一种基于SeqRank图算法的序列聚类方法与流程
本发明涉及生物序列信息研究领域,尤其涉及一种基于seqrank图算法的序列聚类方法。
背景技术:
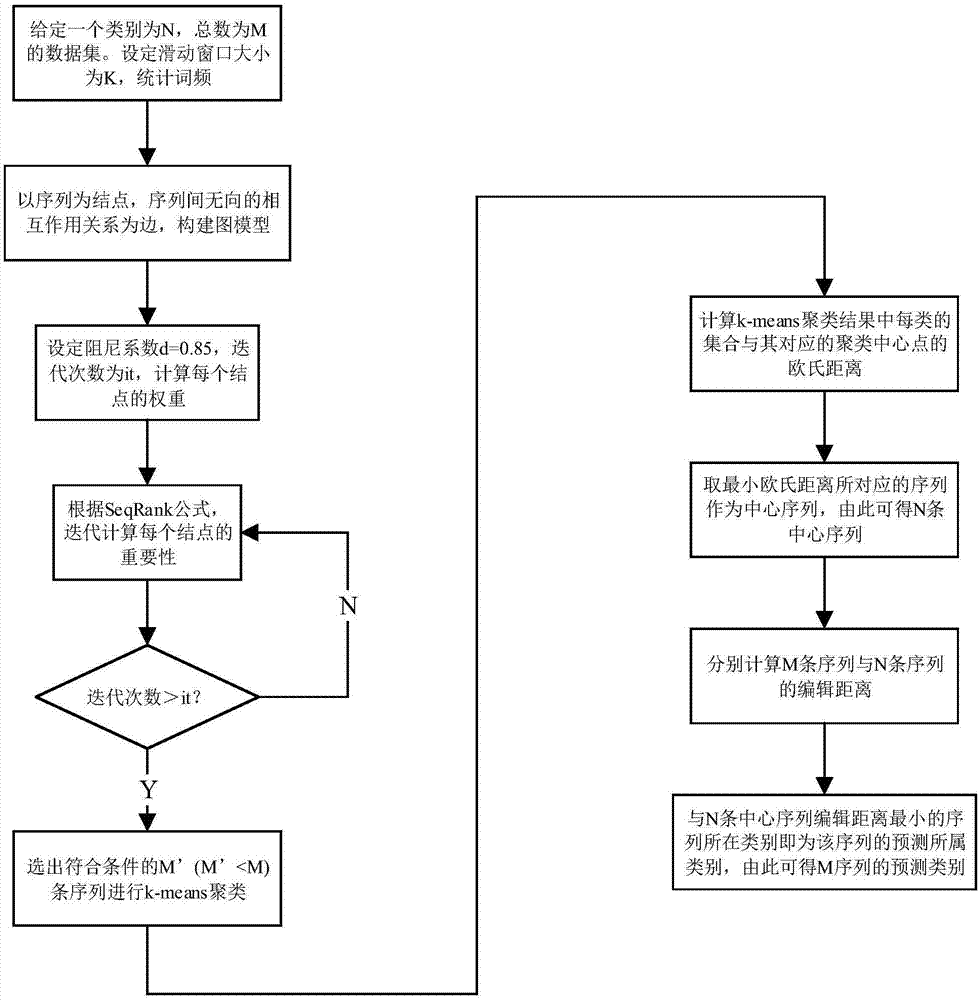
:在生物信息研究领域中,pcr、生物芯片等技术为代表的现代生物技术发展迅猛,并日益影响着人们的生产和生活方式。生物技术迅猛发展的同时,也将产生数量极为巨大的生物序列数据和信息。科研工作如何从这样大量的数据中得到有用的信息,聚类方法为数据的初步分析提供了基本手段。在生物学中,序列比对通过排列生物序列的方式,识别可以描述序列间的功能、结构以及进化关系的相似序列区域。聚类是把相似序列以静态分类的方法划分到不同的组中,使得组内的序列比不同组中的更相近。如果两个序列之间具有足够的相似性,就推测二者在进化上具有同源性,这将会大大节省重新测定未知序列结构和功能的时间和精力。此外,序列比对一般决定了许多生物信息学技术及程序的分析结果,影响着很多序列比较研究的结论和生物解释,是dna聚类分析等研究中的一个重要内容。技术实现要素:本发明的目的在于提供一种基于seqrank图算法的序列聚类方法。本发明采用的技术方案是:一种基于seqrank图算法的序列聚类方法,其包括以下步骤:步骤1,对数据集中的序列进行分割:步骤1.1,设定数据集中的每条序列的长度为m;每个序列进行k-词处理获得m-k+1个子序列,k-词处理的窗口长度为k,1≤k≤m;步骤1.2,统计两序列间共现k-mers的词频;当两序列存在共同k-mers,则认为该两条序列是相邻序列;步骤2,构件图模型:以序列为结点,序列间无向的相互作用关系为边,构建图模型;当两序列存在共同k-mers,则认为该两条序列存在共同的边,边的权重即为共同k-mers的数量;步骤3:计算每个结点的权重:步骤3.1:对于任意两个结点vi和vj,结点vi对vj的权重计算方法如下:wi表示k-merw在结点i中的数量,wj表示k-merw在结点j中的数量;步骤3.2:wjk表示结点vj接收到的来自其它结点的作用,通过以下公式计算wjk:wjk表示每个相邻结点对本结点的贡献程度,m表示序列总数,分母∑i∈m|wj|表示指向当前节点vj的k-mers总和,∑i∈m|wj|为一个加权和;步骤4:计算每个结点的重要性,迭代计算每个结点vi的重要性ws(vi),ws(vi)对应的seqrank计算公式为:ws(vi)表示结点vi的重要性,e(vi,vj)≠0表示结点vi和结点vj存在共同的边,vk是与结点vj存在共同的边的结点,wji表示结点vi指向结点vj的边的权重,即结点vi和结点vj存在的共同k-mers的数量;表示e(vj,vk)≠0时结点vj指向结点vk的边的权重;ws(vj)为上一次迭代后结点vj的重要性;d为阻尼系数,d表示在任意时刻用户从一个结点跳转到另一结点的概率;步骤5:基于seqrank算法对序列进行聚类;对所有序列的迭代结果进行处理,选出重要性大于均值的m′条序列进行k-means聚类;ccate=min(|rcate-kmcate|)1≤cate≤n公式(4)ccate表示k-means聚类的中心序列,rcate表示每一类聚类结果的集合,kmcate是k-means对应的每一类的聚类中心点,n为中心序列的总条数;步骤6:预测序列的所属类别,分别计算各条序列与每条中心序列ccate的编辑距离,选出中心序列ccate中编辑距离最小的序列作为该序列的预测所属类别。进一步地,步骤1中数据集为一个类别为n、总数为m且n<m的序列数据。进一步地,步骤2中构建的图模型为seqrank图模型s=(v,e),s=(v,e)是由结点集和边集组成的一个有向图,其中v为由每一条序列组成的结点集,e代表结点间相互作用关系形成的边集合,e=v*v。进一步地,步骤4中d满足0≤d≤1。进一步地,d的取值为0.85。进一步地,步骤5中k-means聚类的中心序列的确定方法:步骤5.1,计算k-means聚类结果中每类的集合与其对应聚类中心点的欧氏距离;步骤5.2,取最小的欧氏距离对应的序列作为k-means聚类的中心序列。进一步地,步骤6中,设定p为seqrank算法预测出的m条序列的类别集合,则计算公式如下:p(vi)=min(adist(vi,vcate))1≤i≤m公式(5)其中vi为第i个结点,vcate为中心序列,adist(vi,vcate)为对应的编辑距离,m为数据集中序列的总数。本发明采用以上技术方案,通过使用seqrank图算法进行聚类,对生物序列进行聚类分析,试图在聚类分析的层次上从生物序列数据中得到深层信息含义及可靠地结论,有效地解决现有技术中存在的序列间聚类结果不够准确、不够客观的问题。附图说明以下结合附图和具体实施方式对本发明做进一步详细说明;图1为本发明一种基于seqrank图算法的序列聚类方法的流程示意图。具体实施方式根据pagerank算法的主要思想:计算一个网页链接的数量和质量,从而估计这个网页的重要程度,建立起判断多文档相似度的模型;将该方法运用到生物序列聚类工作中,准确有效地得到了精度较高的聚类结果;由于该方法考虑到了每条序列间的局部特征,而这些特征可能是表达该序列之间相似性的关键,这样所获得的序列聚类结果是比较准确、客观的。如图1所示,本发明公开了一种基于seqrank图算法的序列聚类方法,其包括以下步骤:步骤1,对数据集中的序列进行分割:给定一个类别为n、总数为m(n<m)的序列数据集,将每个序列分割,一条长度为m的序列可分割为:x1,x2,…,xk,xm;对于每一条序列,提取滑动窗口大小为k的k-mers子序列。假设序列长度为m,则该序列中含有m个长度为1的元素,含有(m-1)个长度为2的元素,依此类推,该序列中含有1个长度为m的元素,这些元素的滑动窗口的大小为1,该滑动窗口从序列起始位置滑向终止位置进而形成了k-mers。所以该序列含有(m-k+1)个长度为k(1≤k≤m)的k-mers。统计两序列间共现k-mers的词频。若两序列存在共同k-mers,则认为这两条序列是相邻序列,k-mers形如:(x1,x2,…,xk),(x2,x3,…,xk+1)等。步骤2,构件图模型:以序列为结点,序列间无向的相互作用关系为边,构建seqrank图模型s=(v,e),其中v为由每一条序列组成的结点集,e代表结点间无向的相互作用关系,e=v*v;为便于描述,我们令s=(v,e)是由结点集和边集组成的一个有向图。对于任意一个结点vi,都有in(vi)和out(vi)。in(vi)表示指向结点vi的结点集合,out(vi)表示结点vi所指向的节点的集合。步骤3:计算每个结点的权重。对于任意两个结点vi和vj,结点vi是通过连接他们的边e=<vi,vj>向结点vj传递作用的。边权重的大小决定了vi对vj的作用大小。结点vi对vj的作用计算方法如下:wi表示k-merw在结点i中的数量,wj表示k-merw在结点j中的数量。同理,我们用wjk来表示结点vj接收到的来自其它结点的作用,可通过以下公式计算wjk:公式(2)等号右侧的求和表示每个相邻结点对本结点的贡献程度。m表示序列总数。分母为一个加权和,表示的是指向当前节点vj的k-mers总和。步骤4:计算每个结点的重要性。seqrank公式为:ws(vi)表示结点vi的重要性,类似地,wji表示结点vi指向结点vj的边的权重,即结点vi与结点vj的相互作用关系。ws(vj)为上一次迭代后结点vj的重要性。d为阻尼系数(0≤d≤1),表示在任意时刻,用户从一个页面继续转而浏览另一个页面的概率,即每个结点均有(1-d)的概率随机跳转到其他结点,而不要求两个结点间一定存在边的连接,从而确保算法在任意图上均可以收敛。d一般取值为0.85。举例理解公式中的求和部分:假设现在有3条序列如下:序列1acgt序列2acgg序列3ccgg当滑动窗口大小k=2时,各序列的k-mers分布如下:序列1ac,cg,gt序列2ac,cg,gg序列3cc,cg,gg各序列间存在共同k-mers的情况(即序列对序列的权重)如下:序列1序列2序列3序列102(ac,cg)1(cg)序列22(倒三角关系与无向图相对应)02(cg,gg)序列3120对于seqrank公式:以序列1为例,各序列的权重初始值为1。当i=1时,即计算序列1的权重,此时有:所以第一次迭代时,序列1的权重为:具体地,若用来表示结点vi经过it次迭代后的重要性,则公式(3)可表示为:若两个结点vi与vj公用一个k-mer或多个k-mers,则称这两个结点之间存在相互作用,由此可建立一个结点网络,这个结点网络的邻接矩阵为w,其中:w表示消除零行以后的矩阵。由图论中知识可知,矩阵w的第i(i=1,2,…,m)行元素的和表示的是第i个结点的输出度。由w定义可知,w表示的是一个无向图,也就是说是对称矩阵,即w=wt。degi表示第i个结点的输出度,则:在求和的过程中,1/degi保证了每个结点在投票过程中具有同等的影响力。d∈(0,1)是一个固定参数,在本实验中用于描述网络对结点排名的影响。设置d=0对应于网络的无影响与结点间相互作用的全面影响,而设置d=1对应于网络的完全影响,不影响结点间的相互作用。本文采用d=0.85。在公式(1)中,由于计算结点的权重时又需要用到结点本身的权重,因此需要进行迭代计算。seqrank算法会对图模型s=(v,e)进行迭代计算直至收敛。步骤5:基于seqrank算法对序列进行聚类。对m条序列的迭代结果进行处理,选出重要性大于均值的m′(m′<m)条序列进行k-means聚类。ccate=min(|rcate-kmcate|)1≤cate≤n公式(4)rcate表示每一类聚类结果的集合,kmcate是k-means对应的每一类的聚类中心点。在每一类的聚类结果中,我们认为,与中心点的欧氏距离最接近的序列是k-means聚类的中心序列。由公式(6)可得n条中心序列ccate。步骤6:预测m条序列的所属类别。分别计算m条序列与ccate的编辑距离。在ccate中选出编辑距离最小的序列作为该序列的预测所属类别。设定p为seqrank算法预测出的m条序列的类别集合,则公式如下:p(vi)=min(adist(vi,vcate))1≤i≤m公式(5)本发明采用以上技术方案,通过使用seqrank图算法进行聚类,对生物序列进行聚类分析,试图在聚类分析的层次上从生物序列数据中得到深层信息含义及可靠地结论,有效地解决现有技术中存在的序列间聚类结果不够准确、不够客观的问题。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1