一种全基因组高效基因区富集测序方法与流程
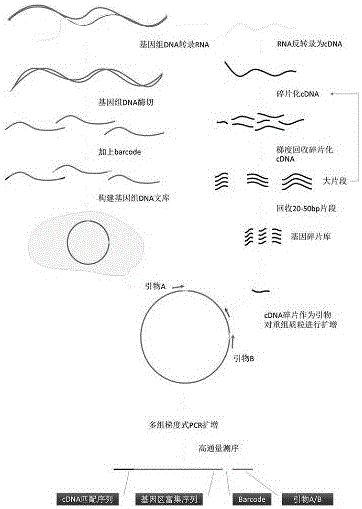
本发明涉及基因组学、生物技术领域,及而言涉及将基因区富集测序、简化重测序、重复序列去除为目的,一种全新的巧妙利用基因组自身基因序列,进行基因区序列的富集测序方案,进而降低测序成本、减少信息处理量,提供特殊基因区文库。对后基因组时代与复杂基因组具有重要意义,应用将及其广泛。
背景技术:
1基因富集的方法
1 .1 cDNA文库(cDNA library)与转录组测序。1976 年Hofstetter 成功的构建了第一个cDNA 文库以来, 构建cDNA 文库已成为研究功能基因组学的基本手段之一。cDNA文库的构建是分子生物学领域的一项重要技术。cDNA是以mRNA为模板,在逆转录酶的作用下,在体外被逆转录为cDNA第一链,再以cDNA为模板,由大肠杆菌DNA聚合酶Ⅰ合成第二链,得到双链cDNA。由于组织或细胞的总RNA或mRNA中,含有该细胞的全部mRNA分子,因而被合成的cDNA产物将是各种mRNA拷贝的群体。当它们与质粒重组后并转化至宿主细胞中,将得到一系列克隆群体,每个克隆仅含有一种mRNA信息,所有克隆的总和则包含细胞内全部mRNA的信息,这种克隆群体则为cDNA文库。目前, 广泛使用的方法是SMART 技术。目前对于大多数物种而言, 全基因组测序是不现实的, 为了快速、经济地获得基因序列、了解基因的功能以及基因组中基因数量等相关信息, 构建cDNA 文库是一种有效、简便且快速的可行方法。所以cDNA 文库的构建已成为当前分子生物学研究和基因工程操作的基础。但是有了最新的测序技术,我们将不再需要构建克隆文库,可以直接对cDNA片段进行测 序。对 RNA进行测序一直以来都被认为是一种发现基因的有效方法,而且这种方法还被认为是对编码基因以及非编码基因进行注释的金标准。与以前的方法相比,大规模平行RNA测序方法(massively parallel sequencing of RNA)极大增强了RNA测序技术的处理能力,使我们得以能够对转录组进行测序。我们现在可以只需要花费几天,仅用以往同类项目科研经费的很少一部分就能够得到一个比较满意的完整的细胞转录组。
1.2外显子捕获技术
外显子捕获测序和转录组测序都是针对基因组上转录区域进行测序,但是外显子捕获测序针对已有基因组信息的物种,而转录组分析既能针对已有基因组信息的物种,也能针对没有基因组信息的新物种,因此,两者的分析存在一定的差异:(1)分析的目标区域有所不同。外显子捕获测序只针对基因组上已知的编码区,而转录组测序不仅针对基因组上已知的编码区,还能够检测非编码RNA等转录组的信息。(2)分析的手段所有不同。外显子捕获测序只需要把测序结果比对基因组,分析序列差异。转录组测序既可以把测序结果比对基因组,也可以进行从头(de Novo)拼接。(3)得到的结果有所不同。外显子捕获测序可以得到序列变异的信息,而转录组测序不仅可以获得已知序列的变异信息和新的转录本信息(针对从头拼接),还可以得到表达谱信息。除此以外,转录组测序还能够分析mRNA的可变剪接,而外显子捕获测序的样品来源是基因组,不能够进行mRNA的可变剪接分析,只能够得到外显子上的序列变化。
1.3 甲基化过滤文库法(Methylation filtration library,MF)迄今为此, 研究学者发现5mC(胞嘧啶5 号位的甲基化)在植物基因组中普遍存在。Rabinowicz 等在《Nature Genetics》上发表论文利用甲基化过滤的方法首次对富含重复序列(占整个基因组序列的80%)的玉米基因组进行测序, 发现与未经甲基化过滤的对照文库(鸟枪法文库)相比,甲基化过滤文库的基因富集率要高出5~7 倍。Timko等使用MF法过滤了豇豆80%的重复序列, 最终得到约151 Mb的富含基因片段的序列, 包含整个豇豆95%的基因,富集效率达4.1 倍。甲基化过滤已经成为快速、高效富集植物基因编码序列高效途径。Yuan等采用跨越甲基衔接物文库法(MSLL) 的HapⅡ和SalⅠ两种甲基化敏感酶对玉米基因组DNA 进行完全酶切后, 将酶切片段连接转化到McrBC- E.coli DH10B 感受态细胞中构建3个BAC 文库(Hap Ⅱ BACs 、Sal Ⅰ BACs(10~15 kb) 、Sal ⅠBACs(15~25 kb))。挑选克隆测序发现: HapⅡBACs,SalⅠBACs(10~15 kb), SalⅠBACs(15~25 kb)3个文库对已知基因发现率分别为5.5%、14%、18%, 加权平均值为10.96%; 而对照EcoRⅠ BAC 文库已知基因发现率与鸟枪法相近仅为1.3%。亚甲基部分限制性文库法(Hypomethylated partialrestriction library, HMPR)在MSLL 方法基础上, Emberton 等发明的HMPR 方法也是采用甲基化敏感的限制性内切酶HpaⅡ(5′-CCGG-3′)和Hpy CH4IV(5′-ACGT-3′)构建HMPR 文库。Bedell 等利用甲基化过滤法(MF)过滤了高梁整个基因组的66%的区域(448 Mb),标记了约96%的基因组序列。
2简化基因组测序
简化基因组测序(Reduced-representation sequenc-ing)是在第二代测序基础上发展起来的一种利用酶切技术、序列捕获芯片技术或其他实验手段降低物种基因组复杂程度, 针对基因组特定区域进行测序, 进而反映部分基因组序列结构信息的测序技术。目前发展起来的简化基因组测序有:复杂度降低的多态序列(Complexity reduction of polymorphic sequences, CRoPS)测序[2], 限制性酶切位点相关的DNA (Re-striction-site associated DNA, RAD)测序[3], 基因分型测序(Genotyping by sequencing, GBS), GBS的发展(Elshire et al., 2011; Poland et al., 2012)和甲基化敏感简化测序AFSM技术(Xia et al., 2014)一个简单的,快速和低成本有效的系统,已经用于在非模式生物的测序。其中运用最为广泛的是限制性酶切位点相关DNA的测序技术, 即RAD-seq。该技术利用限制性内切酶对基因组进行酶切, 产生一定大小的片段, 构建测序文库, 对酶切后产生的RAD标记进行高通量测序。由于RAD标记是全基因组范围的呈现特异性酶切位点附近的小片段DNA标签, 代表了整个基因组的序列特征, 因此通过对RAD标记测序能够在大多数生物中获得成千上万的单核苷酸多态性(Single nucleotide polymorphism, SNP)标记。
其中cDNA文库(cDNA library)和转录组测序都无法获得除了RNA以外的区域,而外显子捕获技术成本较高,只能捕获已知序列。甲基化富集操作复杂成本较高,面临还有不少的重复序列存在。而简化基因组虽然成本较低但是无法对基因区进行富集。
技术实现要素:
发明人面对现有技术的缺陷,设想通过前期处理,经过多方面的选择和研究,得到本发明的技术方案。
用于全基因组高效基因区富集测序的建立DNA基因区富集库的方法,包括以下步骤:
A) 样品进行全基因组DNA的提取;
B) 全基因组mRNA 的提取;
C )将步骤B)得到的全基因组mRNA 反转录为cDNA;
D )将步骤C)得到的全基因组cDNA 的碎片化;
E )分批回收步骤D)的碎片化cDNA得到20-50bp的短序列片段;
F) 将步骤A)得到的全基因组DNA,进行单酶切;
G )对步骤F)得到的酶切片段末端可增加barcode接头连接,以区分不同样品;
H) 将步骤G)产出的DNA酶切片段装入环形质粒中,构建DNA文库;
I )由步骤E)中的cDNA碎片为左引物,根据质粒两端序列分别设计两对引物为右引物;
J )将步骤I)的两对引物对,对H中构建好的DNA文库进行PCR扩增;
K )对步骤J)扩增片段进行高通量测序,获得基因区富集序列。
另外,本发明也可以采用先碎片化mRNA,再反转录为cDNA的方案,具体为:用于全基因组高效基因区富集测序的建立DNA基因区富集库的方法,包括以下步骤:
A) 样品进行全基因组DNA的提取;
B) 全基因组mRNA 的提取;
C )将步骤B)得到的全基因组mRNA 进行碎片化;反转录为cDNA;
D )将步骤C)得到的碎片化mRNA反转录为cDNA;
E )分批回收步骤D)的碎片化cDNA得到20-50bp的短序列片段;
F) 将步骤A)得到的全基因组DNA,进行单酶切;
G )对步骤F)得到的酶切片段末端增加barcode接头连接,以区分不同样品;
H) 将步骤G)产出的DNA酶切片段装入环形质粒中,构建DNA文库;
I )由步骤E)中的cDNA碎片为左引物,根据质粒两端序列分别设计两对引物为右引物;
J )将步骤I)的两对引物对,对H中构建好的DNA文库进行PCR扩增;
K )对步骤J)扩增片段进行高通量测序,获得基因区富集序列。
进一步的:所述cDNA或者mRNA碎片化方式,为物理破碎,或者为酶切破碎。
进一步的:所述步骤E) 中片段化分批回收技术,为普通电泳胶回收方式,或者为E-Gel胶按时间回收方式。
进一步的:所述步骤 F) 中酶切,可选择4-6碱基酶,根据目标基因组大小和特征调整,或者结合甲基化敏感酶进行甲基化识别。
本发明中,将NA酶切片段装入环形质粒,可以解决基因组DNA扩增时cDNA引物没有扩增而是另一端引物的假阳性扩增;此处也可以为其他避免假阳性方式,例如不对称酶切方式以连接一头接头。另外,以cDNA碎片去扩增基因组的序列,将不单是外显子序列,而得到该cDNA碎片附近的有效富基因集序列;对于不同的研究方向,cDNA 碎片可以来源于不同物种,将扩增物种间的同源基因,可以用于物种间的基因进化研究。采用mRNA碎片或者cDNA碎片化,可以通过两组测序确定该位置“基因”在DNA双链中的方向性,具有更加深远意义。
附图说明
图1为基因区富集测序基本原理图;
图2为接头设计原理图。
具体实施方式
下面结合具体实施例和附图对本发明做进一步详细说明。
对木薯基因组进行基因区富集建库测序:基因区富集测序基本原理如图1所示,接头设计原理如图2所示。
使用本发明所述技术对在木薯的样品进行实验:
(1)组织要新鲜,尽可能嫩,取在同一生长条件下生长一致,同生长期、同一部位,且无病虫害的材料提取基因组DNA。长期保存样品需液氮或-70℃以下冰箱。采用DNeasy 96 Plant Kit (QIAGEN)试剂盒提取基因组DNA。
(2)利用RNeasy Plant Mini Kit提取总RNA,取2ul电泳检测纯度和质量。-20℃短时间保存,或-80℃长期保存。
(3)DNA与RNA质量检测及定量:琼脂糖凝胶检测以λmarker为标记,取1μL DNA ,加入2μL l0× 溴酚蓝上样缓冲液,混匀,点入含0.5μg/ml Goldview 染料的0.8% 琼脂糖凝胶中,用1× TAE 缓冲液,90 V 电泳40 m in;凝胶成像分析系统(Tanon4100)观察DNA与RNA条带。
取1-2μL DNA与RNA样品,用NANODROP 2000C 对基因组DNA进行检测。根据260nm处的光吸收值计算DNA浓度,根据OD260/OD280、OD260/OD230比值判断有无多糖、蛋白质、RNA等杂质,从而确定DNA的纯度。所有DNA样品工作液浓度需均一化,精确定量到100ng/μL。Qubit(Invitrogen)进行定量定性分析,保证DNA的高质量,包括完整性和纯度。
(3)酶切
在0.5mL离心管中加入(20μL体系)
对照 样品
模板DNA 2μL(100ng/μL) 2μL
HpaII(10U) 4μL 4μL
HpaII Buffer 2μL 2μL
纯净水 12μL 12μL
混合离心数秒37℃温浴2小时。65℃ 30min,4℃保存。酶切液应不能放置太久,应尽快进行连接。
(4)凝胶检测
取8μL酶切液2%琼脂糖胶检测。
(5)制备Adapters
合成Adapter_1:5’CGXXXXXCAG 3’与 Adapter_2:5’ACTGXXXXX 3’带有Barcodes的 Adapter。
“Barcodes” Adapter制备:
在1.5mL离心管中加入
“Barcodes” Adapter_1 300μL
“Barcodes” Adapter_2 300μL
95℃ 2min,再降温致25℃(-0.1℃/s),25℃ 30min,4℃保存。
(6)连接
在0.5mL离心管中加入(20μL体系)
HpaII酶切液 10μL
“Barcodes” Adapter 1.5μL
Water 6μL
10×Ligase Buffer 2μL
T4 DNA ligase (200 U/μL) 0.5μL
混合离心数秒16℃过夜, 65℃ 20min,4℃保存。
(7)DNA混合池纯化
从连接产物中,各取5μL加入1个2mL离心管,混合离心数秒。用E.Z.N.A. Cycle-Pure Kit (Omega Bio-tek)试剂盒纯化混合基因池。
另取1个2mL离心管,从连接产物中,各取5μL加入2mL离心管,混合离心数秒。用E.Z.N.A. Cycle-Pure Kit (Omega Bio-tek)试剂盒纯化混合基因池。回收300bp以上片段。
(8)DNA混合池文库构建
将纯化后的DNA混合池进行文库构建,
Invitrogen公司Topo TA克隆系统提供一个线性含3`-T突出端的载体用于直接高效地连接DNA片段。系统中也包含感受态细胞和S.O.C培养基(或用LB培养基),T4(Tth)连接酶把PCR片断连接到T载体上,而Topo TA Cloning用的是DNA Topoisomerase(Topo酶)。Topoisomerase的用途一般使用在复制DNA前把超螺旋DNA切割使之解旋后,再连接成线性DNA。
Topo TA克隆即使用Topoisomerase高效连接的特性把含3`A端的PCR扩增片断快速连接到3`T端载体上。Topo TA克隆系统提供Topoisomerase I载体,感受态细胞用LB培养基。
10μl体积反应体系如下:
1)取T载体1μl (50ng),加入等摩尔数PCR 产物 。
2)加入含ATP的10×Buffer 1μl,T4 DNA连接酶合适单位,用ddH2O 补足至10μl 。
3)稍加离心,通常为14-16℃水浴连接8-14hr,或4℃过夜。
4)转染。
(9)cDNA引物制备。
A.反转录试剂盒(TAKARA 6210A)1st-Strand cDNA合成反应
1)配制下列反应混合液。
试剂 使用量
Oligo dT Primer (50 μM) 1 μl
dNTP Mixture (10 mM each) 1 μl
模板RNA Total RNA:5 μg
RNase free dH2O Up to 10 μl
2)65℃保温5 min后,冰上迅速冷却。
(注:上述处理可使模板RNA变性,提高反转录效率。)
3)在上述Microtube管中配制下列反转录反应液,总量为20 μl。
试剂 使用量
上述变性后反应液 10 μl
5×PrimeScript II Buffer 4 μl
RNase Inhibitor (40 U/μl) 0.5 μl (20 U)
PrimeScript II RTase (200 U/μl) 1 μl (200 U)
RNase free dH2O Up to 20 μl
4)缓慢混匀。
5)按下列条件进行反转录反应:
(30℃ 10 min) (使用Random 6 mers时)
42℃ (~50℃ )*2 30~60 min
6)95℃ 5 min*3 (酶失活)后,冰上冷却。
B.碎片化cDNA单链。
95℃解链,加RNA酶消化多余RNA。
利用核酸内切酶CELI,酶切cDNA单链。
酶切1小时后,使用E-Gel®电泳系统,回收20-50bp片段。
剩余单链继续酶切30分钟,回收20-50bp片段。
反复3-5次,收集多次不同酶切时间的20-50bp 的cDNA 碎片。
(10)合成质粒端的引物:
质粒引物F端: TGTAAAACGACGGCCAGT
质粒引物R端: CAGGAAACAGCTATGACC
(11)基因富集选择性PCR扩增
在0.2mL离心管中,按下列方式加入:
体积(50μL体系)
提取的质粒 2μL
2× NEB Taq Master Mix 25μL
5μM Primer 质粒引物R端 4μL
5μM cDNA Primer 4μL
纯净水 15μL
体积(50μL体系)
提取的质粒 2μL
2× NEB Taq Master Mix 25μL
5μM Primer 质粒引物F端 4μL
纯净水 15μL
以上2个离心管分别混匀,离心数秒,按下列参数PCR循环。
1、预变性 95℃ 30s
2、18轮循环扩增参数: 95℃ 30s,55℃ 30s,68℃ 30s
3、延伸 72℃ 5min
PCR产物-20℃保存,待测序。如需长期保存-70℃以下保存。
(9)凝胶检测
取8μLPCR产物,2%琼脂糖胶检测。
(10)测序
将两组96个PCR扩增产物进行混合测序,选择高通量的Hiseq 2000双端测序。Hiseq 2000测序每一组测一条lane得到30G的数据量。
(11)实验结果:
1、在木薯的2份样品中,进行了酶切,2个标签接头连接,构建DNA文库,进行PCR扩增基因富集区。挑单克隆测序结果。
所有测序结果一端涵盖有设计的 Barcodes Adapter。
>ZB06151579(6)M13+_J_B06
ACTGTGTATTCGTAGACTAATTGGATCATACAGCATTCACCCACAACCACAAAATAAAATGCAATGCGACATATTTGTGAACTAATGCAATCAGCCTATTACATGTCATCATGATGCATGAAACATGCTCAAAACATTTAATTGCTTGATTTAAAACATTAAGCTTGTTCCCACTCACCTCTGGCTAGCTCTGACCAGACACTGAAGCAGCTCACTCACTGCTGGGGTCCTCGGTTCCTCGGGTCCGAACCTACACAGGTGGACTCCAATGAGGGACCAAACATATATAAACACAACTCTAATATATCCCCCAAAAACCCCTAAAACACCATGAAAACATCACAGAAAATATGCATGAAATGGCTGGACCAATCCCT
>ZB06151578(5)M13+_J_A06
ACTGTGTATCCTCTCGTACTAGGTTGAATTACCATCGCGACGCGGTCATCAGTAGGGTAAAACTAACCTGTCTCACGACGGTCTAAACCCAGCTCACGTTCCCTATTGGTGGGTGAACAATCCAACACTTGGTGAATTCTGCTTCACAATGATAGGAAGAGCCGACATCGAAGGATCAAAAAGCAACGTCGCTATGAACGCTTGGCTGCCACAAGCCAGTTATCCCTGTGGTAACTTTTCTGACACCTCTAGCTTCAAATTCCGAAGGTCTAAAGGATCGATAGGCCACGCTTTCACGGTTCGTATTCGTACTGGAAATCAGAATCAAACGAGCTTTTACCCTTTTGTTCCACACGAGACCAATCCCT
>ZB06151577(4)M13+_J_H05
ACTGTACGGCTTGTACACTTCGGGCTGCCTATAACAATCCAGAACGAGCAGTAGATTACTTGTACTCTGTATGTCCAAACTCAATGCATGATTTTCACCAACTTCTATTTAGATTGGCTACTTATGTTGTTTGGTGTTCAGGGTATTCCAGAAGCAGCGGAAGTTGCTGTCCCAGTGTCTCATTTCCCTGCAGGTCAGGCAACTGAAACAGGTACAGCTGCACCTGCATCTGGAGCACCCAATACGTCTCCCTTTAATATGTTCCT
>ZB06151576(3)M13+_J_G05
ACTGTACGGCTCGTGCCCTCATCCTCAGGTGGTACTCCGTCAATCTTGCCGATCGACGGGTTCCTCTCATCCTGTTTACTGAAAAACAGCACACATCACATAAACATTAGCATCAAATGGTTCATATGCAAACACATGAACCCACATCACATACATCACAGACATAGCATATCATTAATGCACATGCATATAATCATTGCATTTCACATCATCATTCAAGACAGGACTCTACATCCTATCCTAGTGGACATGATTTTCCTATTGTGCTTGACCTTCTAGAACATCTATGAGCCCGACACTCTAGGTCCGACCATATGAACCTAGGGCTTTGATACCAATCTGTAACGACCCGAAAATCAGACCGCTACCGGCGCTAGGATCCAGATCG
序列登录NCBI,通过blast比对,所有序列都涵盖部分基因区 (E-vaule<10-5) 。
序列表:
<110> 中国热带农业科学院热带生物技术研究所
<120>一种胞嘧啶甲基化挖掘的方法
<160> 6
<210> 1
<211> 18
<212> DNA
<213> 人工序列
<220>
<221>
<222> (1)...(18)
<223>
<400> 1
TGTAAAACGACGGCCAGT
<210> 2
<211> 18
<212> DNA
<213> 人工序列
<400> 2
>CAGGAAACAGCTATGACC
<210> 3
<211> 377
<212> DNA
<213> 人工序列
>ZB06151579(6)M13+_J_B06
ACTGTGTATTCGTAGACTAATTGGATCATACAGCATTCACCCACAACCACAAAATAAAATGCAATGCGACATATTTGTGAACTAATGCAATCAGCCTATTACATGTCATCATGATGCATGAAACATGCTCAAAACATTTAATTGCTTGATTTAAAACATTAAGCTTGTTCCCACTCACCTCTGGCTAGCTCTGACCAGACACTGAAGCAGCTCACTCACTGCTGGGGTCCTCGGTTCCTCGGGTCCGAACCTACACAGGTGGACTCCAATGAGGGACCAAACATATATAAACACAACTCTAATATATCCCCCAAAAACCCCTAAAACACCATGAAAACATCACAGAAAATATGCATGAAATGGCTGGACCAATCCCT
<210> 4
<211> 368
<212> DNA
<213> 人工序列
>ZB06151578(5)M13+_J_A06
ACTGTGTATCCTCTCGTACTAGGTTGAATTACCATCGCGACGCGGTCATCAGTAGGGTAAAACTAACCTGTCTCACGACGGTCTAAACCCAGCTCACGTTCCCTATTGGTGGGTGAACAATCCAACACTTGGTGAATTCTGCTTCACAATGATAGGAAGAGCCGACATCGAAGGATCAAAAAGCAACGTCGCTATGAACGCTTGGCTGCCACAAGCCAGTTATCCCTGTGGTAACTTTTCTGACACCTCTAGCTTCAAATTCCGAAGGTCTAAAGGATCGATAGGCCACGCTTTCACGGTTCGTATTCGTACTGGAAATCAGAATCAAACGAGCTTTTACCCTTTTGTTCCACACGAGACCAATCCCT
<210> 5
<211> 266
<212> DNA
<213> 人工序列
>ZB06151577(4)M13+_J_H05
ACTGTACGGCTTGTACACTTCGGGCTGCCTATAACAATCCAGAACGAGCAGTAGATTACTTGTACTCTGTATGTCCAAACTCAATGCATGATTTTCACCAACTTCTATTTAGATTGGCTACTTATGTTGTTTGGTGTTCAGGGTATTCCAGAAGCAGCGGAAGTTGCTGTCCCAGTGTCTCATTTCCCTGCAGGTCAGGCAACTGAAACAGGTACAGCTGCACCTGCATCTGGAGCACCCAATACGTCTCCCTTTAATATGTTCCT
<210> 6
<211> 388
<212> DNA
<213> 人工序列
>ZB06151576(3)M13+_J_G05
ACTGTACGGCTCGTGCCCTCATCCTCAGGTGGTACTCCGTCAATCTTGCCGATCGACGGGTTCCTCTCATCCTGTTTACTGAAAAACAGCACACATCACATAAACATTAGCATCAAATGGTTCATATGCAAACACATGAACCCACATCACATACATCACAGACATAGCATATCATTAATGCACATGCATATAATCATTGCATTTCACATCATCATTCAAGACAGGACTCTACATCCTATCCTAGTGGACATGATTTTCCTATTGTGCTTGACCTTCTAGAACATCTATGAGCCCGACACTCTAGGTCCGACCATATGAACCTAGGGCTTTGATACCAATCTGTAACGACCCGAAAATCAGACCGCTACCGGCGCTAGGATCCAGATCG
- 还没有人留言评论。精彩留言会获得点赞!