一种适用于Sequel测序的三代全长转录组测序结果分析方法与流程
本发明涉及基因检测领域,具体涉及适用于sequel测序平台的三代全长有参转录组分析方法。
背景技术:
转录组是某个物种或者特定细胞类型产生的所有转录本的集合。转录组研究能够从整体水平研究基因功能以及基因结构,揭示特定生物学过程以及疾病发生过程中的分子机理,已广泛应用于基础研究、临床诊断和药物研发等领域。真核生物的蛋白编码基因在3’末端有一段poly(a)尾,所以对于真核生物,提取总rna后,可以用带有polyt的反转录引物,将rna序列反转录成cdna,再以cdna为模板,制备全长cdna文库,构建好的文库用sequel测序仪进行测序。
测序后的数据需要进行生物信息学分析,获取样品的转录本结构信息,推断生物学意义。通常一个样品可以获得数百万个测序reads,之前的分析方法存在计算机资源消耗大、运行时间慢等缺点。同时,不断的出现新的分析方法和软件,现有的全长转录组分析流程需要优化和补充。
技术实现要素:
为了克服现有技术的上述缺陷,本发明的目的在于提供一种适用于sequel测序平台的三代全长转录组分析方法。
为了实现本发明的目的,所采用的技术方案是:
一种适用于sequel测序平台的三代全长转录组分析方法,包括如下步骤:
步骤一,测序数据过滤步骤:
使用pacbio官方的isoseq3流程对原始数据进行处理:
使用ccs程序对下机的subreads进行处理,得到每个零模波导孔的一致性序列ccs;
使用lima程序对一致性序列进行接头识别,得到全长序列fl;
使用isoseq3refine程序对全长序列进行嵌合去除和polya识别,得到全长非嵌合序列flnc;
使用isoseq3cluster对全长非嵌合序列进行聚类,得到去冗余的高质量转录本hqisoform;
步骤二,测序数据比对步骤:
使用minimap2和cdna_cupcake软件将聚类后的高质量转录本与参考基因组进行比对并进一步去除冗余:
首先从基因组数据库中下载对应物种的参考基因组;
将高质量序列比对到参考基因组上;
根据转录本在基因组上的剪切模式,将剪切模式相同、3’端位点相近、仅5’端不同的转录本合并,作为样品识别到的转录本;
步骤三,转录本注释步骤:
使用squanti2将合并后的转录本与参考转录本注释进行比较,具体为:
将两种转录本的位置和剪切模式进行比较;
步骤四,orf预测步骤:
使用transdecoder软件对转录本进行orf预测,具体是:
使用transdecoder.longorfs识别转录本序列中的长度大于300的orf结构;
对识别的orf进行swissprot同源序列比对和pfam结构域预测,作为orf结果可信度的一个判断依据;
使用transdecoder.predict对orf进行检验,每个转录本保留最优的orf结果,得到相应的cds和蛋白质序列;
步骤五,转录本功能注释步骤:
对预测得到的转录本蛋白质序列进行功能分析,具体是:
使用diamond软件将转录本比对nr数据库,获得同源蛋白信息;
使用blast2go软件,根据nr注释结果,提取go注释;
使用diamond软件将转录本比对到kegg数据库,然后使用kobas预测转录本的kegg编号;
使用diamond软件将转录本比对到eggnog数据库,获得转录本的直系同源信息;
使用diamond软件将转录本比对到swissport数据库,获得转录本的swissprot同源蛋白信息;
步骤六,融合基因分析步骤:
用cdna_cupcake软件包进行融合基因分析:
从转录本与基因组比对结果中挑选出不完全匹配基因后,分别比对到基因组多个位置的转录本,作为融合基因位点;
步骤七,lncrna预测步骤:
使用cpc进行lncrna预测,具体是:
从squanti2结果中挑选出genic、intergenic、antisense的转录本;
使用cpc对挑选出来的转录本进行编码潜能预测,识别潜在的lncrna序列;
步骤八,可变剪切分析步骤:
使用splicegrapher软件进行分析可变剪切分析;
步骤九,可变多聚腺苷酸化分析步骤:
使用tapis进行可变多聚腺苷酸化分析。
在本发明的一个优选实施例中,所述步骤三的比较为根据转录本与参考基因注释的重叠、链方向是否一致、剪切模式是否相同及是否覆盖多个基因区域,将合并后的转录本分成fsm、ism、nic、nnc、genic、intergenic、antisense七种类型。
在本发明的一个优选实施例中,所述步骤六的不完全匹配是指转录本不能完整匹配基因,但是可以分段匹配到基因组不同的位置。
在本发明的一个优选实施例中,所述步骤七的编码潜能预测具体为cpc首先预测转录本的开放阅读框,将预测到的开放阅读框对于的编码序列与uniprot进行同源性比对,根据比对结果建立模型,对转录本进行编码潜能打分,把分值低于0的作为预测到的潜在lncrna序列。
在本发明的一个优选实施例中,所述步骤八的可变剪切分析具体是splicegrapher将同一个基因的转录本进行两两比较,根据外显子剪切位点的不同,识别intronretention、skippedexon、alt.5’、alt.3’四种类型的可变剪切。
在本发明的一个优选实施例中,所述步骤九的可变多聚腺苷酸化分析具体是:tapis根据输入的全长非嵌合序列,调用gmap比对基因组,根据序列比对的位置,对转录本的终止位点进行识别,对于距离小于5bp的终止位点进行合并,选择reads数大于等于2的作为识别到的apa位点。
本发明的有益效果在于:
本发明的运行速度更快,且与常用的matchannot软件相比对转录本的注释更加精细,更加便于分析转录本的类型。
附图说明
图1为本发明的pacbioccs示意图。
图2为本发明的ccs序列类型分布示意图。
图3为本发明的不同类型的转录本序列长度分布示意图。
图4为本发明的转录本在不同的数据库中注释数目分布图。
图5为本发明的基因的转录本可变剪切可视化图。
图6为本发明的flnc终止位点在基因组上分布图。
具体实施方式
一种适用于sequel测序平台的三代全长转录组分析方法,包括如下步骤:
步骤一,测序数据过滤步骤:
使用pacbio官方的isoseq3流程对原始数据进行处理:
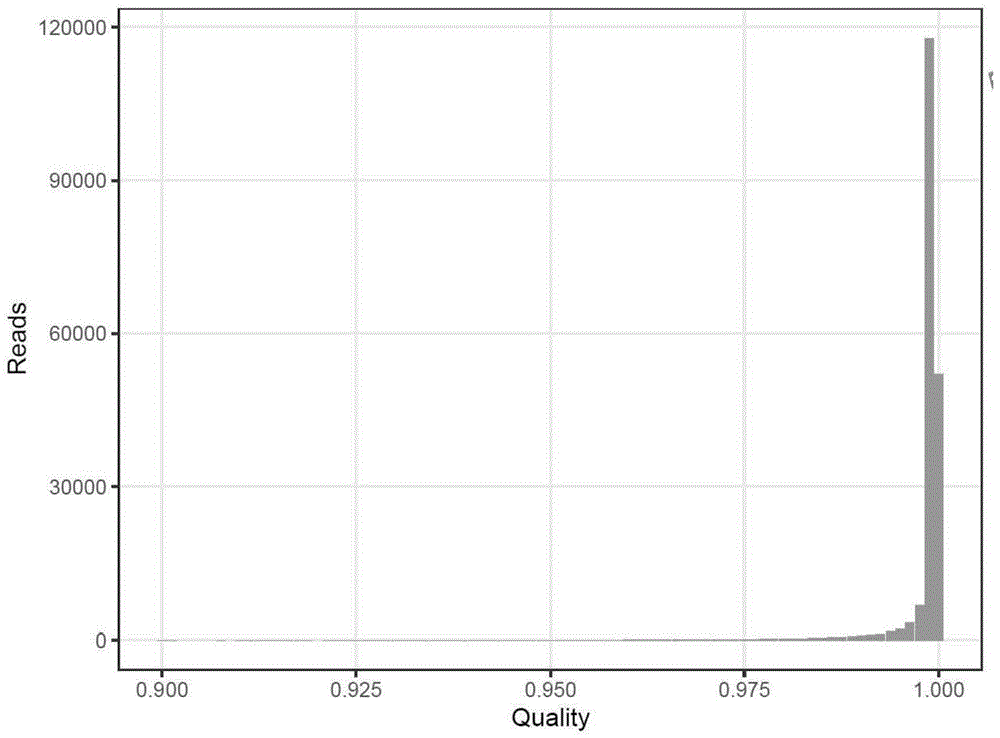
使用ccs程序对下机的subreads进行处理,得到每个零模波导孔的一致性序列ccs,具体如图1所示,pacbioccs(一致性序列)精确性值分布,主要分布在0.99左右,说明测序结果经过处理后质量非常高;
使用lima程序对一致性序列进行接头识别,得到全长序列fl,具体如图2所示,图中fulllengthnonchimericwithpolya(全长非嵌合、含有polya)的序列占绝大部分,说结果中的有效序列比对较高;
使用isoseq3refine程序对全长序列进行嵌合去除和polya识别,得到全长非嵌合序列flnc;
使用isoseq3cluster对全长非嵌合序列进行聚类,得到去冗余的高质量转录本hqisoform,具体参见图4,转录本在不同的数据库中注释数目分布图,可以看到在五种数据库中注释到信息的转录本比例较高,方便鉴别转录本的功能;
步骤二,测序数据比对步骤:
使用minimap2和cdna_cupcake软件将聚类后的高质量转录本与参考基因组进行比对并进一步去除冗余:
首先从基因组数据库中下载对应物种的参考基因组;
将高质量序列比对到参考基因组上;
根据转录本在基因组上的剪切模式,将剪切模式相同、3’端位点相近、仅5’端不同的转录本合并,作为样品识别到的转录本;
步骤三,转录本注释步骤:
使用squanti2将合并后的转录本与参考转录本注释进行比较,具体为:
将两种转录本的位置和剪切模式进行比较(根据转录本与参考基因注释的重叠、链方向是否一致、剪切模式是否相同及是否覆盖多个基因区域),将合并后的转录本分成fsm、ism、nic、nnc、genic、intergenic、antisense七种类型;
步骤四,orf预测步骤:
使用transdecoder软件对转录本进行orf预测,具体是:
使用transdecoder.longorfs识别转录本序列中的长度大于300的orf结构,具体如图3所示,图中不同类型的转录本序列长度分布,可以看出长度大部分在1k以上,说明能检测到更多的长序列;
对识别的orf进行swissprot同源序列比对和pfam结构域预测,作为orf结果可信度的一个判断依据;
使用transdecoder.predict对orf进行检验,每个转录本保留最优的orf结果,得到相应的cds和蛋白质序列;
步骤五,转录本功能注释步骤:
对预测得到的转录本蛋白质序列进行功能分析,具体是:
使用diamond软件将转录本比对nr数据库,获得同源蛋白信息;
使用blast2go软件,根据nr注释结果,提取go注释;
使用diamond软件将转录本比对到kegg数据库,然后使用kobas预测转录本的kegg编号;
使用diamond软件将转录本比对到eggnog数据库,获得转录本的直系同源信息;
使用diamond软件将转录本比对到swissport数据库,获得转录本的swissprot同源蛋白信息;
步骤六,融合基因分析步骤:
用cdna_cupcake软件包进行融合基因分析:
从转录本与基因组比对结果中挑选出不完全匹配(不完全匹配指的是转录本不能完整匹配基因,但是可以分段匹配到基因组不同的位置)、分别比对到基因组多个位置的转录本,作为融合基因位点;
步骤七,lncrna预测步骤:
使用cpc进行lncrna预测,具体是:
从squanti2结果中挑选出genic、intergenic、antisense的转录本;
使用cpc对挑选出来的转录本进行编码潜能预测(cpc首先预测转录本的开放阅读框,将预测到的开放阅读框对于的编码序列与uniprot进行同源性比对,根据比对结果建立模型,对转录本进行编码潜能打分,把分值低于o的作为预测到的潜在lncrna),识别潜在的lncrna序列;
步骤八,可变剪切分析步骤:
使用splicegrapher软件进行分析可变剪切分析(splicegrapher将同一个基因的转录本进行两两比较,根据外显子剪切位点的不同,识别intronretention、skippedexon、alt.5’、alt.3’四种类型的可变剪切),输出的图如图5所示,基因的转录本可变剪切可视化图,便于直观的分析转录本剪切方式和位置;
步骤九,可变多聚腺苷酸化分析步骤:
使用tapis进行可变多聚腺苷酸化分析(tapis根据输入的全长非嵌合序列,调用gmap比对基因组,根据序列比对的位置,对转录本的终止位点进行识别,对于距离小于5bp的终止位点进行合并,选择reads数大于等于2的作为识别到的apa位点,如图6所示,flnc终止位点在基因组上分布,可以看到基因有很多的终止位点,说明基因有多样的apa位点。
- 还没有人留言评论。精彩留言会获得点赞!