皮质激素
17. 抗内源性炎性实体的单克隆抗体活性剂
18. 抗-肿瘤坏死因子(抗-TNF-α)剂
19. 粘附分子抑制剂包括VLA-4拮抗剂
20. 激肽-β1 -和β2 -受体拮抗剂
21. 免疫抑制剂
22. 基质金属蛋白酶抑制剂(MMPs)
23. 速激肽NK1、NK2和NK3受体拮抗剂
24. 弹性蛋白酶抑制剂
25. 腺苷A2a受体激动剂
26. 尿激酶抑制剂
27. 作用于多巴胺受体的化合物,如D2激动剂
28. NFκb路径的调节剂,如IKK抑制剂
29. 可分类为粘液溶解剂的药物或止咳药
30. 抗生素
31. 细胞因子信号传导路径的调节剂,如p38 MAP激酶抑制剂、SYK激酶抑制剂或JAK激酶抑制剂
32. 前列腺素路径的调节剂,包括H-PDGS的抑制剂和DP-1和CRTH2的拮抗剂
33. 趋化因子受体CXCR1和CXCR2的拮抗剂
34. 趋化因子受体CCR3、CCR4和CCR5的拮抗剂
35. 胞质和可溶性磷脂酶A2 (cPLA2和sPLA2)的抑制剂
36. 磷酸肌醇-3-激酶的抑制剂
37. HDAC抑制剂,
38. p38抑制剂和/或
39. CXCR2拮抗剂.
40. 钙调神经磷酸酶抑制剂
41. 抗-白介素17 (抗-IL-17)剂
42. 抗-白介素4受体(抗-IL4R)剂
43. 抗-白介素31 (抗-IL-31)剂
在任何制剂中的一种或多种活性剂的作用可通过使用增强皮肤渗透的试剂而提高。增强皮肤渗透的合适试剂的实例公开于Int J Pharm. 2013 Apr 15; 447(1-2): 12-21中(通过引用结合到本文中)。因此,所述本发明的组合物可包括一种或多种渗透促进剂。渗透促进剂包括但不限于在5-30% (w/w)范围内的2-(2-乙氧基乙氧基)乙醇和二甲基异山梨醇酯。其它渗透促进剂的非-限制性实例包括C8-C22脂肪酸如异硬脂酸、辛酸和油酸;C8-C22脂肪醇如油醇和月桂醇;C8-C22脂肪酸的低级烷基酯如油酸乙酯、肉豆蔻酸异丙酯、硬脂酸丁酯,和月桂酸甲酯;C6-C8二酸的二(低级)烷基酯如己二酸二异丙基酯;C8-C22脂肪酸的单甘油酯如一月桂酸甘油酯;四氢糠醇聚乙二醇醚;聚乙二醇;丙二醇;2-(2-乙氧基乙氧基)乙醇;二乙二醇单甲基醚;聚氧化乙烯的烷基芳基醚;聚氧化乙烯单甲基醚;聚氧化乙烯二甲基醚;二甲亚砜;丙三醇;乙酸乙酯;乙酰乙酸酯;N-烷基吡咯烷酮;萜;大环促进剂如大环酮,例如,3-甲基环十五烷酮、9-环十七烯-1-酮、环十六烷酮和环十五烷酮;大环酯如十五内酯。
本发明的组合物可包括水,但它也可基本上或完全不含水。优选地,所述组合物的水含量为从约0至约80 % (w/w)。
本组合物也可包含通常用于局部制剂的其它组分,包括但不限于抗氧化剂(如α-生育酚、丁羟甲苯、丁羟茴醚)、稳定剂、螯合剂、增稠剂、软化剂、润滑剂、防腐剂、润肤剂、色素、香料、皮肤舒缓剂,皮肤愈合剂和皮肤调理剂如脲、丙三醇、尿囊素或红没药醇(bisabolol)。
所述组合物也可包含表面活性剂或乳化剂包括,但不限于,杏仁油PEG-6酯(Labrafil M1944CS)、鲸蜡硬脂醇聚醚(ceteareth)-12 (Brij C20)、辛酰己酰聚乙二醇(caprylocaproyl macrogol)-8甘油酯(Labrasol)、十六十八醇、单硬脂酸甘油酯、月桂酰聚乙二醇-6甘油酯(Labrafil M2130CS)、聚乙二醇15羟基硬脂酸酯、(聚氧乙烯15羟基硬脂酸酯)、聚乙二醇鲸蜡硬脂醚(聚西托醇(cetomacrogol) 1000)、PEG-100硬脂酸酯(Myrj S100)、聚氧乙烯35蓖麻油(聚乙二醇甘油蓖麻油酸酯)、聚氧乙烯40 氢化蓖麻油、PEG-40硬脂酸酯(聚乙二醇硬脂酸酯、聚氧乙烯硬脂酸酯)、聚山梨醇酯60 (吐温60)、聚山梨醇酯80 (吐温80)、司盘60 (脱水山梨醇单硬脂酸酯)、硬脂醇聚醚(steareth)-2 (Brij S2)、硬脂醇聚醚-20 (Brij S20),和硬脂酸。
所述组合物也可包括一种或多种增粘剂。增粘剂包括,但不限于聚乙烯吡咯烷酮、聚乙烯聚吡咯烷酮(交聚维酮)、甲基纤维素、羟丙基纤维素(HPC)、羟丙基甲基纤维素(hypromellose, HPMC)、羟基乙基纤维素(HEC)、黄原胶、卡波姆(carbomer),和透明质酸钠(透明质酸)。
在特别优选的实施方案中,所述组合物包含:
(a) 式I化合物;
(b) 溶剂;
(c) 共-溶剂;
(d) 渗透促进剂;和
(e) 水。
在另一方面,提供一种用于局部给药的药用组合物,其包含式I化合物:
或其药学上可接受的盐和药学上可接受的稀释剂或载体,用于疗法。
优选地,疗法包括局部给药。
在又一方面,提供一种用于局部给药的药用组合物,其包含式I化合物:
或其药学上可接受的盐和药学上可接受的稀释剂或载体,用于治疗皮肤炎性疾病或病症。
本文还提供式I化合物:
或其药学上可接受的盐在制备用于治疗皮肤炎性疾病或病症的药物中的用途。
要治疗的皮肤炎性疾病或病症优选地是胞质磷脂酶A2α-依赖性或胞质磷脂酶A2α-介导的疾病或病症。
更优选地,皮肤炎性疾病或病症选自结疤、皮炎、增生性疾病或病症、肥大细胞疾病或病症、烧伤或接触过敏原和/或刺激物。
还更优选地,皮肤炎性疾病或病症选自特应性皮炎、大疱性疾病、胶原性疾病、银屑病、银屑病性损伤、脂溢性皮炎或接触性皮炎、湿疹、荨麻疹、瘙痒症、酒渣鼻、结节性痒疹、增生性瘢痕、瘢痕疙瘩形成、硬皮病、项部瘢痕瘤性毛囊炎、川崎病、Sjögren-Larsson综合征、Grover病、一度烧伤、二度烧伤、三度烧伤、四度烧伤、皮肤粘蛋白沉积症、日光性角化病、鳞状细胞癌或黑素瘤、皮脂缺乏性湿疹、盘状湿疹、手部湿疹、重力性/静脉曲张性湿疹(varicose eczema)、湿疹型药疹、单纯地衣、硬化性地衣、刺激性扁平地衣、过敏性接触性皮炎、光敏性/光加重性皮炎、感染性(继发于细菌/病毒/真菌感染)皮炎、瘙痒性疾病包括与慢性全身性疾病有关的那些疾病如尿毒症瘙痒、胆汁淤积性瘙痒、成人blaschkitis、水痛症(aquadynia)、水源性瘙痒、balsam of Peru、胆汁性瘙痒,肱桡肌瘙痒、药物引起的瘙痒、羟乙基淀粉引起的瘙痒、痒点(itchy points)、慢性单纯地衣、神经性皮炎,朊病毒瘙痒、痒疹、色素性痒疹、单纯痒疹、肛门瘙痒、阴囊瘙痒、外阴瘙痒、痒点(puncta pruritica)、称痒、肾性瘙痒、头皮瘙痒、老年性瘙痒、干燥性湿疹、与HIV感染有关的瘙痒、T-细胞淋巴瘤、塞扎里综合征(Sezary syndrome)和蕈样肉芽肿。
最优选地,皮肤炎性疾病或病症是银屑病或特应性皮炎。
在另一方面,提供一种在有需要的患者中治疗炎症和/或水肿的方法,所述方法包括局部给予包含式I化合物或药学上可接受的盐的组合物至需要治疗的区域。
附图简述
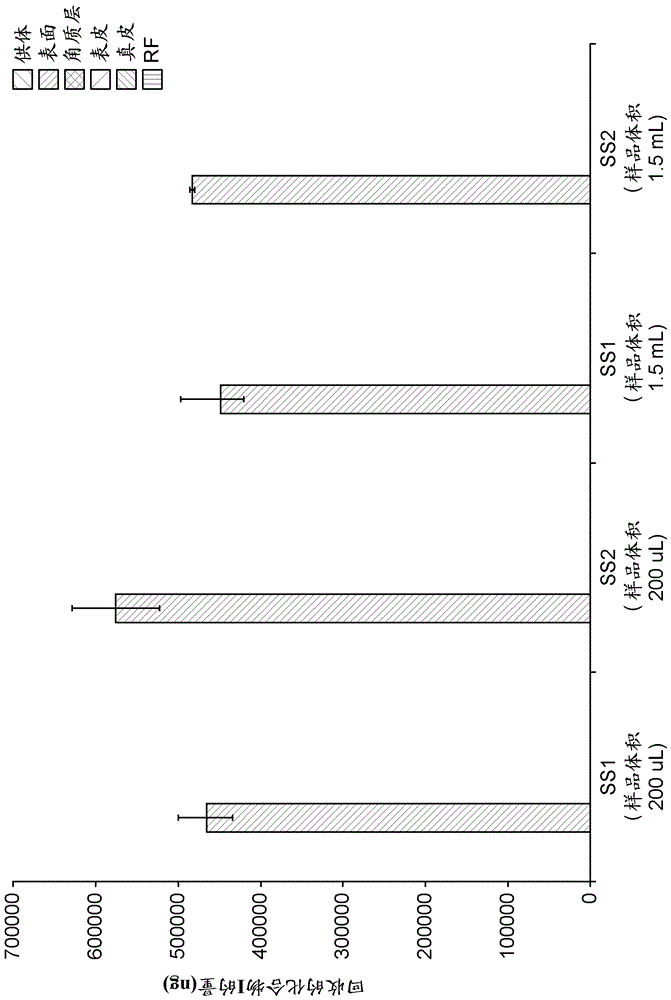
图1显示在应用溶剂系统SS1和SS2至经植皮刀分层的(dermatomed)人类皮肤后的最终时间点(t=48 h)后从供体室、表面、角质层、表皮、真皮和接收液回收的化合物I的量(ng)。每个点代表回收的化合物I的平均水平,误差棒代表范围,n=2-3。
图2显示与图1相同的数据,但为便于数据解释而删除了从表面回收的量。
图3显示在应用溶剂系统FSS1和FSS2后,在48 h实验期间每单位面积接受室中的化合物I的累积量(ng/cm2)。每个点代表在从3个供体穿越皮肤的接收液中检测的化合物I的平均水平±SD,n=9-10。
图4和5显示化合物I从溶剂系统(FSS1和FSS2),穿过经植皮刀分层的来自3个供体的腹部皮肤的体外渗透。每个点代表回收的化合物I的平均水平±SEM,n=9-10。
图6显示对于不同剂量的化合物I,耳朵厚度随时间推移(PMA应用后小时数)的变化。
图7显示化合物I与其它化合物的比较。
实施例
通过参考以下实施例将进一步理解本发明。
实施例1
3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸的制备
式I化合物可如在WO2006/128142中所述制备,其内容通过引用结合到本文中。进行以下步骤:
步骤1:使2-溴-4-氯苯胺(1.0 eq)溶于CH2Cl2 (0.25 M),然后加入三乙胺和三氟乙酸酐(各1.1 eq)。将生成的混合物于室温下搅拌1小时。蒸发溶剂和残留物经快速层析纯化,用CH2Cl2作为洗脱剂,得到97%收率的酰胺。m/z(M-H)- 300.0
步骤2:在N2下,在密封容器中,使N-(2-溴-4-氯代苯基)-2,2,2-三氟乙酰胺(步骤1,1.0 eq)与3-丁炔-1-醇(2.0 eq)、二氯双(三苯膦)钯(ll) (2.5% eq)、三乙胺(3.0 eq)、CuI (5% eq)在DMF (0.2 M)中混合并加热至120℃经4小时。然后反应混合物用乙酸乙酯稀释,用盐水洗涤并经Na2SO4干燥。经使用2% MeOH/CH2Cl2的快速柱层析纯化,以67%收率得到炔。m/z(M-H)- 194.09
步骤3:于室温及搅拌下,使2-(5-氯-1H-吲哚-2-基)乙醇(步骤2, 1.0 eq)和咪唑(2.0 eq)溶于DMF (0.3 M),然后加入叔-丁基氯代二苯基硅烷(1.2 eq)。将生成的混合物于室温下搅拌过夜,然后用饱和碳酸氢钠水溶液猝灭之并用乙酸乙酯提取。有机相用水和盐水洗涤并经Na2SO4干燥。经快速层析纯化,用CH2Cl2作为洗脱剂,以90%以上的收率得到甲硅烷基醚,为棕色胶状物。m/z(M-H)- 433.0
步骤4:使2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚(步骤3,1.0 eq)溶于乙醚(0.4 M)并使溶液冷却至0℃。在剧烈搅拌下,将草酰氯(1.2 eq)加入到上述冷却溶液中。将反应混合物于0℃搅拌1小时,然后加入EtOH,接着加入NEt3。再将生成的混合物用更多的EtOH稀释,然后将其倾入水中并用EtOAc提取。有机相用盐水洗涤,经Na2SO4干燥,并浓缩,以70%收率得到酮酯,为黄色固体。m/z(M-H)- 533.0
步骤5:使[2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚-3-基](氧代)乙酸乙基酯(步骤4, 1 eq)、Ph2CHBr (1.5 eq)和Cs2CO3 (1.5 eq)在无水乙腈(0.1 M)中混合。使混合物加热至回流2小时。使反应混合物冷却至室温,用水稀释并用EtOAc提取。浓缩有机相,残留物经层析,用CH2Cl2作为洗脱剂,以45%收率得到N-二苯甲基吲哚,为橙色胶状物。m/z (M+H)+ 701.3
步骤6:向[1-二苯甲基-2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚-3-基] (氧代)乙酸乙基酯(步骤5,1 eq)的THF (0.1 M)溶液中加入BH3•Me2S (2M在THF中) (2 eq)。将生成的混合物在N2下加热至回流过夜。使反应混合物冷却至室温,然后用1N NaOH缓慢地猝灭,用EtOAc提取,并用盐水洗涤。浓缩得到醇(65%收率)。m/z(M+H)+ 645.0
步骤7:向2-[1-二苯甲基-2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚-3-基]乙醇(步骤6,1 eq)的CH2CI2 (0.08M)溶液中加入1,3-双(二苯基膦基)-丙烷(DPPP, 0.75 eq)。使溶液在N2下冷却至0℃,然后加入CBr4 (1.25 eq)。使反应温度回到室温经2 h。蒸发溶剂,残留物使用短硅胶柱纯化,用CH2Cl2作为洗脱剂,以定量得率得到溴化物。m/z(M+H)+ 708.0
步骤8:使1-二苯甲基-3-(2-溴乙基)-2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚(步骤7,1 eq)与3-(4-缩硫醇苯基)丙酸甲基酯(1.5 eq)和K2CO3 (1.5 eq)在DMF (0.1 M)中混合。将生成的混合物于室温、N2下搅拌2 h,然后用水稀释并用EtOAc提取。有机提取物用盐水洗涤、浓缩,并经快速层析纯化(CH2Cl2作为洗脱剂),以80%收率,得到为浅棕色胶状物的硫醚。m/z(M+H)823.0
步骤9:使3-[4-({2-[1-二苯甲基-2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚-3-基]乙基}硫烷基)苯基]丙酸甲基酯(步骤8,1 eq)溶于乙腈(0.1 M),然后在N2下加入分子筛(粉末,4 A)和4-甲基吗啉N-氧化物(NMO) (4eq)。5 min后,加入n-Pr4NRuO4(TPAP) (5% eq)。将生成的混合物于40℃加热1.5 h。浓缩混合物,残留物经快速层析纯化,使用CH2Cl2,然后使用1 % EtOAc/CH2Cl2作为洗脱剂,以44%收率得到砜,为白色泡沫剂。m/z(M+H)+855.1
步骤10:使3-(4-{2-[1-二苯甲基-2-({[叔丁基(二苯基)甲硅烷基]氧基}乙基)-5-氯-1H-吲哚-3-基]乙氧基}苯基)丙酸甲基酯(步骤9,1 eq)溶于THF (0.1 M)并冷却至0℃,然后用nBu4NF (1 M在THF中) (1.2 eq)处理。将生成的混合物于0℃搅拌5 min,然后温热至室温并搅拌30 min。蒸发溶剂和残留物经快速层析纯化,用EtOAc/CH2Cl2 (1:9至1:4)作为洗脱剂,以90%收率得到醇,为白色泡沫。m/z(M+H)+ 616.20
步骤11:于0℃用MeSO2Cl (2.0 eq)和Et3N (2.5 eq)在CH2Cl2 (0.02 M)中处理3-[4-{2-[1-二苯甲基-5-氯-2-(羟基乙基)-1H-吲哚-3-基]乙基}-磺酰基)苯基]丙酸甲基酯(步骤10,1 eq)并搅拌1小时。移去冰浴并于室温下将反应混合物搅拌1小时,然后用CH2Cl2稀释之,用NaH2PO4、盐水洗涤并经经Na2SO4干燥。蒸发溶剂,以定量得率得到甲磺酸酯。m/z(M+H)+695.0
步骤12:使3-(4-{[2-(1-二苯甲基-5-氯-2-{2-[(甲基磺酰基)氧基]乙基}-1H-吲哚- 3-基)乙基]磺酰基}苯基)丙酸甲基酯(步骤11,1.0 eq)溶于DMF (0.03 M)并用NaN3 (3.0 eq)处理。将生成的混合物加热至60℃并搅拌2小时,然后冷却至室温,用水稀释,用乙酸乙酯提取,用盐水洗涤并用Na2SO4干燥。蒸发溶剂,以定量得率得到叠氮化物。m/z (M+H)+ 641.1
步骤13:使3-[4-({2-[2-(2-叠氮基乙基)-1-二苯甲基-5-氯-1H-吲哚-3-基]乙基}磺酰基)苯基]丙酸甲基酯(步骤12, 1 eq)溶于THF (0.1 M),并用三苯膦(1.1 eq)处理。在加入水2天后,将混合物搅拌过夜、浓缩,并经快速层析纯化,使用4% MeOH:CH2Cl2作为洗脱剂,以71 %收率得到胺。m/z (M+H)+ 615.2
步骤14:于-20℃,经1hr向(2-三氟甲基苯基)甲磺酸(20.3 g, 84 mmol)在THF (1.9 L)和DMF (5.0 mL)中的悬浮液中缓慢滴加入草酰氯(44.7 mL,0.5 mol)。将浴温维持于0℃以下4 h,于该时间点将反应物蒸发至~ 250 mL的体积并用500mL乙酸乙酯稀释。该溶液用盐水在一个分液漏斗中洗涤并经硫酸镁干燥。然后将该溶液蒸发至棕色油。将这种油吸收到500mL的pet醚(30-50°)并用加热枪加热,直至油进入溶液中。然后将该溶液置于干冰丙酮浴中,以冷却导致白色结晶物质的形成。经由过滤收集这种物质并干燥以提供19 g (85%)的(2-三氟甲基苯基)甲烷磺酰氯,为白色固体。
步骤15:向在CH2Cl2 (0.07 M)中的3-[4-({2-[2-(2-氨基乙基)-1-二苯甲基- 5-氯-1H-吲哚-3-基]乙基}磺酰基)苯基]丙酸乙基酯(步骤14, 200 mg, 0.32 mmol)和饱和NaHCO3 (0.14 M)中加入(2-三氟甲基苯基)甲烷磺酰氯(步骤14,110 mg, 0.42 mmol)。在16 h后,将该混合物倾入饱和碳酸氢钠并用CH2Cl2提取。合并的有机相用盐水洗涤,经硫酸钠干燥并经柱层析纯化,以93%收率提供250 mg磺酰胺,为淡黄色泡沫。1HNMR (400 MHz, CDCl3) δ 1.23 (t, J=I.2 Hz, 3 H), 2.62 - 2.71 (m, 2 H), 2.76 - 2.93 (m, 4 H), 2.98 - 3.17 (m, 4 H), 3.27 - 3.38 (m, 2 H), 4.11 (q, J=7.2 Hz, 2 H), 4.35 (s, 2 H), 4.57 (t, J=5.3 Hz, 1 H), 6.43 (d, J=9.1 Hz, 1 H), 6.77 (dd, J=8.8, 2.0 Hz, 1 H), 6.81 (s, 1 H), 7.18 (d, J=2.0 Hz, 1 H), 7.24 - 7.35 (m, 10 H), 7.41 (d, J=8.6 Hz, 3 H), 7.49 (t, J=8.3 Hz, 1 H), 7.60 - 7.77 (m, 2 H), 7.88 (d, J=8.6 Hz, 2 H)。
步骤16:通过与1N NaOH (5 equiv)和足量的MeOH一起在THF (0.07M)中搅拌,水解生成的磺酰胺酯(220 mg, 0.26 mmol),以产生澄清的溶液。反应通过TLC (10% MeOH-CH2Cl2)监测起始原料的消失。当反应完成时,浓缩混合物,用H2O稀释,并使用1M HCl酸化至pH 2-4。含水相用EtOAc提取和有机相用盐水洗涤,经硫酸钠干燥,并浓缩以提供200 mg (92%)的3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸,白色泡沫。1H NMR (400 MHz, DMSO-d6) δ 2.65 (t, J=7.6 Hz, 2 H), 2.91 - 3.13 (m, 8 H), 3.60 (dd, J=9.7, 5.4 Hz1 2 H), 4.46 (s, 2 H), 6.48 (d, J=8.8 Hz, 1 H), 6.83 (dd, J=8.7, 2.1 Hz, 1 H), 7.05 - 7.16 (m, 5 H), 7.19 (d, J=2.3 Hz, 1 H), 7.33 - 7.47 (m, 6 H), 7.53 - 7.72 (m, 6 H), 7.80 (d, J=7.6 Hz, 1 H), 7.94 (d, J=8.3 Hz, 2 H), 12.26 (s, 1 H);HRMS: C42H38ClF3N2O6S2 + H+的计算值, 823.18847;实测值(ESI-FTMS, [M+H]1+), 823.1887;HPLC纯度H2O/CH3CN: 100 %, H2OMeOH: 100 %。
实施例2 体外药物渗入和渗透实验的“概念证明”
通常地,体外皮肤渗透实验涉及使用设计模拟皮肤原位的生理学和解剖学条件的扩散池。用于这种实验的模型是弗兰兹扩散池(Franz diffusion cell) (Finnin等,局部和透皮药物传递(Topical and Transdermal Drug Delivery) 2012: 85-100, Benson和Watkinson (编辑),通过引用结合到本文中)。如下所述制备的皮肤被定位于带有面向允许药物应用的供体室的角质层(Stratum corneum)的两半池之间。所述工作的目的是比较当应用时从不同的制剂/溶剂系统渗入和穿过经植皮刀分层的人类皮肤的药物浓度。在可能时,该方法根据OECD 428指导进行。
皮肤准备
使用来自美容整形手术的人类皮肤。机械切除皮下脂肪并使用谱瓦格刀具(Nouvag cutter)将皮肤分层切割成400 ± 100 µm的厚度。
小规模皮肤渗透研究
可行性实验如下进行:
弗兰兹池的制备
(i) 使用弗兰兹扩散池,其具有大约0.6 cm²的表面积和大约2.0 mL的体积。
(ii) 将皮肤(如上所述准备)固定于供体和接受室之间,而池使用封口膜(Parafilm)和夹子密封在一起。
(iii) 为检查皮肤的完整性,使供体和接受室充满PBS溶液(通过将1粒PBS片溶于100 mL水中制备)并将一个小的磁随器放置在接受室中。
(iv) 使池在确保膜温度在32℃的水浴中平衡30 min (水浴温度37℃)。
(v) 在每个弗兰兹池中的皮肤电阻使用LCR 6401数据桥测量。
(vi) 通过采样臂,将电极放置在接受室和供体室中。
(vii) LCR设置在100 Hz并设定为‘R’电阻。
(viii) 具有在可接受的限度以下的电阻的池被弃去并重新安装。可接受的限度根据植皮刀切下的皮肤对照的测量限定,其中皮肤已被故意地干扰。具有对照品电阻(KΩ)两倍以上的池被认为是可接受的并被选择用于弗兰兹池渗透实验。
(ix) 按照皮肤完整性试验,可接受池的接受室填充有接收液。然后在给药之前(水浴温度37℃),每个池被平衡以确保表面温度为32℃ (皮肤外表面温度)至少30 min。
(x) 也固定另外的弗兰兹池(每个皮肤供体),但不给药(以用作空白对照)以评估样品定量的干扰。
试验溶剂系统的给予和采样程序
选择两个溶剂系统用于小规模体外药物渗透研究并如下所述给予:
(i) 如上所述组装池。
(ii) 组装9个池。两个溶剂系统(n=3)、各自的安慰剂溶剂系统(n=1)和空白池(n=1),在可行性实验期间被评价。
(iii) 用一个正吸液管(导致约10 mg/cm2的剂量),将选择的溶剂系统(6 mg)直接应用于皮肤表面。在给予前,验证吸液管分配的量,以确保剂量的可重复性。分配的量的验证通过将6个复制品称重到玻璃小瓶中进行。记录所应用的重量和体积(以确保6 mg剂量)。
(iv) 在以下时间点t=0、1、2、4、6、24、30和48 h除去接收液(200 µL)。每份样品被分配到2 x LCMS小瓶(每小瓶100 µL),其中一个被掺入内部标准品(1:1)并使用LC-MS/MS分析。第二个小瓶于-20℃贮存作为备份。
(v) 使用新鲜的预热接收液(200 µL)以替换在每个时间点除去的接收液。
(vi) 在48 h时间点后,化合物I按如下所述从弗兰兹池回收。
渗透调查
在渗透实验完成后,在皮肤表面、角质层、表皮和部分真皮上的来自供体室残留的化合物I的定量使用以下程序进行:
从供体室回收
(i) 使用3个棉花拭子以从供体室回收化合物I。
(ii) 在拆解弗兰兹池后,使用一个干棉花拭子以从供体室的所有内表面除去所有残留的溶剂,并将所述拭子置于7 mL小瓶中。
(iii) 然后将第二个拭子浸入提取液中,然后用来擦拭弗兰兹池的内表面,然后将这种拭子置于含有第一个拭子的小瓶中。
(iv) 使用最后的干燥拭子以擦拭弗兰兹池内部,然后置于含有两个其它拭子的玻璃小瓶中。
(v) 使用吸液管将2 mL提取液加入到含有棉花拭子的玻璃小瓶中。
(vi) 在环境温度下,在轨道震摇器上震摇小瓶16-20 h。
(vii) 按照提取程序,从小瓶除去提取溶剂系统并使用Heraeus Labofuge微型离心机(Heraeus Labofuge pico centrifuge),以13,000 RPM (16,060 g-力)离心10 min,以去除未溶解的物质和微粒。
(viii) 将上清液分成2等份(100 µL在小瓶1中,而剩余的在第二个小瓶中),其中小瓶1掺入内部标准品(1:1)并使用LC-MS/MS分析。第二个小瓶于-20℃贮存作为备份。
从皮肤上表面回收
(i) 使用3个棉花拭子以从皮肤表面回收化合物I。
(ii) 在从弗兰兹池拆除供体室后,使用一个干燥棉花拭子以从皮肤表面除去所有残留的溶剂并将拭子置于7 mL小瓶中。
(iii) 然后,将第二个拭子浸没在提取溶剂中并用来擦拭皮肤表面,然后,将这种拭子置于含有洗涤溶剂和第一个拭子的小瓶中。
(iv) 使用最后的干燥拭子以擦拭皮肤表面,然后置于含有两个其它拭子的玻璃小瓶中。
(v) 来自皮肤表面的初始带条也与来自步骤(iv)的棉花拭子放置在一起。
进行从供体室回收的方法的步骤(v)-(viii)以完成从皮肤表面回收化合物I。
从角质层回收
(i) 从皮肤表面除去至多5个带条以从表皮分离角质层并置于7 mL小瓶中。
(ii) 进行从供体室回收的方法的步骤(v)-(viii)以完成从角质层回收化合物I。
从表皮膜和部分真皮分离和回收的方法
如下处理剩余的表皮和部分真皮:
(i) 将表皮和真皮置于60℃的恒温箱中2 min。
(ii) 然后从恒温箱取出表皮和真皮并使用戴着手套的手人工分离。
(iii) 将表皮和真皮层置于各Precellys 24匀浆器小瓶中并加入1 mL提取溶剂。
(iv) 将来自步骤(iii)的Precellys小瓶置于Precellys 24中并于2-8℃将内容物以5,800 RPM匀化2 x 20 s。
(v) 来自步骤(iv)的Precellys小瓶的内容物被清空到一个7 mL玻璃小瓶中。
(vi) 将提取稀释液(1 mL)加入到步骤(v)的空Precellys小瓶中并将小瓶涡流混合约30 s,然后将内容物清空到来自步骤(v)的玻璃小瓶中
(vii) 进行从供体室回收的方法的步骤(vi)-(viii),以完成从表皮和真皮对化合物I的回收。
全量渗入和渗透研究
全量体外药物渗入和渗透实验如关于小规模实验所述进行,但有以下修改:
(i) 研究了含有接近于饱和的化合物I的两个溶剂系统:一个含有渗透促进剂(n=4活性剂,n=1安慰剂)和一个无渗透促进剂(n=4活性剂,n=1安慰剂)
(ii) 使用得自3个供体的皮肤,即总共有33个池(n=12个池/每活性剂,n=3个池/每安慰剂每制剂和n=3个池作为空白剂/每皮肤供体)。
数据分析
在体外皮肤渗透实验期间生成的样品的数据分析和统计学比较如下进行:
(i) 在每个样品中检测的化合物I的浓度(ng/mL)从在相同时间,在LC-MS/MS上分析的校准标准来定量。
(ii) 计算采样的每体积的化合物I的总量(ng) (总量/采样的体积 = ng/mL x 采样的体积)。
(iii) 然后计算在每个时间点回收的化合物I的总量(ng) (总量= ng/mL x 每个弗兰兹池的总体积)。
(iv) 化合物I的累积量(ng)通过将每个时间点的总量(ng, 步骤(iii))与从先前的每个时间点(步骤(ii))收回的总量(ng)相加来计算。
(v) 累积量/每单位面积化合物I (ng/cm2)通过将累积量(ng, 步骤(iv))除以扩散面积(ng/cm2 = 累积量(ng) /扩散面积)来计算。
(vi) 根据内部程序排除任何异常值,但报告所有的数据。
从在接收液中随时间的推移测量的化合物I浓度和从真皮回收的浓度分别计算通量率(在可能时)和渗透。
异常值的鉴定
渗入/渗透实验中的疑似异常值被证实并从进一步的数据分析中排除,如果它们的渗透数据超过了以下定义的限制的话:
平均值 + (2*标准差) < 异常值 < 平均值 – (2*标准差)
统计学分析
使用SPSS对来自供体、表面、角质层、表皮、真皮和接收液的渗透和回收数据进行统计学分析。使用Levene's检验分析回收数据,以证实每个组的方差是否相等。如果数据具有相等或不相等的方差,确定的p值分别是或者p > 0.05或者p ≤ 0.05。随后,采用独立的T-检验,假定相等(即p > 0.05)或不相等的方差(即p < 0.05),并对从皮肤基质回收的化合物I的量进行统计学比较。为归一化,使用Shapiro-Wilk检验分析从相同制剂的角质层、表皮和真皮回收化合物I之间的比较,以确定数据是否为参数或者非参数的,其中确定的p值分别是p > 0.05或者p ≤ 0.05。如果数据是参数的(即正态分布,使用Shapiro-Wilk检验确定p > 0.05),统计学比较使用单向ANOVA Tukey’s HSD进行。然而,如果已确定数据是非-参数的(即使用Shapiro-Wilk检验确定不相等的方差p ≤ 0.05),数据的统计学比较使用单向ANOVA和事后Tamhane (Post Hoc Tamhane)进行。
结果
小规模渗入/渗透实验
小规模渗入实验使用以下程序进行:
使用来自单一供体的皮肤(经植皮刀分层的人类皮肤),评价在PEG-400 (SS1)中的化合物I和在PEG-400 (75 % w/w)和Transcutol P (25 % w/w) (SS2)中的化合物I,n=3个池/每溶剂系统
剂量:约6 mg (即10 mg/cm2)
在水中的EtOH (20 % v/v)被用作接收液
接收液样品在t=0、1、2、4、6、24、30和48 h时采集。由于化合物I在潜在的接收液中的低溶解性,研究了2个样品体积;200 µL和1.5 mL。
在最后的时间点后,使用组织均质和溶剂提取(稀释剂:90:10,EtOH:水),从供体室、表面、角质层、表皮和真皮回收化合物I。
采用小规模实验以证实用于全量渗入/渗透实验的方案。不管样品体积如何,在48 h实验的持续时间过程中在接收液中未检出化合物I。
为便于数据解释,在应用溶剂系统SS1和SS2后回收化合物I的值在图1中用在图2中除去的从表面回收的量来说明。对于每种应用的溶剂系统,总回收的化合物I范围在所应用剂量的从90.00至109.72 %。从维持在表面的溶剂系统回收最高量的化合物I。也以类似的量从角质层、表皮和真皮回收化合物I。
全量体外渗入和渗透实验
使用来自3个皮肤供体的腹部皮肤评价化合物I从两个溶剂系统的渗入和渗透。使在应用溶剂系统通过经植皮刀分层的皮肤后渗透的化合物I的平均累积量(ng/cm2)对时间(h)作图。溶剂系统是:在PEG-400 (FSS1)中的化合物I和在PEG-400 (60 % w/w)、Arlasolve DMI (15 % w/w)和Transcutol P (25 % w/w) (FSS2)中的化合物I。从所有3个皮肤供体呈现来自供体、表面、角质层、表皮、真皮和接收液的化合物I的量(ng,均值 + SEM)。
经48 h实验期,化合物I从2个溶剂系统(FSS1和FSS2),越过来自3个供体的经植皮刀分层的腹部皮肤的体外渗透在图3中说明。在24 h实验期间,化合物I从两个溶剂系统的渗透在低水平时被观察到,在应用溶剂系统(FSS1和FSS2)后的1小时后,在接收液中首次检测到化合物I。化合物I对于两个溶剂系统的渗透曲线,遵循存在于t=1 h接收液中的化合物I的类似趋势,接着是一个平稳高原水平状态(plateau),接近该曲线末段时的下降在误差内,并认为是一个被量化的低水平化合物和供体对供体变异性的结果。不管在接收液中检测的低水平的化合物I,在应用FSS1后化合物I的渗透被观察到显著地高于(p < 0.05)在应用FSS2后化合物I的渗透。化合物I从溶剂系统(FSS1和FSS2),越过来自3个供体的经植皮刀分层的腹部皮肤的体外渗透在图4和5中说明。在最后的时间点后化合物I的回收被观察到对于溶剂系统FSS1和FSS2分别是所应用剂量的101.47 %和87.39 %。在应用FSS1和FSS2后,以最高量从皮肤表面回收化合物I,回收在所应用剂量的约82 % (FFS2)-约97 % (FSS1)之间。
为便于数据解释,从表面回收的化合物I的量在图5中已被去除,以突出在溶剂系统(FSS1和FSS2)之间,从角质层、表皮、真皮和接收液回收的化合物I。在应用FSS1后回收的化合物I的量在表皮中是最高的(基于平均量),即表皮(3281 ± 1435 ng) > 角质层(1976 ± 729 ng) > 真皮(1593 ± 609 ng) > 接收液(4.67 ± 0.58 ng)。在应用FSS2后从皮肤各层和接收液回收的化合物I的量是较低的(虽然在皮肤各层之间未观察到显著的差异,p > 0.05,从接收液中回收的化合物I的接受量显著地低于(p < 0.05))在应用FSS1后回收的量。从皮肤基质回收的化合物I的排序不同于用FSS1观察到的,从真皮回收的化合物I的最高量(基于平均量) (1126 ± 543 ng) > 角质层(931 ± 310 ng) > 表皮(352 ± 113 ng) > 接收液(0.89 ± 0.27 ng)。然而,排序的差异并不认为是一个问题,因为在角质层、表皮和真皮回收的水平对于各溶剂系统没有显著的不同(p > 0.05)。
在表皮和真皮内的化合物I的大约浓度使用以下程序计算:
a. 表皮和部分真皮的厚度被假定分别在30-130和250-350 µm之间(一系列的皮肤厚度被用来提供在表皮中检测的化合物I的水平最好的和最坏的情况)。
b. 所用皮肤的面积是约0.6 cm2,因而表皮和部分真皮的体积分别是0.0018-0.0078和0.015-0.021 cm3。
c. 假定皮肤的密度是1和1 cm3 = 1 g,表皮和部分真皮的重量将分别是0.0018-0.0078和0.015-0.021 g。
d. 然后将从表皮和部分真皮回收的化合物I的总量除以所述质量,得到和接近回收的化合物I的量(ng/g)。
e. 接着将回收的量(ng/g)除以分子量(823.34 Da),以提供纳摩尔/g的浓度,随后将其转化为从表皮和真皮回收的化合物I的微摩尔/kg (重量微摩尔浓度(micromolality)) (相当于当假定皮肤的密度是1 g/cm3时的重量微摩尔浓度(µmol/L;µM))。
计算的结果概述于表2中。化合物I是一种在细胞和酶分析中的cPLA2α活性的亚低纳摩尔浓度抑制剂。在表皮和真皮中检测的浓度表明它们在这些位点足以显著地抑制cPLA2α生物学活性。
表2. 在应用FSS1和FSS2后,在48 h时间点化合物I (µM)在表皮和真皮中的浓度范围(基于平均量和皮肤厚度的范围)。所述值表示基于皮肤厚度和回收的平均量的范围,n=9-10。
实施例3 – 对小鼠中PMA-诱导的炎症的作用
佛波醇12-十四酸酯13-乙酸酯(PMA)通常被用来诱导作为临床前炎症模型的实验动物的皮肤炎症和水肿。
在本实施例中,使PMA溶于10%乙醇溶液至0.4 µg每µl的最终浓度。在以0.3 %或者3 % w/w局部应用3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸(化合物I)的10%乙醇溶液后1小时,将10 µl应用于Balb/C实验小鼠的耳朵。作为阴性对照,应用无活性化合物的媒介物。阳性对照是吲哚美辛(一种非-甾体抗-炎药物)和CAY10650 (一种已知的cPLA2α抑制剂)。
水肿的水平通过在1、3、6和24小时的间隔,用卡尺测量耳朵厚度来监测。
图6显示对于不同剂量的3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸(称为化合物I),耳朵厚度随时间推移(在PMA应用后小时数)的变化。
图7显示3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸(称为化合物I)与其它化合物的比较。
已发现局部应用的3-{4-[2-{5-氯-1-(二苯基甲基)-2-[2-({[2-(三氟甲基)苄基]磺酰基}氨基)乙基]-1H-吲哚-3-基}乙基]磺酰基}苯基}丙酸显示PMA-介导的皮肤水肿的剂量-依赖性减少,其效果与口服给予的吲哚美辛相当。
这证实该药物当局部给予时在体内是有效的。
实施例4 – PEG基软膏的制备
如在表3中描述的几种软膏剂根据以下程序制备:
(i) 将PEG-400称重到合适大小的玻璃容器中。
(ii) 将化合物I称重(在适用时)到重量船形器皿中,然后转移到来自步骤(i)的系统中并搅拌直至观察到完全溶解。
(iii) 将以下赋形剂(适用时),丙二醇、乙醇、水和丙三醇顺序称重到来自步骤(ii)的容器中,插入磁随器并搅拌内容物直至溶液被观察到视觉上的同质化。
(iv) 将PEG 400称重到一个独立的合适大小的玻璃容器中并在先前已校准于65℃的水浴中加热。
(v) 搅拌PEG 400直至观察到一个透明的熔体,此时将其加入到已加热至61℃的溶剂系统(步骤(iii))中。
(vi) 搅拌制剂(步骤(iv))直至目测到混合,然后从水浴移出。
(vii) 混合制剂直至它达到环境温度。
表3. 软膏剂制剂的组成(% w/w)
实施例5 – 霜剂和洗剂制剂的制备
如在表4、5和6中描述的霜剂和洗剂制剂如下制备:
(i) 将PEG-400称重到合适大小的玻璃容器中。
(ii) 将化合物I称重(在适用时)到重量船形器皿中,然后转移到来自步骤(i)的系统中并搅拌过夜。
(iii) 将剩余的含水相赋形剂称重到来自步骤(ii)的容器中并搅拌内容物。
(iv) 将胶凝剂(如果适用时)加入到来自步骤(iii)的小瓶中并匀化30 s或直至观察到胶凝剂完全分散。
(v) 在一个独立的合适大小的玻璃容器中,称重油相赋形剂并将油相在先前已校准于65℃的水浴中加热。
(vi) 搅拌油相直至观察到一个透明的熔体。
(vii) 当来自步骤(iii)的油相的一个透明的熔体被观察到时,将来自步骤(v)的含水相在先前已校准于61℃的水浴中预热最多5 min。
(viii) 将油相缓慢转移到含水相中并将来自步骤(vii)的制剂在最大速度设置(10,000 RPM)上匀化2 min。在匀化制剂前,预热匀浆器头。
(ix) 然后通过手动搅拌制剂直至它冷却至室温。
表4:实施例吐温/司盘霜剂
表5:霜剂(聚西托醇)制剂的实施例
表6:实施例洗剂制剂
实施例6 – 凝胶剂制剂的制备
如在表7中描述的几种凝胶剂制剂如下制备:
凝胶剂制剂
(i) 将PEG-400称重到一合适大小的玻璃容器中。
(ii) 将化合物I称重(在适用时)到重量船形器皿中,然后转移到来自步骤(i)的系统中并搅拌直至观察到完全溶解。
(iii) 将以下赋形剂(乙醇、水、丙二醇、Transcutol P、Arlasolve DMI、苄醇和苯氧基乙醇,当适用时)顺序称重到来自步骤(ii)的玻璃小瓶中。
(iv) 将一个磁随器插入来自步骤(iii)的小瓶中并搅拌小瓶中的内容物直至目测到同质化。
(v) 在不停的搅拌下,将胶凝剂(HPC/卡波姆980)称重到重量船形器皿中并缓慢转移到(减轻胶凝剂的聚集风险)来自步骤(iv)的小瓶中。
(vi) 对于卡波姆基凝胶剂,核查pH以确保其约为pH 6,其中pH调节将不是必要的。
(vii) 搅拌制剂直至观察到胶凝剂完全水合。
乳化凝胶剂制剂
(i) 将PEG-400称重到合适大小的玻璃容器中。
(ii) 将化合物I称重(在适用时)到重量船形器皿中,然后转移到来自步骤(i)的系统中并搅拌直至观察到完全溶解。
(iii) 将以下赋形剂(乙醇、水、丙二醇、Transcutol P、Arlasolve DMI、苄醇、苯氧基乙醇和泊洛沙姆407,在适用时)顺序称重到来自步骤(ii)的玻璃小瓶中。
(iv) 将一个磁随器插入来自步骤(iii)的小瓶中并搅拌小瓶中的内容物直至目测到同质化。
(v) 在不停的搅拌下,将胶凝剂(卡波姆980)称重到重量船形器皿中并缓慢转移到(减轻聚集形成的风险)来自步骤(iv)的小瓶中。
(vi) 核查pH以确保其约为pH 6,其中pH调节将不是必要的。
(vii) 将环甲硅油称重到来自步骤(v)的玻璃小瓶中并将系统以10,000 RPM匀化2 min,直至观察到环甲硅油完全分散。
(viii) 随后搅拌制剂直至观察到胶凝剂完全水合。
表7:实施例凝胶剂制剂
虽然已经详细并参照其具体实施方案描述了本发明,但对本领域技术人员显而易见的是,可在本文进行各种改变和修饰,而不背离其精神和范围。此外,本文描述的所有实施方案被认为是广泛适用的,并在合适时可与任何和所有其它一致的实施方案组合。
- 还没有人留言评论。精彩留言会获得点赞!