一种基于特定片段交叉的蛋白质结构预测方法与流程
本发明涉及生物信息学、智能信息处理、计算机应用领域、蛋白质结构预测,尤其涉及的是一种基于特定片段交叉的蛋白质结构预测方法。
背景技术:
蛋白质是生命体的重要组成部分,是生命活动的承担者。蛋白质的基本组成单元是氨基酸,自然界中常见的氨基酸有20多种,氨基酸是由中心碳原子及其相连的氨基、羧基、氢原子以及氨基酸的侧链组成,氨基酸经过脱水缩合形成肽键,由肽键连接起来的氨基酸形成一条长链,即为蛋白质。
蛋白质分子在生物细胞化学反应过程中起着至关重要的作用。它们的结构模型和生物活性状态对我们理解和治愈多种疾病有重要的意义。蛋白质只有折叠成特定的三维结构才能产生其特有的生物学功能。要了解蛋白质的功能,就必须获得其三维空间结构。因此,获得蛋白质的三维结构对人类来说是至关重要的,1961年,anfinsen提出了氨基酸序列决定蛋白质三维结构这一开创新的理论。而三维结构直接决定了蛋白质的生物性功能,所以人们对蛋白质的三维结构产生了浓厚兴趣并展开研究。国外学者肯德鲁和佩鲁茨对肌血蛋白和血红蛋白进行了结构分析,得到其蛋白质三维结构,是人类第一次测定蛋白质的三维结构,二人借此夺得年诺贝尔化学奖。此外,英国晶体学家bernal与1958年提出了蛋白质四级结构的概念,将其定义为蛋白质一级结构、二级结构以及三级结构的延伸发展。多维核磁共振方法和射线晶体方法是近些年来发展起来的两个最主要的测定蛋白质结构的实验方法。多维核磁共振方法是将蛋白质放在水中,利用核磁共振直接测定其三维结构的方法。而射线晶体方法是目前为止最有效的蛋白质三维结构测定手段。到前为止,使用这两种方法测定的蛋白质占了已测蛋白质中的绝大比例。由于釆用实验方法的条件有限、时间有限,需要花费大量的人力和物力,而且测定的速度远远跟不上序列的测定速度,所以急需一种既不依赖化学实验,又具有一定准确率的预测方法。这样如何简便、快速、高效地对未知蛋白质进行三维结构预测,成为研究者的棘手问题。在理论探索和应用需求的双重推动下,依据提出的蛋白质一级结构决定蛋白质三维结构的理论,利用计算机设计适当的算法,以序列为起点,三维结构为目标的蛋白质结构预测自20世纪末蓬勃发展。
因此,根据蛋白质的氨基酸序列,从理论上预测其相应空间结构就成为蛋白质研究领域科学家们的奋斗目标!预测蛋白质结构不仅是解开第二遗传密码的一把金钥匙,而且是设计出新型蛋白质分子的基础。理论计算方法(也称热力学方法)是一种常用的蛋白质结构预测方法,由于它仅利用一级序列信息进行预测,而不需要任何其它已知蛋白质结构信息,所以该方法也是一种较理想的预测方法。其基本假设是:一定环境中天然蛋白质的三维结构是整个系统自由能最小的结构。要实现这一方法有两个关键:一是要有一个合理的势函数,势函数的全局极小对应于蛋白质的天然结构;二是要有一个好的算法,保证在有效的计算时间内找到势函数的全局最小。尽管热力学方法建立在物理理论基础之上,但是目前这种方法的预测结果并不理想,主要障碍就是势函数的准确性和多重极小问题。因为目前的优化方法还不能确定性地求出势函数的全局极小,从而又制约着势函数的发展。所以研究有效的优化方法,解决多重极小问题是蛋白质结构预测中的当务之急。四十年来,人们发展了许多用于解决蛋白质结构预测中多重极小问题的方法,大致分为:分子动力学方法、系统搜索方法(包括格点搜索、树搜索)和随机搜索方法(包括monetacarlo方法、模拟退火方法、禁忌搜索、遗传算法等等)。随着数学和计算机技术的发展,这些方法也在不断地改进,并提出了其它一些新的算法。
因此,现在的蛋白质结构预测方法在预测精度和构象搜索方面存在着缺陷,需要改进。
技术实现要素:
为了克服现有的蛋白质结构预测方法的构象搜索能力较低和预测精度较低的缺陷,本发明提供一种构象搜索能力较高和预测精度较高的基于特定片段交叉的蛋白质结构预测方法。
本发明解决其技术问题所采用的技术方案是:
一种基于特定片段交叉的蛋白质结构预测方法,所述方法包括以下步骤:
1)输入查询序列,利用psipred(http://bioinf.cs.ucl.ac.uk/psipred/)预测查询序列的二级结构信息;
2)设置种群规模np、最大迭代次数g、交叉概率cr、设置变异失败次数m,允许最大变异次数n,玻尔兹曼温度因子kt,输入片段库,预测的二级结构信息,迭代次数g=0;
3)对种群所有构象进行初始化,对种群中每个构象进行片段组装,直到构象的每个残基二面角至少被替换过一次;
4)构象交叉,操作如下:
4.1)选第i,i∈[1,np]个构象ci为目标构象,产生一个随机数r,r∈[0,1],如果r小于cr,则跳到4.2),否则跳至步骤5);
4.2)随机选择一个构象cj,j≠i,利用计算二级结构算法dssp获取构象ci的二级结构信息;
4.3)根据ci残基位置随机选择一个交叉点p,判断交叉点p对应的残基被预测的二级结构的类型;
4.4)针对ci和cj,从交叉点p开始依次互换二面角对直到从交叉点p起预测的二级结构类型和交叉点p处对应的二级结构类型不同为止,产生一个构象c′i,并用rosetta能量函数“score3”计算其能量值;
5)构象变异,对构象c′i变异过程如下:
5.1)对构象c′i进行9残基片段组装,生成构象c″i,并用rosetta能量函数“score3”计算其能量值,若变异后的能量值比变异前能量值变小,则接收变异构象c″i,若能量值变大,则以boltzmann概率
5.2)如果拒绝接收变异后的构象c″i,则变异失败次数t加一;
5.3)如果m等于允许最大变异次数n,则直接接收变异后的构象c″i,否则返回步骤5.1);
6)基于按比例的适应度分配方法进行选择,过程如下:
6.1)对构象c″i求适应度值
其中l是查询序列长度,
6.2)对种群中每个构象ci,求适应度值
6.3)计算构象c″i被选择的概率pi:
6.4)产生一个随机数r′,r′∈[0,1],如果r′小于pi,则用构象c″i替换构象ci实现种群更新,否则保持种群不变;
7)g=g+1,判断是否达到最大迭代次数g,若不满足条件终止条件,则遍历种群执行步骤4),否则输出最后预测结果。
本发明的技术构思为:一种基于特定片段交叉的蛋白质结构预测方法,包括以下步骤:首先预测查询序列的二级结构信息,构建片段库;其次设计基于特定片段交叉的策略,建立二级结构信息的适应度函数,设计交叉变异策略;最后根据按比例的适应度分配方法更新种群,利用设计基于特定片段交叉的策略能够有效地提高算法构象搜索能力和预测精度,预测的三级结构有很好的二级结构。
本发明的有益效果为:构象空间搜索能力强较强、能够有效地提高蛋白质的二级结构的准确性和精度较高的三级结构。
附图说明
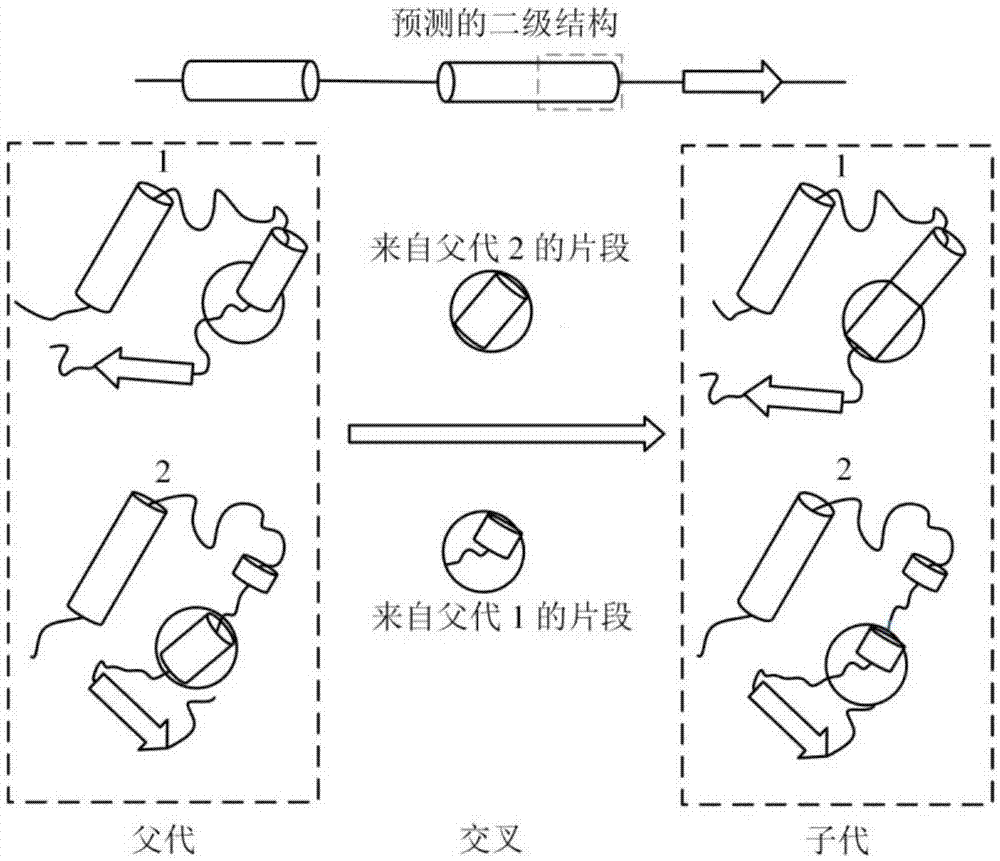
图1是蛋白质1tig特定片段的交叉示意图。
图2是蛋白质1tig利用基于特定片段交叉的蛋白质三级结构预测方法预测得到的三维结构示意图。
具体实施方式
下面结合附图对本发明做进一步描述。
参照图1和图2,一种基于特定片段交叉的蛋白质结构预测方法,包括以下步骤:
1)输入查询序列,利用psipred(http://bioinf.cs.ucl.ac.uk/psipred/)预测查询序列的二级结构信息;
2)设置种群规模np、最大迭代次数g、交叉概率cr、设置变异失败次数m,允许最大变异次数n,玻尔兹曼温度因子kt,输入片段库,预测的二级结构信息,迭代次数g=0;
3)对种群所有构象进行初始化,对种群中每个构象进行片段组装,直到构象的每个残基二面角至少被替换过一次;
4)构象交叉,操作如下:
4.1)选第i,i∈[1,np]个构象ci为目标构象,产生一个随机数r,r∈[0,1],如果r小于cr,则跳到4.2),否则跳至步骤5);
4.2)随机选择一个构象cj,j≠i,利用计算二级结构算法dssp获取构象ci的二级结构信息;
4.3)根据ci残基位置随机选择一个交叉点p,判断交叉点p对应的残基被预测的二级结构的类型;
4.4)针对ci和cj,从交叉点p开始依次互换二面角对直到从交叉点p起预测的二级结构类型和交叉点p处对应的二级结构类型不同为止,产生一个构象c′i,并用rosetta能量函数“score3”计算其能量值;
5)构象变异,对构象c′i变异过程如下:
5.1)对构象c′i进行9残基片段组装,生成构象c″i,并用rosetta能量函数“score3”计算其能量值,若变异后的能量值比变异前能量值变小,则接收变异构象c″i,若能量值变大,则以boltzmann概率
5.2)如果拒绝接收变异后的构象c″i,则变异失败次数t加一;
5.3)如果m等于允许最大变异次数n,则直接接收变异后的构象c″i,否则返回步骤5.1);
6)基于按比例的适应度分配方法进行选择,过程如下:
6.1)对构象c″i求适应度值
其中l是查询序列长度,
6.2)对种群中每个构象ci,求适应度值
6.3)计算构象c″i被选择的概率pi:
6.4)产生一个随机数r′,r′∈[0,1],如果r′小于pi,则用构象c″i替换构象ci实现种群更新,否则保持种群不变;
7)g=g+1,判断是否达到最大迭代次数g,若不满足条件终止条件,则遍历种群执行步骤4),否则输出最后预测结果。
本实施例以序列长度为88的α/β折叠蛋白质1tig为实施例,一种基于特定片段交叉的蛋白质结构预测方法,所述方法包括以下步骤:
1)输入查询序列,利用psipred(http://bioinf.cs.ucl.ac.uk/psipred/)预测查询序列的二级结构信息;
2)设置种群规模50、最大迭代次数1000、交叉概率0.5、设置变异失败次数0,允许最大变异次数150,玻尔兹曼温度因子2,输入片段库,预测的二级结构信息,迭代次数g=0;
3)对种群所有构象进行初始化,对种群中每个构象进行片段组装,直到构象的每个残基二面角至少被替换过一次;
4)构象交叉,操作如下:
4.1)选第i,i∈[1,np]个构象ci为目标构象,产生一个随机数r,r∈[0,1],如果r小于0.5,则跳到4.2),否则跳至步骤5);
4.2)随机选择一个构象cj,j≠i,利用计算二级结构算法dssp获取构象ci的二级结构信息;
4.3)根据ci残基位置随机选择一个交叉点p,判断交叉点p对应的残基被预测的二级结构的类型;
4.4)针对ci和cj,从交叉点p开始依次互换二面角对直到从交叉点p起预测的二级结构类型和交叉点p处对应的二级结构类型不同为止,产生一个构象c′i,并用rosetta能量函数“score3”计算其能量值;
5)构象变异,对构象c′i变异过程如下:
5.1)对构象c′i进行9残基片段组装,生成构象c″i,并用rosetta能量函数“score3”计算其能量值,若变异后的能量值比变异前能量值变小,则接收变异构象c″i,若能量值变大,则以boltzmann概率
5.2)如果拒绝接收变异后的构象c″i,则变异失败次数t加一;
5.3)如果m等于允许最大变异次数n,则直接接收变异后的构象c″i,否则返回步骤5.1);
6)基于按比例的适应度分配方法进行选择,过程如下:
6.1)对构象c″i求适应度值
其中l是查询序列长度,
6.2)对种群中每个构象ci,求适应度值
6.3)计算构象c″i被选择的概率pi:
6.4)产生一个随机数r′,r′∈[0,1],如果r′小于pi,则用构象c″i替换构象ci实现种群更新,否则保持种群不变;
7)g=g+1,判断是否达到最大迭代次数g,若不满足条件终止条件,则遍历种群执行步骤4),否则输出最后预测结果。
以序列长度为88的α/β折叠蛋白质1tig为实施例,运用以上方法得到了该蛋白质的近天然态构象,最小均方根偏差为
以上说明是本发明以1tig蛋白质为实例所得出的优良效果,显然本发明不仅适合上述实施例,在不偏离本发明基本内容所涉及范围的前提下对其做各种变形和改进,不应排除在本发明的保护范围之外。
- 还没有人留言评论。精彩留言会获得点赞!