布洛芬立方液晶前体溶液、立方液晶纳米粒及其制备方法与流程
本发明涉及药物制剂领域,特别是涉及一种布洛芬立方液晶前体溶液、立方液晶纳米粒及其制备方法。
背景技术:
布洛芬(ibuprofen,IBU)是一种非甾体类抗炎药物,具有很好的解热镇痛及抗炎效果。用于缓解轻至中度疼痛如头痛、关节痛、偏头痛、牙痛、肌肉痛、神经痛、痛经。也用于普通感冒或流行性感冒引起的发热。
布洛芬属于生物药剂学分类系统的Ⅱ类药物,溶解度低、渗透性高。药物的溶出是其吸收的限速阶段,严重影响了药物的口服生物利用度。此外,布洛芬生物半衰期短(~1.8h),为保持有效的治疗浓度,患者需频繁大量服药,这会对胃肠道有一定的刺激性。溶解度差和半衰期短严重地限制了布洛芬的临床应用。因此,有必要研发新的给药剂型克服布洛芬在口服吸收过程中溶解度差的问题,以提高布洛芬口服的生物利用度。
技术实现要素:
基于此,本发明提供了一种布洛芬立方液晶前体溶液,用于制备用于口服的布洛芬立方液晶纳米粒,以克服布洛芬口服给药过程中溶解度低,体内半衰期短的缺点,以提高布洛芬口服的生物利用度。
具体技术方案如下:
一种布洛芬立方液晶前体溶液,主要由布洛芬、液晶材料和稳定剂制备而成,所述布洛芬、液晶材料和稳定剂的质量比为0.01-2:10:0.5-2.5;所述液晶材料为植烷三醇;所述稳定剂选自脂肪酸甘油酯、多元醇型非离子表面活性剂、聚氧乙烯型非离子表面活性剂、泊洛沙姆和卵磷脂中的至少一种。
在其中一些实施例中,所述布洛芬、液晶材料和稳定剂的质量比为0.4-1.6:10:0.8-2。
在其中一些实施例中,所述稳定剂选自泊洛沙姆。
在其中一些实施例中,所述泊洛沙姆选自泊洛沙姆407、泊洛萨姆188、泊洛萨姆338中的至少一种。
在其中一些实施例中,所述布洛芬立方液晶前体溶液,主要由布洛芬、植烷三醇和泊洛沙姆407制备而成,所述布洛芬、植烷三醇和泊洛沙姆407的质量比为1-1.4:10:0.8-1.2。
本发明还提供了上述布洛芬立方液晶前体溶液的制备方法。
具体技术方案如下:
一种上述的布洛芬立方液晶前体溶液的制备方法,包括以下步骤:将布洛芬、稳定剂和植烷三醇于40-70℃加热熔融,涡旋,充分混合均匀,室温下静置,即得所述布洛芬立方液晶前体溶液。
本发明还提供了上述布洛芬立方液晶前体溶液的应用。
具体技术方案如下:
上述的布洛芬立方液晶前体溶液在制备布洛芬立方液晶纳米粒中的应用。
本发明还提供了一种布洛芬立方液晶纳米粒,可显著提高布洛芬口服的生物利用度。
具体技术方案如下:
一种布洛芬立方液晶纳米粒,主要由上述的布洛芬立方液晶前体溶液分散在水中制备而成。
本发明还提供了上述布洛芬立方液晶纳米粒的制备方法。
具体技术方案如下:
一种上述的布洛芬立方液晶纳米粒的制备方法,包括以下步骤:
在布洛芬立方液晶前体溶液中加水,用超声波粉碎仪进行超声分散,得布洛芬立方液晶粗分散溶液;
将布洛芬立方液晶粗分散溶液进行高压均质,均质压力为600-1500bar,循环次数为3-13次,即得布洛芬立方液晶纳米粒溶液。
在其中一些实施例中,所述均质压力为1100-1300bar,循环次数为8-10次。
在其中一些实施例中,所述水的加入量为布洛芬立方液晶前体溶液总重量的20-50倍。
在其中一些实施例中,所述超声分散的条件为:分散时间为10-15min,功率为150-400w,工作时间为4-6s,间断时间为8-12s。
在其中一些实施例中,所述布洛芬立方液晶纳米粒的制备方法还包括以下步骤:在所述布洛芬立方液晶纳米粒溶液中加入冻干保护剂,充分溶解后,进行冷冻干燥,即得布洛芬立方液晶纳米粒的冻干粉末。
在其中一些实施例中,所述冻干保护剂的加入量与立方液晶纳米粒溶液的质量体积比为:0.04-0.06g:1mL。
在其中一些实施例中,所述冻干保护剂选自甘露醇、蔗糖、葡糖糖或乳糖。
在其中一些实施例中,所述冷冻干燥包括:先于-18~-22℃的冰箱中预冻10-14h,再于冷冻干燥机中冻干22-26h,冷冻干燥机的条件为:-48~52℃冷阱温度,真空度为0.08-0.12Mbar。
立方液晶是一种新型脂质药物载体,是由两亲性脂质分散在水性环境中自发形成各种几何形态构成的一个中介相形态(Mesophase)。因其独特的三维网络结构而具有良好的药物传递性能。其三维网络结构主要由亲水域中的双连续水通道和亲脂域中的脂质双分子层构成的晶格单元在空间上延伸折叠,堆叠成具有三维,循环排列和最小表面积特点的紧密结构。难溶性药物具有较强的疏水性,与脂质结构极性相近,在亲脂域中具有较高的溶解度。
本发明的布洛芬立方液晶前体溶液、立方液晶纳米粒及其制备方法具有以下优点和有益效果:
本发明的发明人通过大量实验研究发现将布洛芬与特定的液晶材料以及稳定剂以特定比例配合制备的立方液晶前体溶液,可用于制备药物包封率高、粒径较小且分布均匀的布洛芬立方液晶纳米粒。立方液晶纳米粒是一种新型纳米脂质药物载体,其巨大的膜表面积和独特的三维晶格结构对难溶的药物具有良好的增溶效果和缓释性能。因而本发明的布洛芬立方液晶纳米粒能显著提高布洛芬的口服生物利用度。
药物颗粒大小,药物与溶出介质的接触面积,以及溶出层与介质之间的药物浓度差是影响难溶性药物溶出度的关键参数。本发明的布洛芬立方液晶纳米粒,其粒径较小(小于250nm),且粒径均一性好,有利于肠上皮细胞内吞并转运到细胞内;布洛芬包载进液晶结构中后,由晶体颗粒转变成无定型的分子态,以稳定的无定型的分子态分散在晶格单元的脂质双分子层中,均匀分布于亲脂域中,双连续水通道与脂质双分子层的巨大交界面积显著增加了布洛芬的溶出表面积;同时布洛芬水溶性差疏水性强,在亲脂域中有较大的溶解度,在溶出层和介质之间形成较大的药物浓度差,可进一步增大布洛芬的溶出度;此外,布洛芬立方液晶纳米粒三维网络结构中的脂质双分子层,具有类细胞结构,与细胞有很强的亲和性,具有良好的生物相容性,可以显著增加立方液晶纳米粒对胃肠道上皮细胞的黏附性和穿透能力,延长载布洛芬的纳米粒在胃肠道的滞留时间并促进载药纳米粒在胃肠道中的跨膜转运吸收,改善布洛芬在体内的循环和分布。因此,本发明的布洛芬立方液晶纳米粒能够克服布洛芬口服给药过程中的溶解障碍,促进药物在胃肠道的黏附和跨膜转运,且能缓慢释放药物,延长药物释放时间,延长布洛芬在体内的半衰期,改善药物在体内的循环分布,有效提高布洛芬的口服生物利用度。
本发明的布洛芬立方液晶纳米粒,能够有效的将难溶性药物布洛芬包埋于液晶结构中,药物的包封率高,克服了其口服生物利用度低的缺点,同时纳米粒溶液也有利于口服,能够提高患者的顺应性。
本发明的布洛芬立方液晶纳米粒显著提高了布洛芬的口服相对生物利用度,同时能使布洛芬缓慢释放,具有24小时缓释的效果,为布洛芬提供了一种能够缓慢释放、吸收良好、生物利用度高的布洛芬新型口服制剂。
本发明的布洛芬立方液晶纳米粒的制备方法工艺简单,包封率高,使制备得到的布洛芬立方液晶纳米粒粒径小且分布均匀,生产成本较低,有利于工业化生产,具有良好的应用前景,同时有利于推进液晶纳米的工业化发展。
附图说明
图1为实施例1中超声分散后未经高压均质的布洛芬立方液晶粗分散溶液中纳米粒的粒径;
图2为实施例1中高压均质后制备的布洛芬立方液晶纳米粒的粒径;
图3为实施例1中布洛芬含量对立方液晶纳米粒粒径的影响;
图4为实施例1中布洛芬含量对立方液晶纳米粒的包封率的影响;
图5为实施例2中F127和F68对布洛芬立方液晶纳米粒粒径的影响;其中A为PYT:F127=10:1均质后第一天,B为PYT:F127=10:1均质后第3天,C为PYT:F68=10:1均质后第1天,D为PYT:F68=10:1均质后第3天;
图6为实施例5中布洛芬立方液晶纳米粒的粒径图;
图7为实施例5中布洛芬立方液晶纳米粒的形态表征图;其中A为原子力学显微镜图,B为冷冻透射电镜图;
图8为实施例6中的DSC图;其中a为布洛芬原料药,b为布洛芬和空白立方液晶纳米粒的物理混合物,c为布洛芬立方液晶纳米粒,d为空白立方液晶纳米粒;
图9为实施例7中布洛芬立方液晶纳米粒的粒径和包封率;
图10为实施例8中布洛芬原料药和布洛芬立方液晶纳米粒的在pH 1.2(A)和pH 7.4(B)的体外释放曲线;
图11为实施例9中布洛芬原料药和立方液晶纳米粒在比格犬灌胃给药后的平均血药浓度-时间变化曲线。
具体实施方式
以下通过具体实施例以及附图对本发明的布洛芬立方液晶前体溶液、立方液晶纳米粒及其制备方法做进一步详细的说明。
实施例1
本实施例的布洛芬立方液晶纳米粒的制备方法,包括如下步骤:
按表1所示的原料处方称取布洛芬、F127和植烷三醇置于100mL塑料离心管中,于水浴60℃加热熔融,涡旋,充分混合均匀,室温静止放置2天,即得布洛芬立方液晶前体溶液。
在布洛芬立方液晶前体溶液中加入30mL水,用超声波粉碎仪进行超声分散,时间为10min,所用超声功率为200W,超声工作时间5s,间断时间10s,得布洛芬立方液晶粗分散溶液,该粗分散溶液中的粒子粒径分布不均匀,而且有微米级的粒子(见图1)。
将布洛芬立方液晶粗分散溶液进行高压均质(均质压力为1200bar,循环次数为9次),高压均质后可以使纳米粒粒径减小,得到粒径分布均匀的立方液晶纳米粒(见图2),即得布洛芬立方液晶纳米粒溶液。
表1布洛芬立方液晶前体溶液的原料处方
测定本实施例制备的布洛芬立方液晶纳米粒的粒径以及包封率。
粒径测定方法如下:用超纯水稀释立方液晶纳米粒后,取1mL纳米粒溶液加入到样品池中,25℃平衡2min,采用粒径分析仪Zetasizer Nano ZS90测定立方液晶纳米粒的粒径和多分散系数(PDI)。
包封率的测定采用超滤离心法。精密移取所制备布洛芬立方液晶纳米粒溶液0.5mL于超滤离心管的内管中,4000r/min离心20min,收集外管中游离的布洛芬溶液,用甲醇洗涤后,并用甲醇定容至10mL容量瓶中,采用高效液相色谱方法测定游离的布洛芬含量Cfree。同时,精密移取载布洛芬的立方液晶纳米粒溶液0.5mL于10mL容量瓶中,加入甲醇破乳并定容,同法测定立方液晶纳米粒总药量Ctotal。使用下列包封率计算公式,计算立方液晶纳米粒的包封率。
高效液相色谱条件如下,色谱柱:Odyssil C18(250×4.6mm,5μm);流动相:甲醇:0.01mol·L-1磷酸二氢钾:磷酸=400:100:0.05,(v/v);柱温:35℃;检测波长:263nm;流速:1.0mL·min-1;进样量:20μL。
粒径和包封率结果分别见图3和图4。布洛芬立方液晶纳米粒的粒径分别为235.1±3.7nm、231.26±6.9nm、237.5±2.8nm、238.5±5.1nm,287.4±6.1nm,结果表明在一定范围内随着布洛芬与植烷三醇的质量比增大,布洛芬立方液晶纳米粒的粒径未发生明显增大,但当载药量超过一定比例时(2.5:10),粒径骤然增大。同时,随着布洛芬与植烷三醇的质量比增大,布洛芬立方液晶纳米粒的包封率也稍有增加,当布洛芬与植烷三醇的质量比为16:100时,包封率略有下降,当比例增加至25:100时,包封率的下降已经较为明显,这表明已经达到了纳米粒载药的上限。综合考虑,布洛芬与植烷三醇的优选质量比为12:100。
实施例2
以表2所示原料处方,按实施例1的制备方法制备稳定剂种类和用量不同的布洛芬立方液晶纳米粒溶液。
测定本实施例制备的布洛芬立方液晶纳米粒的粒径,测定方法同实施例1,并通过对比第1天和第3天布洛芬立方液晶纳米粒的粒径以及PDI判断其稳定性。结果见图5、表2和表3。由结果可知,在缺乏稳定剂的情况下,纳米粒容易发生聚集,粒径增大;不同用量的F68(泊洛萨姆188)制备的布洛芬立方液晶粗分散溶液效果欠佳,易团聚;而不同用量的F127制备的布洛芬立方液晶粗分散溶液无团聚、易于分散,当PYT:F127为10:1时,制备的布洛芬立方液晶纳米粒的粒径小、分布均匀而且稳定,放置3天后,均未出现聚集现象,PYT:F127的比例过小难以起到稳定剂的作用,过大则自身容易破坏立方液晶中不连续的双水通道以及封闭的脂质双层结构。由于立方液晶纳米粒的粒径大小是影响口服吸收的重要参数,它影响着粒子在肠上皮细胞的摄取,从而会导致不同的口服吸收效果,主要因为粒径小的粒子能被肠上皮细胞内吞转运到细胞内,而粒径大的粒子不易被细胞内吞,因此优选F127为最佳稳定剂,最佳比例为F127:PYT=1:10。
表2稳定剂对布洛芬立方液晶粗分散溶液的影响
表3稳定剂对立方液晶纳米粒的影响
实施例3
按质量比12:100:10称取布洛芬、植烷三醇和F127,按实施例1的方法制备布洛芬立方液晶纳米粒,区别在于所采用均质压力分别为670bar,800bar,1200bar和1500bar;均质次数分别为3,5和9次。
测定本实施例制备的布洛芬立方液晶纳米粒,测定方法同实施例1。结果见表4。结果表明,不同的均质压力和次数的条件下,布洛芬立方液晶纳米粒都具有较小的粒径和PDI,其中,当均质压力为1200bar,均质次数为9次时,粒径最小。因此,制备布洛芬立方液晶纳米粒的高压均质的最佳条件为:均质压力1200bar,均质循环次数9次。
表4布洛芬立方液晶纳米粒的粒径(n=3)
实施例4:布洛芬立方液晶纳米粒冻干粉的制备
按质量比12:100:10称取布洛芬、植烷三醇和F127,按实施例1的方法制备布洛芬立方液晶纳米粒溶液,取立方液晶纳米粒溶液2mL,加入0.1g冻干保护剂(甘露醇),充分溶解后,放入-20℃冰箱预冻12h,然后放置冷冻干燥机中-50℃冷阱温度,真空度为0.1Mbar条件下冻干24h,即得布洛芬立方液晶纳米粒冻干粉末。所得粉末表面光滑,色泽均匀,无花斑,重分散性好。
在其他实施例中,冻干保护剂还可以是蔗糖、葡糖糖或乳糖。
实施例5:布洛芬立方液晶纳米粒的形态与粒径表征
按质量比12:100:10称取布洛芬、植烷三醇和F127,按实施例1的方法制备布洛芬立方液晶纳米粒溶液,采用Zetasizer Nano ZS90测制备的布洛芬立方液晶纳米粒的粒径,并采用原子力学显微镜(Atomic force microscopy,AFM)和冷冻透射电镜(Cryo-TEM)表征其形态和粒子大小。
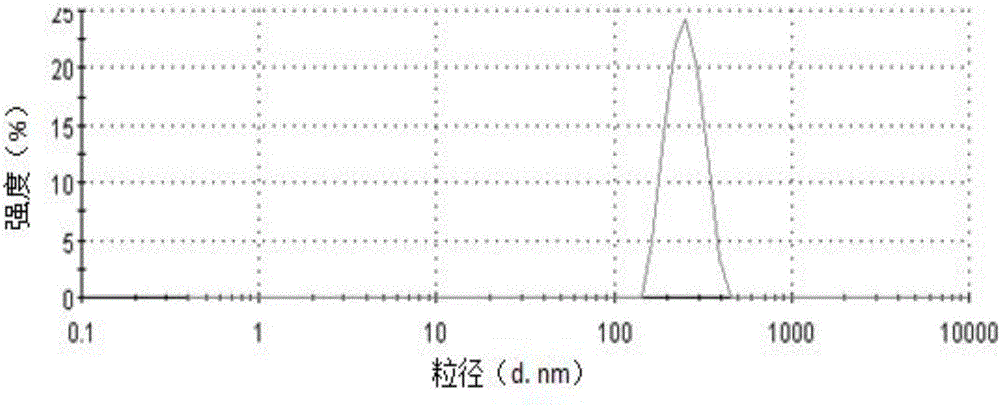
粒径测量方法同实施例1。结果(见图6)表明,其粒径为238.1±2.8nm,PDI为0.056±0.007。
原子力学显微镜表征方法:将布洛芬立方液晶纳米粒滴在干净的云母片上,孵化5min,然后用去离子水冲洗没有被吸附的纳米粒,自然晾干后用原子力学显微镜扫描成像、观察。结果(见图7A)表明,布洛芬立方液晶纳米粒呈球形或类球形,粒径大小分布均匀,与所测粒径分布基本一致。
冷冻透射电镜(Cryo-TEM)表征方法:将布洛芬立方液晶纳米粒溶液滴加到铜网上,直至立方晶纳米粒完全吸附在铜网上形成薄膜,用磷钨酸负染色,迅速放到液态乙烷中速冻,速冻后的铜网放置在冷阱,-172℃下透射电镜观察立方液晶纳米粒形态,CCD摄像机(Gatan 832)拍摄照片。结果(见图7B)表明,布洛芬立方液晶纳米粒平均粒径在230nm左右,与Zetasizer Nano ZS90测得粒径大小基本一致。大量有序的立方液晶纳米粒,呈现圆整的形状,显示纳米粒子具有典型的立方液晶结构。
实施例6:布洛芬在立方液晶纳米粒体系中的晶型测定
采用DSC表征布洛芬在立方液晶纳米粒中的存在状态,分别取适量(3-5mg)布洛芬原料药、空白立方液晶纳米粒冻干粉、空白立方液晶纳米粒冻干粉和药物的物理混合物及布洛芬立方液晶纳米粒冻干粉(实施例4)进行DSC分析,升温速度10℃/min,测量范围30-150℃,空铝盘为空白对照,炉内气体为氮气。
DSC结果见图8,布洛芬原料药在77.8℃有一个明显的熔融吸热峰;布洛芬原料药和空白立方液晶纳米粒物理混合物在77.8℃有一个明显的熔融吸热峰;而布洛芬立方液晶纳米粒在77.8℃的熔融吸热峰基本上消失,与空白立方液晶纳米粒图谱一样只显示载体熔融峰。DSC结果显示布洛芬在立方液晶纳米粒中可能以分子状态或无定形状态存在,也由此说明药物包封在立方液晶纳米粒中。由Noyes-Whitney方程可知,药物与溶出介质的接触面积是影响难溶性药物溶出速率的关键参数。药物包载进液晶结构中后,药物由晶体颗粒转变成无定型的分子态,均匀分布于亲脂域中,双连续水通道与脂质双分子层的巨大交界面积显著增加了药物的溶出表面积。因此,布洛芬以无定形态存在有利于其均匀分布于亲脂域中,增加与溶出介质的接触面积,提高生物利用度。
实施例7:布洛芬立方液晶纳米粒的稳定性
考察实施例5中所制备的布洛芬立方液晶纳米粒的稳定性。将布洛芬立方液晶纳米粒溶液在室温下放置2个月,每月取样一次,测定布洛芬立方液晶纳米粒的粒径和包封率。粒径测定方法同实施例1,包封率测定方法同实施例1。
布洛芬立方液晶纳米粒粒径和包封率结果见图9。布洛芬立方液晶纳米粒溶液在室温下放置2个月,外观观察呈乳白均匀溶液,未出现聚集、絮凝、结块,布洛芬立方液晶纳米粒的粒径和包封率基本没有变化。结果表明,本发明的布洛芬立方液晶纳米粒溶液具有良好的物理稳定性。
实施例8:布洛芬立方液晶纳米粒的释放行为
采用人工模拟胃液(pH1.2,不含酶)和肠液(pH7.4,不含酶)作为释放介质,研究布洛芬立方液晶纳米粒的释药行为。分别取实施例1处方3、实施例1处方5(投药量过大)、实施例2处方6(过量稳定剂)所制备的布洛芬立方液晶纳米粒溶液和布洛芬原料药溶液(药物溶于0.3%CMC-Na溶液中)置于透析袋(截留相对分子质量14000Da)中,分别编号为实验组1、对照组1、2和3,扎紧透析袋两端,浸入盛有250mL pH1.2模拟胃液(不含酶)中释放2h,pH7.4肠液(不含酶)中释放24h,释放条件为37±0.5℃,100rpm,于0.5、1.0、2.0、3.0、4.0、5.0、6.0、8.0、12.0和24.0h定时取样2mL,并补充等量等温释放介质。所取样品经0.45μm微孔滤膜过滤,采用HPLC(条件同实施例1)测定释放液中布洛芬的含量,计算不同时间药物的累积释放百分率,绘制累积释放曲线。
体外释放结果见图10,布洛芬立方液晶纳米粒相对布洛芬原料药具有明显的缓释作用。在人工模拟胃液(pH1.2,不含酶)中布洛芬原料药2h内释放了34.24%,而实验组1的布洛芬立方液晶纳米粒释放了3.22%(见图10A)。在人工模拟肠液(pH7.4,不含酶)中布洛芬原料药6h内释放了95%,而实验组1的布洛芬立方液晶纳米粒释放了56.46%,24h内实验组1的布洛芬立方液晶纳米粒释放了大约85%(见图10)。对照组2和3由于投药量过大、稳定剂含量过高,使得液晶纳米粒的结构有一定程度的破坏,释放效果没有实验组1的理想,前期有一定程度的突释,后期释放不完全,24小时释放小于70%。结果表明,本发明的布洛芬立方液晶纳米粒具有良好的缓释效果,具有良好的24h缓释作用,这是由布洛芬立方液晶纳米粒的独特脂质双层结构决定的。药物的释放是从脂质双层结构扩散到内水道层,这是药物释放的限速步骤,再从内水道层结构扩散到释放介质。
布洛芬立方液晶纳米粒的释放机制。分别以零级动力学方程、一级动力学方程和Higuchi方程(扩散方程)对实验组1的布洛芬立方液晶纳米粒溶液体外释放数据进行处理,得到回归方程见表5。按Higuchi方程拟合,相关系数(R2>0.99)大,拟合效果较好,表明布洛芬立方液晶纳米粒释放是以扩散为主的释药方式,药物分子从立方液晶纳米粒的独特脂质双层部位慢慢地扩散到立方液晶内水道层,再到释放介质。可能由于布洛芬脂溶性和脂质材料之间相容性好,导致药物释放更加缓慢。结果表明,相对于布洛芬原料药,布洛芬立方液晶纳米粒呈现明显的缓释效果,可显著延长药物在体内的滞留时间,提高布洛芬口服生物利用度。
表5布洛芬立方液晶纳米粒体外释放模型(n=3)
缩写:y,累积释放百分比;t,释放时间;R,相关系数.
实施例9:布洛芬立方液晶纳米粒的药代动力学实验
本实施例考察布洛芬立方液晶纳米粒的药代动力学。取健康的比格犬12只,给药前禁食12h,自由饮水。12只比格犬随机平均分成4组:第一组以15mg/kg剂量的布洛芬立方液晶纳米粒(处方组成和制备方法同实施例5)给予比格犬灌胃;第二组以15mg/kg剂量的布洛芬溶液(布洛芬溶于0.3%CMC-Na溶液中)给予比格犬灌胃作为对照组1;第三组给予实施例1处方5制备的布洛芬立方液晶纳米粒给予比格犬灌胃作为对照组2;第四组给予实施例2处方1制备的布洛芬立方液晶纳米粒给予比格犬灌胃作为对照组3。
分别在给药0.17、0.33、0.67、1.0、1.5、2.0、3.0、4.0、5.0、6.0、8.0、12.0和24.0h后从比格犬后肢静脉血管取血约3mL,全血置于涂有肝素的管中,5000rpm离心l0min分离血浆,-20℃保存;精密吸取空白血浆0.1mL于1.5mL离心管中,加入甲醇0.3mL,涡旋混合2min,12000rpm离心10min,取上清液20μL进样,HPLC测定。
HPLC条件,色谱柱:Odyssil C18(250×4.6mm,5μm);流动相:甲醇:0.01mol·L-1;磷酸二氢钾:磷酸=400:100:0.05,(v/v);柱温:35℃;检测波长:263nm;流速:1.0mL·min-1;进样量:20μL。
血药浓度数据采用3P87药代动力学软件包进行处理。布洛芬立方液晶纳米粒(IBU-NPs)的相对生物利用度按下式计算:
F=AUCIBU-NPs/AUCIBU×100%。
AUCIBU-NPs为布洛芬立方液晶纳米粒的药时曲线下面积;AUCIBU为布洛芬溶液的药时曲线下面积。
两种制剂均是以单室模型和权重为1/C2时拟合后与实际曲线最相符。模型的药物动力学参数和相对生物利用度结果见表6。其中,Cmax(血浆峰浓度)和Tmax(血浆浓度达峰时间)采用实测值,AUC(药时曲线下面积)由梯形法计算而得,其他参数由3p87软件计算而得。
两种制剂灌胃给药后的平均药时曲线见图11。根据血药浓度,计算出布洛芬口服后的Cmax、Tmax、t1/2(半衰期)、AUC0–∞等动力学参数,见表6。比格犬体内血药浓度结果显示布洛芬立方液晶纳米粒实验组口服给药后与同剂量的布洛芬溶液对照组相比,相对生物利用度为222%。布洛芬立方液晶纳米粒实验组血药中布洛芬的达峰时间由布洛芬溶液对照组口服给药后的0.84±0.19h提高到布洛芬立方液晶纳米粒口服给药后的1.82±0.31h。布洛芬血药检测时间由布洛芬溶液对照组口服给药后的8h延长到布洛芬立方液晶纳米粒口服给药后的24h,布洛芬立方液晶纳米粒的半衰期和滞留时间分别是布洛芬原料药的3.09倍和5.39倍,显示布洛芬立方液晶纳米粒在体内具有良好的缓释作用,能明显改变药物在比格犬体内的吸收过程,促进药物的吸收,可显著提高布洛芬口服吸收的生物利用度。而对照组2和3虽然也可以一定程度上起到增大生物利用度的作用,但是由于没有添加稳定剂或者投药量过高,造成了液晶结构的不稳定以及药物的泄露,因此其提高程度比实验组的低很多。
表6布洛芬口服后的药代动力学参数(n=3)
缩写:AUC,药时曲线下面积;T1/2,消除半衰期;Cmax,血浆峰浓度;Tmax,血浆浓度达峰时间;MRT,平均驻留时间。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!