用于非入侵性性染色体非整倍性确定的统计分析的制作方法
本申请是申请日为2014年02月19日,申请号为201480019119.1,发明名称为“用于非入侵性性染色体非整倍性确定的统计分析”的申请的分案申请。
优先权
本申请要求于2013年6月13日提交的美国专利申请序列号13/917,329的权益,美国专利申请序列号13/917,329为于2013年3月8日提交的美国专利申请序列号13/790,642的继续部分申请,并为于2011年12月28日提交的美国专利申请序列号13/338,963的继续部分申请,美国专利申请序列号13/338,963为于2011年12月9日提交的美国专利申请序列号13/316,154的继续部分申请,美国专利申请序列号13/316,154要求于2011年1月25日提交的美国临时专利申请序列号61/436,135的优先权,其中每一个专利申请被转让给本发明的受让人。
发明领域
本发明涉及鉴于母体混合样品中胎儿贡献百分比,通过检测和确定来自x和y染色体的遗传序列的相对贡献,非入侵性性别确定胎儿或x和y染色体频率异常的统计分析。
发明背景
在以下讨论中,为了背景和介绍的目的将描述某些文章和方法。本文包含的任何事物不被解释为是对现有技术的“承认”。申请人明确地保留在适当时表明本文提及的文章和方法基于适用的法定条款不构成现有技术的权利。
遗传异常造成大量病理学,包括由染色体非整倍性引起的综合征(比如,唐氏综合征)和由导致单基因或多基因疾病或紊乱的种系突变引起的那些综合征。检测总染色体异常,例如三体、易位和大的插入或缺失和单基因性状,例如与rh血型状态、常染色体显性或x连锁紊乱或常染色体隐性紊乱有关的单基因突变或多态性在检测可以影响胎儿的真实和潜在病理学和紊乱方面是有用的。例如,染色体异常(诸如三体13、18和21)、robertsonian易位和较大缺失(例如在digeorge综合征中染色体22上发现的那些)全部对胎儿健康有明显地影响。
尽管常规的技术提供用于这些不同遗传异常的检测方法,但是直到最近不同遗传异常要求不同技术来询问不同种类的突变。例如,用于染色体非整倍性的产前诊断测试的常规方法要求利用通常地在11和14周妊娠之间的绒毛膜绒毛取样(cvs)或通常地在15周之后的羊膜穿刺术从子宫直接地取出胎儿细胞的样品用于遗传分析。但是,这样的入侵性程序携带约百分之一的流产的风险(参见,mujezinovic和alfirevic,obstet.diagn.,110:687-694(2007))。胎儿细胞的其他分析通常包括人类染色体核型分析或原位荧光杂交(fish)并且不提供关于单基因性状的信息;因此,需要另外的用于识别单基因疾病和紊乱的测试。
非入侵性检测在母体基因组中缺乏的父系遗传的dna序列,比如用于胎儿性别检测的y染色体序列和用于血型基因定型的rhd基因自从20世纪90年代中期已经是可能的。但是,最近出现的单分子计数技术-例如数字聚合酶链式反应和特定地大规模平行测序-已经允许循环中的胎儿dna用于非入侵性产前诊断胎儿染色体非整倍性和单基因疾病,但用于测试的其他胎儿异常和/或质量控制参数仍未解决。
发明概述
在本领域中存在对精确确定胎儿性别、x染色体频率和y染色体频率的需求。本发明解决此需求。
提供该概述以以简化的形式介绍在以下在详细描述中进一步描述的观念的选择。该概述并非意图标识所要求保护的主题的关键或本质特征,也并非意图用来限制所要求保护的主题的范围。根据以下书写的包括在所附的附图中表明的以及在所附的权利要求中限定的那些方面的详细描述,所要求保护的主题的其它特征、细节、效用、和优势将是明显的。
在一个方面中,所述方法利用多路扩增和检测性染色体和一个或更多个常染色体(即,常染色体)上选择的核酸区域以计算母体混合样品中与胎儿核酸贡献百分比有关的x和y染色体的频率。利用如本文描述的分析方法确定感兴趣的基因组区域(比如,性染色体序列和来自一个或更多个染色体序列的序列)的选择的核酸区域的相对量。这样的方法被用以确定胎儿的性别、可能的x和y染色体非整倍性和雌雄间性镶嵌现象(intersexmosaicisms)以及评估母体混合样品的污染的似然。
这些和其它的方面、特征和优势将更详细的被提供,如本文描述的。
本文还描述了以下内容:
1.一种计算母体样品中x或y染色体非整倍性的风险的方法,其中所述方法的步骤在计算机上来执行,所述方法包括以下的步骤:
询问一个或更多个y染色体基因座;
询问一个或更多个x染色体基因座;
询问至少第一常染色体上一个或更多个基因座;
估计y染色体、x染色体和所述第一常染色体的染色体频率;
计算所述母体样品中所述y染色体以无拷贝、一个拷贝、或两个或更多个拷贝存在的似然值;
通过比较所述似然值与假定无拷贝的所述y染色体的第一数学模型、假定一个拷贝的所述y染色体的第二数学模型和假定两个或更多个拷贝的所述y染色体的第三数学模型来计算所述母体样品中所述y染色体的非整倍性的风险;
计算所述母体样品中所述x染色体以一个拷贝、两个拷贝、或三个或更多个拷贝存在的似然值;以及
通过比较所述似然值与假定一个拷贝的所述x染色体的第一数学模型、假定两个拷贝的所述x染色体的第二数学模型和假定三个或更多个拷贝的所述x染色体的第三数学模型来计算所述母体样品中所述x染色体的非整倍性的风险。
2.如项目1所述的方法,其中所述x或y染色体非整倍性是胎儿非整倍性。
3.如项目1所述的方法,其中所述方法还包括通过分析至少一个常染色体上多态性的频率来计算所述母体样品中的胎儿核酸比例。
4.如项目3所述的方法,所述方法还包括确定所述母体样品中的所述胎儿核酸比例是否足以可靠地执行分析。
5.如项目1所述的方法,其中至少十个或更多个多态性基因座被询问。
6.如项目5所述的方法,其中至少两个常染色体上的至少十个或更多个多态性基因座被询问。
7.如项目6所述的方法,其中通过分析来自至少两个常染色体的多态性的频率来计算所述母体样品中的胎儿核酸比例。
8.如项目6所述的方法,其中至少三个常染色体上的至少十个或更多个多态性基因座被询问。
9.如项目8所述的方法,其中通过分析来自至少三个常染色体的多态性的频率来计算所述母体样品中的所述胎儿核酸比例。
10.如项目1所述的方法,其中所述x染色体、所述y染色体和所述至少一个常染色体的每一个上至少24个基因座被询问。
11.如项目10所述的方法,其中所述x染色体、所述y染色体和所述至少一个常染色体的每一个上至少32个基因座被询问。
12.如项目11所述的方法,其中每一个基因座被测量至少100次。
13.如项目3所述的方法,其中至少96个多态性基因座被测量以计算所述胎儿核酸比例百分比。
14.如项目2所述的方法,其中使用关于现有风险的外来信息调整胎儿非整倍性的计算的风险。
15.如项目2所述的方法,其中利用xff计算x染色体非整倍性的风险。
16.如项目2所述的方法,其中利用yff计算y染色体非整倍性的风险。
17.如项目1所述的方法,其中通过bootstrap抽样来执行计算所述母体样品中所述y染色体以无拷贝、一个拷贝和两个或更多个拷贝存在的似然值和计算所述母体样品中所述x染色体以一个拷贝、两个拷贝或三个或更多个拷贝存在的似然值。
18.如项目1所述的方法,其中使用log10比数比执行所述计算风险步骤。
19.一种计算母体样品中x染色体非整倍性的风险的方法,其中所述方法的步骤在计算机上来执行,所述方法包括以下的步骤:
询问一个或更多个x染色体基因座;
询问至少第一常染色体上的一个或更多个多态性基因座;
估计x染色体和所述第一常染色体的染色体频率;
计算所述母体样品中所述x染色体以一个拷贝、两个拷贝或多于两个拷贝存在的似然值;以及
通过比较似然值与假定一个拷贝的所述x染色体的第一数学模型、假定两个拷贝的所述x染色体的第二数学模型和假定三个拷贝的所述x染色体的第三数学模型来计算所述母体样品中所述x染色体的非整倍性的风险。
20.如项目19所述的方法,其中所述x染色体非整倍性是胎儿非整倍性。
21.如项目19所述的方法,其中所述方法还包括通过分析至少一个常染色体上的多态性基因座的频率来计算所述母体样品中的胎儿核酸比例。
22.如项目20所述的方法,所述方法还包括确定所述母体样品中的所述胎儿核酸比例是否足以可靠地执行分析。
23.如项目19所述的方法,其中至少十个或更多个多态性基因座被询问。
24.如项目23所述的方法,其中至少两个常染色体上的至少十个或更多个多态性基因座被询问。
25.如项目24所述的方法,其中通过分析来自至少两个常染色体的多态性基因座的频率来计算所述母系样品中的胎儿核酸比例。
26.如项目24所述的方法,其中至少三个常染色体上的至少十个或更多个多态性基因座被询问。
27.如项目26所述的方法,其中通过分析来自至少三个常染色体的多态性的频率来计算所述母体样品中的所述胎儿核酸比例。
28.如项目19所述的方法,其中所述x染色体上至少24个基因座被询问。
29.如项目28所述的方法,其中所述x染色体和所述至少一个常染色体的每一个上至少32个基因座被询问。
30.如项目28所述的方法,其中每一个基因座被测量至少20次。
31.如项目21所述的方法,其中至少96个多态性基因座被测量以计算所述胎儿核酸比例。
32.如项目20所述的方法,其中使用关于现有风险的外来信息调整胎儿非整倍性的计算的风险。
33.如项目20所述的方法,其中利用xff计算x染色体非整倍性的风险。
附图简述
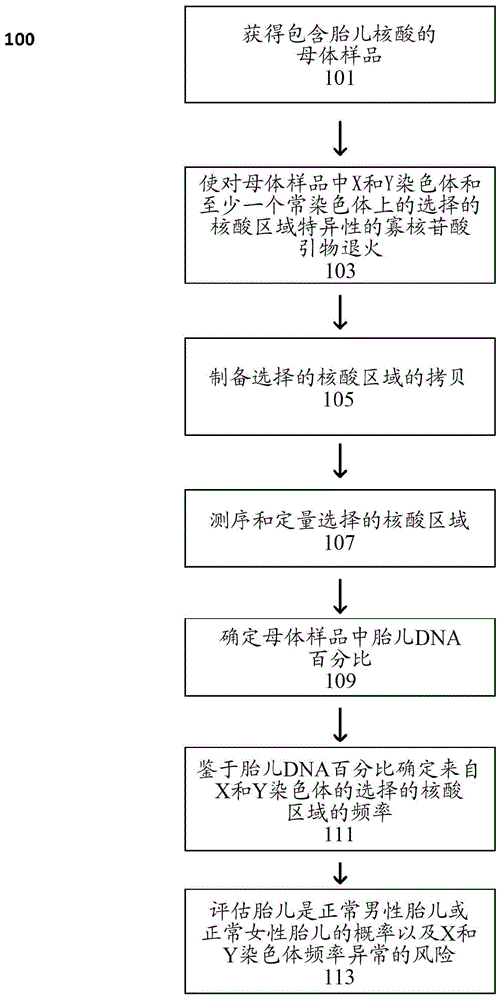
图1是根据本发明的一种测定方法的简化的流程图。
图2阐明了用于检测两个或更多个选择的核酸区域的多路测定系统。
图3阐明了用于检测两个或更多个选择的核酸区域的可选的多路测定系统。
图4阐明了用于检测两个或更多个选择的核酸区域的又另一个可选的多路测定系统。
图5阐明了用于检测两个或更多个选择的核酸区域的又另一个可选的多路测定系统。
图6阐明了用于检测选择的核酸区域的又另一个可选的多路测定系统。
图7阐明了用于检测选择的核酸区域的又另一个可选的多路测定系统。
图8是用于执行根据本发明的统计分析的示例性方法的简化的流程图。
图9是用于执行根据本发明的统计分析的示例性方法的又另一个简化的流程图。
发明详述
除非另有说明,本文描述的方法可使用分子生物学(包括重组技术)、细胞生物学、生物化学、和微阵列和测序技术的常规技术和说明,其在本领域熟练技术人员能力范围内。此类常规技术包括聚合物阵列合成、寡核苷酸的杂交和连接、寡核苷酸的测序、和利用标记检测杂交。可通过参考本文的实例获得合适的技术的特别说明。但是,当然也可使用等价的常规程序。此类常规技术和说明可以在实验室手册中被发现,例如green等人编辑,genomeanalysis:alaboratorymanualseries(vols.i-iv)(1999);weiner等人编辑,geneticvariation:alaboratorymanual(2007);dieffenbach,dveksler编辑,pcrprimer:alaboratorymanual(2003);bowtell和sambrook,dnamicroarrays:amolecularcloningmanual(2003);mount,bioinformatics:sequenceandgenomeanalysis(2004);sambrook和russell,condensedprotocolsfrommolecularcloning:alaboratorymanual(2006);以及sambrook和russell,molecularcloning:alaboratorymanual(2002)(全部来自coldspringharborlaboratorypress);stryer,l.,biochemistry(第4版)w.h.freeman,newyork(1995);gait,“oligonucleotidesynthesis:apracticalapproach”irlpress,london(1984);nelson和cox,lehninger,principlesofbiochemistry,第3版,w.h.freemanpub.,newyork(2000);以及berg等人,biochemistry,第5版,w.h.freemanpub.,newyork(2002),所有这些出版物通过引用以其整体为了所有目的被并入本文。在描述本发明的组合物、研究工具和方法之前,要理解本发明不局限于描述的特定的方法、组合物、目标和用途,因为这些当然可变化。还要理解,本文使用的术语只是为了描述特定方面的目的并且不是意图限制本发明的范围,本发明的范围将只由所附的权利要求限制。
应注意,如本文以及在所附的权利要求中使用的,单数形式“一(a)”、“一(an)”和“该(the)”包括复数指代对象,除非上下文另有清楚的指示。因此,例如,提及“核酸区域”指一个、多于一个此类区域、或其混合物,并且提及“方法”包括提及本领域技术人员已知的等价步骤和方法,等等。
在提供值的范围的情况下,要理解,在该范围的上限和下限之间的每个介于中间的值以及在该陈述范围中的任何其它陈述或介于中间的值被包括在本发明内。在陈述的范围包括上限和下限的情况下,排除这些限值中的任一个的范围也被包括在本发明中。
为了包括描述和公开制剂和方法的目的所有目的,通过引用将本文提到的所有出版物并入,并且其可与本文描述的发明关联使用。
在以下的说明中,列出了很多具体细节以提供本发明的更彻底的理解。但是,对于本领域技术人员来说将清楚的是,本发明可以被实施而无这些具体细节中的一个或更多个。在其它的情形中,为了避免模糊本发明,本领域技术人员熟知的特征和程序未被描述。
定义
本文使用的术语意图具有如由本领域普通技术人员所理解的清楚且普通的意义。以下定义意图帮助读者理解本发明,但并非意图改变或者以其他方式限制此类术语的含义,除非另有声明。
术语“扩增的核酸”为其量与其起始量相比已通过体外进行的任何核酸扩增或复制方法增加了至少两倍的任何核酸分子。
术语“染色体异常”指的是染色体的全部或部分的任何遗传变异。遗传变异可以包括但不局限于诸如加倍或缺失、易位、倒位、和突变的任何拷贝数变化。该术语还包括胎儿或母体组织中的染色体镶嵌现象。
如本文使用的术语“诊断工具”指的是例如如在系统中使用的本发明的任何组合物或方法以对患者样品进行诊断测试或分析。
术语“雌雄间体镶嵌现象”或“性染色体镶嵌现象”或“性染色体镶嵌”指的是在一个个体中存在具有不同性染色体基因型的两种或更多种细胞群。当在一个个体中一些细胞具有,比如两个x染色体(xx)并且在该个体中其他细胞具有一个x染色体和一个y染色体(xy)时;当在一个个体中一些细胞具有一个x染色体(xo)并且在该个体中其他细胞具有一个x染色体和一个y染色体(xy)时;或当在一个个体中一些细胞具有两个x染色体和一个y染色体(xxy)并且在该个体中其他细胞具有一个x染色体和一个y染色体(xy)时,雌雄间体镶嵌现象出现。
术语“杂交”通常地意指互补的核酸链藉以发生配对的反应。dna通常地是双链的,并且当链是分开的时,在适当的条件下其将重新杂交。杂合体可在dna-dna、dna-rna或rna-rna之间形成。其可在短链和含有与短链互补的区域的长链之间形成。不完全杂合体也可形成,但是其越不完全,其将越不稳定(并且越不可能形成)。
术语“似然”指的是通过直接地计算似然得到的任何值或可以与似然相关或以其他方式指示似然的任何值。
如本文使用的术语“基因座(locus)”和“基因座(loci)”指的是基因组中已知位置的核酸区域。
如本文使用的术语“母体样品”指的是取自含有胎儿和母体两者的核酸(例如,dna)的妊娠女性的任何样品。优选地,用于在本发明中使用的母体样品通过相对地非入侵性的方法例如静脉切开术或用于从受试者提取外周样品的其它标准技术获得。
“微阵列”或“阵列”指的是具有表面的固相支持物,所述表面优选地但不排他地平面的或基本上平面的表面,其载有包含核酸的位点的阵列,使得阵列的每个位点包含寡核苷酸或多核苷酸的基本上相同或相同的拷贝并且被空间上限定且不与阵列的其它成员位点重叠;即,位点是空间上离散的。阵列或微阵列还可包含具有平面的非平面可询结构,诸如珠或孔。阵列的寡核苷酸或多核苷酸可以共价地结合至固体支持物,或可以非共价地结合。常规的微阵列技术被综述在,比如schena编辑,microarrays:apracticalapproach,irlpress,oxford(2000)中。“阵列分析”、“通过阵列分析”或“通过微阵列分析”指的是利用微阵列的分析,诸如例如特定核酸的分离或一种或更多种生物分子的序列分析。
当关于检测选择的核酸区域使用时,通过“非多态的”或“多形性不可知论的(polymorphism-agnostic)”来意指这样的核酸区域的检测:可以包含一种或更多种多态性,但是其中检测不依赖于区域内的特定多态性的检测。因此选择的核酸区域可以包含多态性,但是利用本发明的方法检测该区域是基于该区域的存在而非在该区域中特定的多态性的存在或不存在。
如本文使用的术语“寡核苷酸(oligonucleotides)”或“寡核苷酸(oligos)”指的是天然的或修改的核酸单体的线性寡聚物,包括脱氧核糖核苷酸、核糖核苷酸、其异头形式、肽核酸单体(pna)、锁核苷酸单体(lna)等,或其组合,其能通过单体与单体相互作用的规则模式的方式特异地与单链多核苷酸结合,所述单体与单体相互作用诸如碱基配对的watson-crick类型、碱基堆积、碱基配对的hoogsteen或反式hoogsteen类型等。通常地单体通过磷酸二酯键或其类似物连接以形成大小范围从几个单体单位例如8-12个到几十个单体单位例如100-200或更多个的寡核苷酸。
如本文使用的术语“聚合酶”指的是利用另一条链作为模板将单独核苷酸连接在一起成长链的酶。有两种常见类型的聚合酶-合成dna的dna聚合酶和合成rna的rna聚合酶。在这两种类型中,取决于什么类型的核酸能起模板的作用以及形成什么类型的核酸,有很多亚型的聚合酶。
如本文使用的“聚合酶链式反应”或“pcr”指的是用于体外复制靶dna的特定片段的技术,甚至在额外的非特异的dna的存在下。引物被添加至靶dna,在该处引物利用核苷酸和通常地taq聚合酶等起始靶dna的复制。通过循环温度,靶dna重复地被变性和复制。即使与其它随机dna混合,靶dna的单拷贝能被扩增以获得数十亿的复制物。聚合酶链式反应能被用来检测并测量很小量的dna并被用来生成dna的定制片段。在一些实例中,线性扩增方法可被用作pcr的可以替代选择。
如本文使用的术语“多态性”指的是可以指示该特定的基因座的基因座中的任何遗传变化,包括但不局限于单核苷酸多态性(snp)、甲基化差异、短串联重复(str)等。
通常地,“引物”是诸如在聚合酶链式反应的合成步骤中,或在某些测序反应中使用的引物延伸技术中被用来例如引发dna延伸、连接和/或合成的寡核苷酸。引物也可以在杂交技术中被用作提供核酸区域与捕获寡核苷酸的互补性的方法,用于特定核酸区域的检测。
如本文使用的术语“研究工具”指的是被用于学术或商业性质的科学探究的本发明的任何方法,包括药物和/或生物治疗的开发。本发明的研究工具并非意图是治疗性的或要受制于监管机构批准;而是,本发明的研究工具意图有利于研究且有助于此类开发活动,包括目的是产生支持监管机构提交的信息而执行的任何活动。
如本文使用的术语“选择的核酸区域”指的是与个体染色体对应的核酸区域。选择的核酸区域可被从用于检测的样品直接地分离并基于例如杂交和/或其它基于序列的技术富集,或在检测序列之前其可使用样品作为模板而被扩增。
术语“选择性扩增”和“选择地扩增”等指的是整体上或部分的取决于寡核苷酸与选择的核酸区域中的序列的杂交的扩增过程。在某些选择性扩增中,用于扩增的引物与选择的核酸区域是互补的。在其它的选择性扩增中,用于扩增的引物是通用引物,但是如果用于扩增的核酸区域与感兴趣的选择的核酸区域是互补的,则其只产生一种产物。
如本文使用的术语“测序”和“序列确定”等通常地指的是可以被用来确定核酸中的核苷酸碱基的顺序的任何和所有的生化方法。
当提及在指定的测定条件下导致生成统计上显著的阳性信号的结合配偶体(例如,核酸探针或引物、抗体等)时,如本文使用术语“特异性结合”、“特异的结合”等。通常地,相互作用将随后导致作为不期望的相互作用(背景)的结果生成的任何信号的标准偏差的至少两倍的可检测的信号。
当被用来描述扩增过程时,术语“通用的”指的是使用单个引物或一组引物用于多个扩增反应。例如,在检测96种不同的靶序列时,所有的模板可共用同一通用引发序列,允许利用单组引物多路扩增96种不同的序列。这样的引物的使用大大地简化了多路化,因为只需要两个引物来扩增多个选择的核酸序列。当被用来描述引发位点时,术语“通用的”是通用引物将与其杂交的位点。还应注意可使用通用引发序列/引物的“组”。例如,在高度地多路的反应中,使用若干组通用序列可以是有用的,而不是单个组;例如,96种不同的核酸可具有第一组通用引发序列,和第二96不同组的通用引发序列等。
一般发明
本发明提供了用于鉴定x和y染色体的拷贝数变体的改进的方法。本发明的方法可用于确定胎儿的性别,评估胎儿中x染色体非整倍性、y染色体非整倍性或性染色体镶嵌现象的可能性,或用于确定母体样品的可能的污染。在一些方面中,本发明的方法也可用于检测母亲的x染色体非整倍性或镶嵌现象。
本发明的测定方法包括选择性富集来自x染色体和y染色体和一个或更多个非性别参考染色体(常染色体)的选择的核酸区域。本发明的明显优势是,选择的核酸区域可利用多种检测和定量技术被进一步分析,所述多种检测和定量技术包括但不局限于杂交技术、数字pcr和优选地高通量测序确定技术。引物可以针对除了x和y染色体的任何染色体的任何数目的选择的核酸区域设计。尽管在选择的核酸区域的鉴定和定量之前扩增是非强制性的,在检测之前有限的扩增是优选的。
本发明是在例如大规模平行鸟枪测序的更随机技术(比如,随机测序)或使用最近已经被用以检测母体样品,例如母体血液中拷贝数变化的随机数字pcr之上的改进。前面提及的方法依靠样品中dna片段的全部或统计学上显著的群体的测序,随后映射片段至其合适的染色体或另外将片段与其合适的染色体关联或对齐。然后将鉴定的片段彼此比较或与一些其他参考(比如,具有已知整倍体染色体互补物的样品)比较以确定特定染色体的拷贝数变化。随机或鸟枪测序方法相比于本发明是固有地无效的,因为在感兴趣的染色体区域上产生的数据仅由产生的很少一部分数据组成。
取决于样品中dna的非常广泛的取样的技术提供分析的dna的广泛覆盖,但是事实上是以1x或更少的基础取样样品内包含的dna(即,二次取样)。与此相比,本方法中使用的选择性扩增和/或富集技术(例如,杂交)提供仅覆盖选择的核酸区域的深度;并且如此提供具有优选地2x或更大的平均序列覆盖率的选择的核酸区域的“超取样”,更优选地100x或更大、200x或更大、250x或更大、500x或更大、750x或更大的覆盖率或甚至更优选地选择的核酸区域的1000x或更大的序列覆盖率。
因此,本发明的测定中,基本上大多数用于定量的分析的序列提供x和y染色体和一个或更多个常染色体上一个或更多个选择的核酸区域的存在的信息。本发明的方法不需要分析大量不是来自感兴趣的染色体的序列和不提供有关感兴趣的染色体的相对量的信息的序列。
检测和定量性染色体
本发明提供用于确定母体样品中x和y序列的频率的方法。这些频率可以被使用,比如以确定胎儿性别和/或用于鉴定x染色体非整倍性、y染色体非整倍性和/或性染色体镶嵌现象。样品是包含母体和胎儿dna二者的母体样品,例如母体血液样品(即,全血、血清或血浆)。所述方法富集和/或分离和扩增母体样品中对应于x染色体和y染色体以及一个或更多个常染色体的选择的核酸区域的一个或优选地若干个至许多个,鉴于该样品中存在胎儿dna的百分比,所述核酸区域用于确定x和y染色体序列的存在或不存在和/或相对量或频率。如以上详细描述的,本发明的方法优选地利用一个或更多个选择性扩增、连接或富集(比如,使用与选择的核酸区域特异性杂交的一种或更多种核酸)步骤以提高样品中选择的核酸区域的含量。选择性扩增、连接和/或富集步骤通常包括将选择的核酸区域的拷贝工程化用于进一步分离、扩增和分析的机制。此选择性方法与其他技术,比如大规模平行鸟枪测序法使用的随机扩增方法直接不同,因为此类技术通常包括随机扩增基因组的全部或基本部分。
在某些方面中,本发明的方法可以鉴定母体非整倍性,包括母体镶嵌现象。鉴于母体样品中的胎儿dna,母体x和y染色体材料的频率必须被分析。这样的母体非整倍性的确定的风险特征可以取决于特定的非整倍性而不同。例如,在其中母亲是xx/xo嵌合体的情况下,确定的可能性将取决于母体镶嵌现象的水平、受影响的组织以及母体样品中胎儿百分比。
以上实例表明,如果人们将通过本文描述的方法强有力地检测以这样低的百分比存在的特定核酸,测量额外染色体中的变化必须显著小于额外染色体的升高百分比。
图1是根据本发明的一个方法100的简化的流程图。在第一步骤中,获得母体样品101。母体样品包含母体和胎儿核酸二者。母体样品可以是取自包含胎儿和母体核酸(比如,dna)二者的妊娠女性的任何样品。优选地,用于在本发明中使用的母体样品是无细胞的,并且通过相对地非入侵性的方法例如静脉切开术或用于从受试者提取外周样品的其它标准技术获得。
在下一个步骤103中,将对x染色体和y染色体和至少一个常染色体(并且优选地多于一个常染色体)上选择的核酸区域特异性的寡核苷酸引物退火至母体样品中选择的核酸。在步骤105中,寡核苷酸引物被用以选择性扩增选择的核酸区域以产生选择的核酸区域的拷贝。如以下详细描述的,选择的核酸区域经历选择性扩增步骤,但在选择性扩增步骤之前或者优选地在选择性扩增步骤之后也可以经受通用扩增步骤。此外,可以如以下描述的执行一个或更多个富集步骤。又,作为扩增的可替代选择,富集步骤可以被执行,例如通过选择性杂交,这将选择的核酸区域与样品中的其他核酸分离。
在步骤107中,然后将扩增的或拷贝的选择的核酸区域测序和定量。优选的实施方案利用高通量或下一代测序技术,尽管其他技术可以任选地被使用,如以下描述的。高通量测序允许测序确定和定量步骤的大规模平行化。
在步骤109中,确定母体样品中胎儿dna的百分比。下一步,在步骤111中,鉴于步骤109中确定的胎儿dna百分比,确定来自x染色体和y染色体的选择的核酸区域的频率。如本文详细描述的,鉴于胎儿百分比的来自x染色体和y染色体的选择的核酸区域的频率允许在步骤113评估x染色体频率异常的风险,例如由x染色体非整倍性、x染色体镶嵌现象或x染色体污染引起的那些和/或y染色体频率异常的风险,例如由y染色体非整倍性、y染色体镶嵌现象或来自怀有女性胎儿的女性的母体样品的y染色体污染。
因此,通常与x染色体上多重基因座对应的选择的核酸区域被检测并且被总计以确定母体样品中x染色体的相对频率。与一个或更多个常染色体上多重基因座对应的选择的核酸区域被检测并且被求和以确定母体样品中一个或更多个另外的染色体的相对频率,母体样品中一个或更多个另外的染色体的相对频率允许计算胎儿百分比。确定胎儿百分比之后,根据胎儿百分比观察x染色体的频率以便评估x染色体异常是否存在。类似地,与y染色体上多重基因座对应的选择的核酸区域被检测并且被求和以确定母体样品中y染色体的相对频率并且根据胎儿百分比观察y染色体的频率以便评估y染色体异常是否存在。
本发明的方法分析代表至少三个染色体上选择的基因座的多重选择的核酸区域:x染色体、y染色体和至少一个常染色体,并且分析和单独定量每一个选择的核酸区域的相对频率以确定样品中每一个选择的核酸区域的相对频率。样品中选择的核酸区域的总和被用以确定样品中胎儿dna百分比并且被比较以统计学上确定染色体非整倍性或其他染色体异常是否与x和/或y染色体序列相关地存在。
在另一方面中,每一个染色体上选择的核酸区域的亚组被分析以确定染色体异常或染色体频率异常是否存在。特定染色体的选择的核酸区域频率可以求和,并且选择的核酸区域的总和可以被用以确定异常。本发明的此方面对来自每一个染色体的单独的选择的核酸区域的频率求和并且然后比较x染色体与一个或更多个非性染色体上选择的核酸区域的和并且比较y染色体与一个或更多个常染色体上选择的核酸区域的和。在确定染色体异常是否存在时,选择的核酸区域的亚组可以被随机地选择但具有足够的数目以获得统计上显著的结果。可以对母体样品进行选择的核酸区域的不同亚组的多重分析以获得更大的统计功效。例如,如果对于染色体y有100个选择的核酸区域且对于染色体2有100个选择的核酸区域,则对染色体中的每一个可进行评价少于100个区域的一系列分析。例如,可以进行评价少于50个区域的一系列分析,例如少于30个区域、少于或等于16个区域、少于10个区域或8个区域。在另一个方面中,特定的选择的核酸区域可以在已知具有较少样品之间的变化的每一个染色体上来选择,或用于确定染色体频率的数据可以被限制,比如通过忽略样品内来自具有非常高或非常低频率的选择的核酸区域的数据。
在又另一个方面中,将选择的核酸区域的频率的比与已经对遗传学上整倍体受试者的统计学上显著群体确定的参考平均比比较,所述遗传学上整倍体受试者即为不具有x染色体异常或y染色体异常的受试者。
本领域中技术人员应该理解,鉴于母体样品中胎儿dna百分比确定x和y染色体的频率的方法可以与其他非入侵性产前诊断技术联合,例如评估常染色体的胎儿非整倍性的风险的那些技术或检测胎儿中多态性序列的技术。
测定方法
在本发明中可以利用大量不同测定方法,包括利用仅由固定的寡核苷酸组成的寡核苷酸的组的测定,或由固定的寡核苷酸和一种或更多种桥接寡核苷酸组成的寡核苷酸的组的测定。另外,组中的寡核苷酸可以在它们可以被连接的地方与直接彼此毗邻的选择的核酸序列杂交,或组中的寡核苷酸可以不与直接彼此毗邻的选择的核酸序列杂交,并且因此在连接组中的寡核苷酸之前使用利用聚合酶和dntp的引物延伸反应。图2至7阐明了一些示例性测定方法。
图2阐明了一种示例性方法的实施方案,该实施方案中在单串联反应测定中检测两个不同的选择的核酸区域。这样的方法的实施方案、测定系统和相关的实施方案被详细描述在,比如2011年1月25日提交的ussn13/013,732;2011年9月26日提交的ussn13/245,133;2011年8月8日提交的ussn13/205,570;2011年11月10日提交的ussn13/293,419;2011年8月8日提交的ussn13/205,409;2011年8月8日提交的ussn13/205,603;2012年2月29日提交的ussn13/407,978;2011年10月15日提交的ussn13/274,309;2011年12月9日提交的ussn13/316,154;和2011年12月28日提交的ussn13/338,963,其全部以其整体被并入本文。与两个不同的选择的核酸区域215、231特异性地杂交的两组固定序列寡核苷酸(201和203、223和225)被引导202至遗传样品并且被允许与各自选择的核酸区域杂交204。固定序列寡核苷酸的每一组包括具有序列特异性区域205、227,通用引物区域209和标志区221、235的寡核苷酸201、223。组中其他固定序列寡核苷酸包括序列特异性区域207、229和通用引物区域211。固定序列寡核苷酸大小通常在从约30-200个核苷酸长度的范围,或从约30-150个核苷酸长度,或从约35-120个核苷酸长度,或从约40-70个核苷酸长度。如果使用桥接寡核苷酸,桥接寡核苷酸大小通常在从约4至约80个核苷酸长度的范围,或从约4至约60个核苷酸长度,或从约5至约50个核苷酸长度,或从约7至约40个核苷酸长度,或从约10至约40个核苷酸长度,或从约12至约30个核苷酸长度,或从约15至约25个核苷酸长度。
在杂交之后,优选地从样品的剩余物中分离未杂交的固定序列寡核苷酸(步骤未显示)。桥接寡核苷酸213、233被引导至固定序列寡核苷酸/核酸区域的杂交的对并且被允许与这些区域杂交206。尽管在图2中显示为两个不同桥接寡核苷酸,事实上相同的桥接寡核苷酸可以适于两个杂交事件(假定序列是相同的或实质上相似的),或它们可以是来自简并序列寡核苷酸的库的两种寡核苷酸。连接208杂交的寡核苷酸以产生跨越感兴趣的每一个选择的核酸区域并且与感兴趣的每一个选择的核酸区域互补的连续的核酸。应该注意的是,尽管此特定实施方案例证了利用两个固定序列寡核苷酸和一个桥接寡核苷酸扩增每一个选择的核酸区域的方法,可以使用仅使用与彼此毗邻杂交的两个固定序列寡核苷酸的方法,或可以使用仅使用不与彼此毗邻杂交,但其中使用聚合酶和dntp填充“空隙”的两个固定序列寡核苷酸的方法。
在连接之后,通用引物217、219被引入以扩增210连接的寡核苷酸以产生212包含感兴趣的选择的核酸区域的序列的扩增产物237、239。这些扩增产物237、239被分离(任选地)、被检测(即,测序)和定量以提供关于样品中选择的核酸区域的存在和量的信息。
大量扩增方法可以被用于选择性扩增本发明的方法中分析的选择的核酸区域,以允许保留起始样品中选择的核酸区域的相对量的方式增加选择的核酸区域的拷贝数。尽管本文未详细描述扩增和分析的所有组合,但是利用与该说明书一致的不同的、可比较的扩增和/或分析方法来分析选择的核酸区域充分在本领域技术人员能力范围内,因为在阅读本公开内容后这样的变化对本领域技术人员将是明显的。
可用于本发明的扩增方法包括但不限于聚合酶链式反应(pcr)(美国专利号4,683,195和4,683,202;和描述在pcrtechnology:principlesandapplicationsfordnaamplification,ed.h.a.erlich,freemanpress,ny,n.y.,1992)中;连接酶链式反应(lcr)(wu和wallace,genomics4:560,(1989);landegren等人,science241:1077(1988));链置换扩增(sda)(美国专利号5,270,184和5,422,252);转录介导扩增(tma)(美国专利号5,399,491);线性连接扩增(lla)(美国专利号no.6,027,923),自主序列复制(guatelli等人,pnasusa,87:1874(1990)和wo90/06995);靶多核苷酸序列的选择性扩增(美国专利号6,410,276);共有序列引发的聚合酶链式反应(cp-pcr)(美国专利号4,437,975);随机引发的聚合酶链式反应(ap-pcr)(美国专利号5,413,909和5,861,245);和基于核酸的序列扩增(nasba)(参见美国专利号5,409,818、5,554,517和6,063,603,其每一个通过引用被并入本文)。可以被使用的其他扩增方法包括:pct专利申请号pct/us87/00880中描述的qβ复制酶;walker等人,nucleicacidsres.20(7):1691-6(1992)中描述的等温扩增方法,例如sda;和美国专利号5,648,245中描述的滚环扩增。又,可以被使用的其他扩增方法被描述在美国专利号5,242,794、5,494,810、4,988,617和美国序列号09/854,317和美国公布号20030143599中,其每一个通过引用被并入本文。在优选的方面中,通过多路基因座特异的pcr扩增dna。在一些方面中,利用适配体连接和单引物pcr来扩增dna。扩增的其他可得的方法包括平衡pcr(makrigiorgos,等人,natbiotechnol,20:936-39(2002))和自主序列复制(guatelli等人,pnasusa,87:1874(1990))。基于这样的方法,本领域技术人员能容易地在对感兴趣的选择的核酸区域5'和3'的任何合适区域中设计引物。这样的引物可被用以扩增任何长度的dna,只要dna在其序列中含有感兴趣的选择的核酸区域。
被选择的选择的核酸区域的长度是足够长以提供区分选择的核酸区域彼此的足够的序列信息。通常地,选择的核酸区域长度是至少约16个核苷酸,并且更通常地,选择的核酸区域长度至少是约20个核苷酸。在本发明的优选的方面中,选择的核酸区域长度是至少约30个核苷酸。在本发明的更优选的方面中,选择的核酸区域长度是至少约32、40、45、50或60个核苷酸。在本发明的其它方面中,选择的核酸区域长度可以是约100、150或多达200。
在一些方面中,选择性扩增过程利用一轮或几轮扩增,所述一轮或几轮扩增用含有与选择的核酸区域互补的核酸的引物对(即,序列特异性扩增过程)。在其它方面中,选择性扩增包括初始的线性扩增步骤(也是序列特异性扩增过程)。如果dna的初始量是有限的,线性扩增方法可以是特别有用的。线性扩增以代表原始dna含量的方式提高dna分子的量,这种方式帮助在例如本发明的情况下在需要选择的核酸区域的精确定量时降低抽样误差。
因此,在优选的方面中,对含有无细胞dna的起始母体样品执行有限的循环数的序列特异的扩增。循环数通常地小于用于通常pcr扩增的循环数,例如5-30个循环或更少。
寡核苷酸的组中的寡核苷酸被设计以序列特异的方式与样品杂交并且扩增选择的核酸区域。用于选择性扩增的引物优选地被设计以1)有效扩增来自感兴趣的染色体的选择的核酸区域;2)具有来自不同母体样品中母体和/或胎儿来源的表达的可预测的范围;以及3)对选择的核酸区域是与众不同的,即不扩增非选择的核酸区域。可以用末端标签在5’端处(例如,用生物素)或沿着引物或探针的其它位置修改引物或探针,使得扩增产物能被纯化或附接至固体基质(例如,珠或阵列),用于进一步的分离或分析。在优选的方面中,引物被工程化以具有比如将在多路反应中使用的相容的解链温度,允许扩增许多选择的核酸区域,使得单反应产生来自不同选择的核酸区域并且优选地所有选择的核酸区域的多重dna拷贝。然后来自选择性扩增的扩增产物可以用标准pcr方法或用线性扩增被进一步扩增。
无细胞dna能从比如,来自妊娠妇女的全血、血浆、或血清分离,并用被工程化以扩增与感兴趣的染色体对应的一组数目的选择的核酸区域的引物孵育。优选地,用于初始扩增x染色体特异序列的引物对的数目(以及因此x染色体上选择的核酸区域的数目)将是8或更多,例如16或更多、32或更多、48或更多或96或更多。类似地,用于初始扩增y染色体特异序列(以及因此y染色体上选择的核酸区域的数目)和一个或更多个常染色体参考染色体的每一个上的引物对的数目将是8或更多,例如16或更多、32或更多、48或更多或96或更多。引物对的每一个与单选择的核酸区域对应,并且引物对任选地被加标签用于鉴定(比如,通过使用如以上描述的标志(indices)或标志(indexes))和/或分离(比如,包含用于捕获的核酸序列或化学部分)。执行有限数目的扩增循环,优选地10个或更少。通过本领域中已知的方法任选地随后分离扩增产物(扩增的选择的核酸区域)。例如,当引物被连接至生物素分子时,扩增产物能经由在固体基质上与亲和素或链霉亲和素结合被分离。然后可以对扩增产物进行进一步的生化过程,例如用其它引物(例如,通用引物)另外扩增和/或例如序列确定和杂交的检测技术。
扩增的效率在选择的核酸区域之间和在循环之间可以变化,使得在某些系统中,归一化(如以下描述的)可以被用以确保来自扩增选择的核酸区域的产物代表样品的核酸含量。实践本发明的方法的人们能开采关于扩增的产物的相对频率的数据以确定选择的核酸区域中的变化,包括样品内选择的核酸区域中和/或不同样品(特定地来自不同样品中相同的选择的核酸区域)中选择的核酸区域之间的变化以使数据归一化。
作为对选择性括增的可替代选择,选择的核酸区域可以通过杂交技术(比如,捕获杂交或杂交至阵列),任选地,随后一轮或更多轮循环扩增来富集。任选地,杂交的或捕获的选择的核酸区域在扩增和序列确定之前被释放(比如,通过变性)。可以使用允许选择性富集分析中使用的选择的核酸区域的多种方法从母体样品分离选择的核酸区域。分离可以是移除分析中未使用的母体样品中的dna和/或移除初始富集或扩增步骤中使用的任何多余寡核苷酸。例如,可以使用杂交技术(富集)从母体样品分离选择的核酸区域,比如利用将选择的核酸区域与例如珠或阵列的固体基质上互补的寡核苷酸结合,随后移除来自样品的未结合核酸的捕获的技术。在另一个实例中,当预环(precircle)型探针技术被用于选择性扩增(参见,比如barany等人,美国专利号6,858,412和7,556,924以及图7)时,可以从线性核酸分离环化的(circularized)核酸产物,环化的核酸产物经历选择性降解。分离的其他有用的方法在阅读本说明书之后对本领域中的技术人员将是明显的。
选择的核酸区域的选择性扩增的拷贝任选地可以在选择性扩增(或富集步骤)之后,在检测步骤之前或者在检测步骤期间(即,测序或其他检测技术)在通用括增步骤中被扩增。在执行通用扩增时,加至选择性扩增步骤中的拷贝的选择的核酸区域的通用引物序列被用以在单个通用扩增反应中进一步扩增选择的核酸区域。如描述的,通过使用用于具有通用引物序列的选择性扩增步骤的引物,通用引物序列可以在选择性扩增过程(如果执行)期间被加至拷贝的选择的核酸区域,使得将选择的核酸区域的扩增的拷贝并入通用引发序列。可选地,在扩增或富集(如果执行)之后,含有通用扩增序列的适体可以被连接至选择的核酸区域的末端,并且从母体样品分离选择的核酸区域。
在dna扩增期间可以向样品引入偏倚和变化性,并且这已知在聚合酶链式反应(pcr)期间发生。在其中扩增反应是多路的情形中,存在选择的核酸区域将以不同速率或效率扩增的可能,因为对于每一组给定的选择的核酸区域的引物可以基于引物和模板dna的碱基组成、缓冲条件或其他条件而表现不同。用于多路的分析系统的通用dna扩增通常引入较少的偏倚和变化性。另一种最小化扩增偏倚的技术包括改变用于不同的选择的核酸区域的引物浓度以限制选择性扩增步骤中序列特异性扩增循环的数目。在扩增步骤中可以使用相同或不同的条件(比如,聚合酶、缓冲液以及类似物)以例如确保偏倚和变化性不因为实验条件而被非故意地引入。
在优选的方面,执行少的循环数(比如1-10、优选地3-5)的选择性扩增或核酸富集,随后利用通用引物通用扩增。使用通用引物的扩增循环数将是变化的,但将优选地是至少5个循环、更优选地至少10个循环,甚至更优选地20个循环或更多。一个或几个选择性扩增循环之后,通过移至通用扩增,某些选择的核酸区域以比其它的更大的速率扩增的偏倚被减少。
任选地,方法包括选择性扩增和通用扩增之间的步骤以移除在选择性扩增中非特异地扩增的任何多余的核酸。来自选择性扩增的整个产物或产物的等份可被用于通用扩增。
在方法中使用的引物的通用区域被设计为与在一个容器中的一个反应中同时地分析大量的核酸的常规多路方法是相容的。这样的“通用的”引发方法允许存在于母体样品中的核酸区域的数量的有效、高容量分析,并且允许在用于确定非整倍性的这样的母体样品内核酸区域的存在的综合定量。
通用扩增方法的实例包括但不局限于用以同时地扩增和/或基因分型多个样品的多路方法,例如在oliphant等人,美国专利号7,582,420中描述的那些,其通过引用被并入本文。
在某些方面中,本发明的测定系统利用以下组合的选择性和通用扩增技术之一:(1)与聚合酶链式反应("pcr")偶联的连接酶检测反应("ldr");(2)与偶联至ldr的二级pcr偶联的初级pcr;以及(3)与二级pcr偶联的初级pcr。这些组合中的每一个对于最佳检测具有特定效用。但是,这些组合中的每一个使用多路检测,其中来自测定系统的早期的寡核苷酸引物含有用于测定系统的后期的序列。
barany等人,美国专利号6,852,487、6,797,470、6,576,453、6,534,293、6,506,594、6,312,892、6,268,148、6,054,564、6,027,889、5,830,711、5,494,810描述了连接酶链式反应(lcr)用于检测多个核酸样品中的核苷酸的特定序列分析的用途。barany等人,美国专利号7,807,431、7,455,965、7,429,453、7,364,858、7,358,048、7,332,285、7,320,865、7,312,039、7,244,831、7,198,894、7,166,434、7,097,980、7,083,917、7,014,994、6,949,370、6,852,487、6,797,470、6,576,453、6,534,293、6,506,594、6,312,892和6,268,148描述了与pcr偶联的ldr用于核酸检测的用途。barany等人,美国专利号7,556,924和6,858,412描述了预环探针(precircleprobe)(也称“挂锁探针”或“多倒置探针(multi-inversionprobes)”)与ldr和pcr偶联用于核酸检测的用途。barany等人,美国专利号7,807,431、7,709,201和7,198,814描述了组合的核酸内切酶裂解和连接反应用于核酸序列的检测的用途。willis等人,美国专利号7,700,323和6,858,412描述了预环探针在多路核酸扩增、检测和基因分型中的用途。ronaghi等人,美国专利号7,622,281描述了用于使用含有独特的引物和条形码的适配体标记和扩增核酸的扩增技术。可用于扩增和/或检测选择的核酸区域的示例性过程包括但不局限于本文描述的方法,其每一个通过引用以其整体被并入本文,为了教导可以在本发明的方法中使用的多种要素的目的。
除了多种扩增技术之外,很多序列确定方法与本发明的方法是相容的。优选地,这样的方法包括“下一代”测序方法。用于序列确定的示例性方法包括但不限于基于杂交的方法,例如在drmanac,美国专利号6,864,052、6,309,824、6,401,267和美国公布号2005/0191656中公开的,其全部通过引用被并入本文;通过合成方法测序,比如nyren等人,美国专利号7,648,824、7,459,311和6,210,891;balasubramanian,美国专利号7,232,656和6,833,246;quak,美国专利号6,911,345;li等人,pnas,100:414-19(2003);焦磷酸盐测序,如描述在ronaghi等人,美国专利号7,648,824、7,459,311、6,828,100和6,210,891中的;和基于连接的测序确定方法,比如drmanac等人,美国公布号2010/0105052和church,美国公布号2007/0207482和2009/0018024。
可以使用能够以高数量级的多路(highordersofmultiplexing)平行测序的任何合适的测序设备执行测序,例如miseq(illumina)、ionpgmtm(lifetechnologies)、iontorrenttm(lifetechnologies)、hiseq2000(illumina)、hiseq2500(illumina)、454平台(roche)、illumina基因组分析仪(illumina)、solid系统(appliedbiosystems)、实时smrttm技术(pacificbiosciences)和合适的纳米微孔和/或纳米通道测序仪。
可选地,可以利用杂交技术选择和/或鉴定选择的核酸区域。用于实施用于检测的多核苷酸杂交测定的方法已经在本领域中良好开发。杂交测定程序和条件将取决于应用而不同并且根据已知的一般结合方法来选择,所述一般结合方法包括在以下中提到的那些:maniatis等人,molecularcloning:alaboratorymanual(第二版coldspringharbor,n.y.,1989);berger和kimmel,methodsinenzymology,152卷;guidetomolecularcloningtechniques(academicpress,inc.,sandiego,calif.,1987);以及young和davispnas,80:1194(1983)。在美国专利号5,871,928、5,874,219、6,045,996、6,386,749和6,391,623中已描述了用于实施重复和控制的杂交反应的方法和设备。
在某些优选的方面本发明还涵盖了配体之间的杂交的信号检测;参见,美国专利号5,143,854、5,578,832、5,631,734、5,834,758、5,936,324、5,981,956、6,025,601、6,141,096、6,185,030、6,201,639、6,218,803和6,225,625,在ussn60/364,731中以及在pct申请pct/us99/06097(作为wo99/47964公布)中。
用于强度数据的信号检测和处理的方法和设备公开于,例如美国专利号5,143,854、5,547,839、5,578,832、5,631,734、5,800,992、5,834,758、5,856,092、5,902,723、5,936,324、5,981,956、6,025,601、6,090,555、6,141,096、6,185,030、6,201,639、6,218,803和6,225,625,在ussn60/364,731中以及在pct申请pct/us99/06097(作为wo99/47964公布)中。
在图3中,使用包含大致上相同的序列特异性区域305、307但包含不同标志321、323的两组固定序列寡核苷酸。用来自相同遗传样品300的材料,但在具有不同等位基因特异性寡核苷酸组的分离管中实施连接反应。与选择的核酸区域313、333中两个可能的snp对应的桥接寡核苷酸313、333被用以检测每一个连接反应中选择的核酸区域。指示snp的两个等位基因标志321、323可以被用以鉴定扩增产物,使得感兴趣的核酸和snp的实际序列的序列确定不一定是必须的,尽管这些序列可以仍旧被确定以鉴定和/或提供等位基因的确证。固定序列寡核苷酸的每一个包含与选择的核酸区域305、307互补的区域和在起始选择和/分离之后被用以扩增不同选择的核酸区域的通用引物序列309、311。通用引物序列位于固定序列寡核苷酸301、303和323的末端并且位于标志和与感兴趣的核酸互补的区域的侧面,因此将核酸特异性序列和等位基因标志保留在扩增产物中。在步骤302将固定序列寡核苷酸301、303、323引至遗传样品300的等份并且允许杂交至选择的核酸区域315或325。杂交之后,优选地将未杂交的固定序列寡核苷酸从遗传样品的剩余物分离(未显示)。
与a/tsnp313或g/csnp333对应的桥接寡核苷酸在步骤304被引入并且允许在固定序列寡核苷酸的第一305和第二307核酸互补区域之间的选择的核酸区域315或325的区域中结合。可选地,可以用固定序列寡核苷酸将桥接寡核苷酸313、333同时引至样品。在步骤306在反应混合物中连接结合的寡核苷酸以产生跨越感兴趣的核酸区域并且与感兴趣的核酸区域互补的连续的寡核苷酸。又,应该注意的是在一些测定中,一些桥接寡核苷酸将是多态性特异的并且一些将不是多态性特异的,因为感兴趣的非多态性(或多态性不可知论的)核酸区域和感兴趣的多态性核酸区域可以在单一测定中被询问。
连接之后,分离反应优选地被组合用于通用扩增和检测步骤。在步骤308将通用引物317、319引至组合的反应以扩增连接的寡核苷酸并且在步骤310产生包含代表选择的核酸区域中的snp的感兴趣的核酸区域的序列的产物327、329。通过鉴定等位基因标志,,通过测序产物或产物的一部分检测并且定量产物327、329,产物的该区域含有来自选择的核酸区域的snp或二者。优选地,通过等位基因标志的下一代测序检测并且定量图3的方法的产物,因此,避免确定与选择的核酸区域互补的产物的区域或整个产物的实际序列的需要。但是,在另一方面中,可以期望确定标志和与选择的核酸区域的序列互补的产物的区域二者的序列以提供结果的确证。
在图3的方法中(和在其他图中阐明的方法中),已经描述了等位基因标志。但是,在321和323显示的标志可以是等位基因标志、样品标志、组合等位基因和样品标志、基因座标志或本文描述的或本领域中另外使用的任何其他标志或标志的组合。
此外,在有区别的核苷酸位于固定序列寡核苷酸而不是桥接寡核苷酸时使用方法。因此在这样的示例性测定系统中,等位基因标志与等位基因特异性固定序列寡核苷酸有关,并且等位基因检测由测序等位基因标志造成。等位基因标志可以嵌入或者等位基因特异性第一序列寡核苷酸或者第二固定序列寡核苷酸。在特定的方面中,等位基因标志存在于第一和第二固定序列寡核苷酸二者上以检测选择的核酸区域内的两种或更多种多态性。在这样的方面中使用的固定序列寡核苷酸的数目可以与对选择的核酸区域评估的可能的等位基因的数目对应,并且等位基因标志的序列确定可以检测遗传样品中特定等位基因的存在、量或不存在。
图4阐明了本发明的这方面。在图4中,使用三种固定序列寡核苷酸401、403和423。两种固定序列寡核苷酸401、423是等位基因特异性的,包含与分别含有例如a/t或g/csnp的核酸区域中的等位基因互补的区域。等位基因特异性固定序列寡核苷酸401、423中的每一个还包含对应的等位基因标志421、431和通用引物序列409。第二固定序列寡核苷酸403具有第二通用引物序列411,并且这些通用引物序列被用以扩增选择的核酸区域,以将寡核苷酸组杂交和连接至来自遗传样品的选择的核酸区域。通用引物序列位于固定序列寡核苷酸401、403、423的末端,位于标志和与感兴趣的选择的核酸区域互补的固定序列寡核苷酸中的区域的侧面;因此捕获任何通用扩增方法的产物中核酸特异性序列和标志。
在步骤402,将固定序列寡核苷酸401、403、423引至遗传样品400并且允许与选择的核酸区域415、425杂交。杂交之后,优选地将未杂交的固定序列寡核苷酸从遗传样品的剩余物分离(未显示)。桥接寡核苷酸413被引入并且被允许与核酸415在第一等位基因特异性固定序列寡核苷酸区域405和另一固定序列寡核苷酸区域407之间的区域中杂交404或与和第二等位基因特异性固定序列寡核苷酸区域435和另一固定序列寡核苷酸区域407之间的区域互补的核酸425杂交。可选地,可以将桥接寡核苷酸413用固定序列寡核苷酸组同时引至样品。如在关于图3之前陈述的,应该注意的是,在一些测定中寡核苷酸的一些组将是多态性特异的并且一些将不是多态性特异的,因为感兴趣的非多态性(或多态性不可知论的)核酸区域和感兴趣的多态性核酸区域二者可以在单一测定中被询问。
在步骤406将与选择的核酸区域杂交的寡核苷酸连接以产生跨越感兴趣的选择的核酸区域并且与感兴趣的选择的核酸区域互补的连续的寡核苷酸。仅当等位基因特异性固定序列寡核苷酸的等位基因特异性末端与选择的核酸区域中的snp互补时连接主要发生。连接之后,在步骤408将通用引物417、419引入扩增连接的寡核苷酸以在步骤410产生包含感兴趣的核酸区域的序列的产物427、429。通过序列确定产物的全部或产物的一部分检测并且定量这些产物427、429,并且特定地产物的该区域含有选择的核酸区域中的snp和/或等位基因标志。这里,等位基因特异性核苷酸显示为是在等位基因特异性固定序列寡核苷酸的末端,然而等位基因特异性核苷酸不需要这样位于等位基因特异性固定序列寡核苷酸的末端。但是为了使连接是等位基因特异性的,等位基因特异性核苷酸必须接近连接的末端。通常,等位基因特异性核苷酸必须在连接的末端的5个核苷酸内。在优选的方面中,等位基因特异性核苷酸是倒数第二的或终端(末端)核苷酸。
在本发明的测定的又另一个实例中,等位基因检测从阵列的基因座标志的杂交获得。通过等位基因特异性标记步骤检测每一个等位基因,其中在通用扩增期间标记每一个等位基因,比如用光谱上有区别的荧光标记。图5阐明了本发明的这方面。在图5中,使用三种固定序列寡核苷酸501、503和523。两种固定序列寡核苷酸501、523是等位基因特异性的,并且每一个包含在相同选择的核酸区域中匹配不同等位基因的区域、基因座标志521和等位基因特异性通用引物序列509、539。第三、非等位基因特异性固定序列寡核苷酸503包含另一个通用引物序列511。杂交和连接寡核苷酸之后,通用引物序列被用以扩增选择的核酸区域。将标记并入区分每一个等位基因的扩增产物。如在之前的实例中,通用引物序列位于固定序列寡核苷酸501、503、523的近端并且因此捕获任何通用扩增方法的产物中的等位基因特异性序列和标志。在步骤502中将固定序列寡核苷酸501、503、523引至遗传样品500并且允许与选择的核酸区域515、525特异地结合。杂交之后,优选地将未杂交的固定序列寡核苷酸从遗传样品的剩余物分离(未显示)。在步骤504将桥接寡核苷酸513引入并且允许结合至第一(等位基因特异性的)505和第二(非等位基因特异性的)507固定序列寡核苷酸之间选择的核酸区域515、525的区域和第一(等位基因特异性的)535和第二507(非等位基因特异性的)固定序列寡核苷酸之间选择的核酸区域515、525的区域。可选地,可以用固定序列寡核苷酸同时将桥接寡核苷酸513引至样品。
在步骤506将结合的寡核苷酸连接以产生跨越感兴趣的选择的核酸区域并且与感兴趣的选择的核酸区域互补的连续的寡核苷酸。当等位基因特异性末端匹配时连接主要发生。连接之后,在步骤508将通用引物517、519、537引入以扩增连接的寡核苷酸以在步骤510产生包含感兴趣的选择的核酸区域的序列的产物527、529。通用引物517和537具有光谱上有区别的荧光标记,使得等位基因特异性信息被捕获并且可以通过这些荧光标记来读出。通过将基因座标志521杂交至阵列和并且成像来检测并且定量产物527、529。如关于图4描述的,重要的是注意连接506优选地是等位基因特异性的;因此,有区别的核苷酸位于从等位基因特异性固定序列寡核苷酸的末端的至少5个核苷酸并且优选地位于倒数第二或终端核苷酸。显示在图5中、其中基因座标志用于杂交至阵列的实例可以在本文描述的多种方法中的任一中被使用,例如其中固定序列寡核苷酸和桥接寡核苷酸不毗邻杂交并且聚合酶和dntp被用以关闭寡核苷酸之间的“空隙”、随后连接的方法。类似地,基因座标志/杂交方法可以在方案中被使用,该方案中仅固定序列寡核苷酸被使用-即不存在桥接寡核苷酸-并且该方案中固定序列寡核苷酸毗邻杂交并且通过连接作用来连接或该方案中固定序列寡核苷酸与它们之间的空隙杂交并且使用聚合酶和dntp来连接、随后连接。
在可选的方面中,等位基因标志存在于第一和第二固定序列寡核苷酸二者上以使用对于给定等位基因对每一个固定序列寡核苷酸的对应的光学上有区别的荧光标记在每一个固定序列寡核苷酸的末端检测多态性。在此方法中,固定序列寡核苷酸的数目与对选择的核酸区域评估的可能的等位基因的数目对应。在以上的图和实例中,固定序列寡核苷酸被表示为两个有区别的寡核苷酸。在另一个方面中,固定序列寡核苷酸可以是相同的寡核苷酸的相对的末端(参见,比如以上图7)。
在以上描述的方面中,使用的桥接寡核苷酸杂交至与和固定序列寡核苷酸互补的区域毗邻的感兴趣的核酸的区域,使得当固定序列和桥接寡核苷酸特异性杂交时,它们直接彼此毗邻用于连接。但是,在其他方面中,将桥接寡核苷酸杂交至不与和固定序列寡核苷酸的一个或两个互补的区域直接毗邻的区域,并且在连接之前需要延伸寡核苷酸的一个或更多个的中间步骤。例如,如图6中阐明的,寡核苷酸的每一组优选地含有两个固定寡核苷酸601、603和一个或更多个桥接寡核苷酸613。固定序列寡核苷酸中的每一个包含与选择的核酸区域605、607,并且优选地通用引物序列609、611互补的区域;即与通用引物互补的寡核苷酸区域。通用引物序列609、611位于或接近固定序列寡核苷酸601、603的末端,并且因此捕获任何通用扩增方法的产物中的核酸特异性序列。
在步骤602将固定序列寡核苷酸601、603引至遗传样品600并且允许与和感兴趣的选择的核酸区域615的互补部分特异性结合。杂交之后,优选地将未杂交的固定序列寡核苷酸从遗传样品的剩余物分离(未显示)。然后将桥接寡核苷酸引入并且允许在步骤604结合至第一601和第二603固定序列寡核苷酸之间的选择的核酸区域615的区域。可选地,可以将桥接寡核苷酸用固定序列寡核苷酸同时引至样品。在这里显示的示例性方面中,将桥接寡核苷酸杂交至与第一固定序列寡核苷酸区域605直接毗邻的区域,但与第二固定序列寡核苷酸607的互补区域间隔一个或更多个核苷酸。固定序列和桥接寡核苷酸杂交之后,在步骤606将桥接寡核苷酸613延伸,比如使用聚合酶和dntp,以填充桥接寡核苷酸613和第二固定序列寡核苷酸603之间的空隙。延伸之后,在步骤608将杂交的寡核苷酸连接以产生跨越感兴趣的选择的核酸区域615并且与感兴趣的选择的核酸区域615互补的连续的寡核苷酸。连接之后,在步骤610将通用引物617、619引入以扩增连接的寡核苷酸以在步骤612产生包含感兴趣的核酸区域的序列的产物623。将这些产物623分离、检测和定量以提供关于遗传样品中选择的核酸区域的存在和量的信息。优选地,通过等位基因标志621的下一代测序检测和定量产物,或可选地在扩增产物623中测序确定与感兴趣的选择的核酸615互补的扩增产物的部分。
图7阐明了固定序列寡核苷酸如何可以是相同分子的一部分。在特定的方面中,单个固定序列寡核苷酸701与选择的核酸区域715在两个末端是互补的。当将此单个固定序列寡核苷酸701杂交至选择的核酸区域715时,它形成预环寡核苷酸703,其中末端被若干核苷酸分开。然后桥接寡核苷酸713结合预环寡核苷酸703的互补区域705、707以填充此空隙。然后将与遗传样品715结合的预环寡核苷酸703的寡核苷酸区域705、707与桥接寡核苷酸713连接在一起,形成完整的圆。如本文示例的其他方法,使用桥接寡核苷酸不是必需的,并且在这样的实施方案中固定序列寡核苷酸可以毗邻杂交或如果固定序列寡核苷酸不毗邻杂交,可以使用聚合酶和dntp填充空隙。优选地使用通用引物位点的一个或更多个裂解并且扩增圆形模板。在特定的方面中,利用如公开在lizardi等人美国专利号6,558,928中的技术,例如滚环复制,使用单通用引物区域复制模板。
如图7中阐明的,固定序列寡核苷酸在圆形模板上具有两个通用引发位点709、711和任选地与选择的核酸区域是互补的构建体的末端之间的一个或更多个标志721。这里显示,裂解位点723存在于两个通用引发位点之间。在步骤702将构建体701引至遗传样品,允许将其杂交至感兴趣的选择的区域,并且在步骤704将桥接寡核苷酸引入并且允许将其杂交至选择的核酸区域。然后在步骤706将构建体通过连接环化至桥接寡核苷酸713,并且可以使用核酸酶移除全部或大部分未环化的寡核苷酸。在移除未环化的寡核苷酸之后,将环化的寡核苷酸裂解,保存并且在一些方面中将通用引发位点709、711暴露。在步骤708加入通用引物717、719并且通用扩增发生710以产生712包含感兴趣的选择的核酸区域的序列的产物725。通过,比如下一代测序与选择的核酸区域或可选地标志互补的产物的部分检测并且定量产物725,这省却了测序整个构建体的需要。但是,在其他方面中期望确定包含标志和选择的核酸区域二者的序列的产物,例如,以提供结果的内部确认或其中标志提供样品信息并且不提供选择的核酸区域的信息。如以上提及的,此单个固定序列寡核苷酸方法可以被应用于图2-7中的实例中的任一。再次,还应该注意的是在一些测定中,寡核苷酸的一些组将是多态性特异性的并且一些将不是多态性特异性的,因为感兴趣的非多态性(或多态性不可知论的)核酸区域和感兴趣的多态性核酸区域可以在单个测定中被询问。
标志在本发明的方法中的使用
如以上关于图2-7描述的,在某些方面中,组中的固定序列寡核苷酸包含一个或更多个标志(indexes)或标志(indices),比如鉴定选择的核酸区域(基因座标志)、选择的核酸区域内的snp(等位基因标志)和/或被分析的特定的样品(样品标志)。例如,一个或更多个基因座标志的检测可以作为用于整个选择的核酸区域的替代检测,如以下描述的,或如果标志的序列和与核酸区域自身互补的寡核苷酸产物的序列两者均被确定则标志的检测可以作为特定的选择的核酸区域的存在的确认。优选地,在利用包含标志和特异杂交至选择的核酸区域的区域(即,选择的核酸区域特异性序列)二者的引物选择性扩增步骤期间,标志是与选择的核酸区域有关的。
通常标志是在扩增引物内使用的非互补、独特的序列以提供与利用引物分离和/或扩增的选择的核酸区域有关的信息。标志的顺序和布置以及标志的长度可以变化,并且标志可以多种组合的形式被使用。可选地,在初始的选择性扩增之后,利用连接包含这些序列的适配体,标志和/或通用扩增序列可被添加至选择性地扩增的选择的核酸区域。使用标志的优势是,能获得选择的核酸区域的存在(和最终地量或频率)而不需要测序选择的核酸区域的整个长度,尽管在某些方面测序选择的核酸区域的整个长度可以是期望的。但是,通常地,通过鉴定一个或更多个标志来鉴定并定量选择的核酸区域的能力将减少所需的测序的长度,尤其如果标志序列在接近测序引物可以位于的地方的所分离的选择的核酸区域的3'或5'端被捕获。因为较长的测序读取更容易引入错误,所以标志作为选择的核酸区域的鉴定的替代的使用还可减少测序错误。又,如以上关于图5描述的,基因座标志-与比如荧光标记连同-可以被用于鉴定并且定量通过杂交至阵列的选择的核酸区域。
在标志的一个实例中,用于选择的核酸区域的选择性扩增的引物被设计为在与选择的核酸区域互补的区域和通用扩增引物位点之间包括基因座标志。通常地基因座标志对于每一个选择的核酸区域是独特的,使得特定的基因座标志在样品中发生的次数的数目的定量可以与对应的单核酸区域和含有单核酸区域的特定染色体的拷贝的相对数目有关。通常地,基因座标志足够长以独特地标记每一个已知的单核酸区域。例如,如果方法使用192个已知的单核酸区域,那么有至少192个独特的基因座标志,每一个独特地鉴定来自染色体上特定基因座的单核酸区域。在本发明的方法中使用的基因座标志可以指示样品中单独染色体上的不同单核酸区域以及在不同染色体上存在的已知单核酸区域。基因座标志可包含允许鉴定和修正测序错误的另外的核苷酸,所述鉴定和修正包括检测在测序期间一个或更多个碱基的缺失、取代或插入以及可以在测序之外诸如寡核苷酸合成、扩增、或方法的任何其它方面发生的核苷酸改变。
在另一个实例中,用于选择的核酸区域的扩增的引物可以被设计为在与选择的核酸区域互补的区域和通用扩增引物位点之间提供等位基因标志(作为对基因座标志的可替代选择)。等位基因标志对于选择的核酸区域的特定的等位基因是独特的,使得特定的等位基因标志在样品中发生的次数的数目的定量可以与该等位基因的拷贝的相对数目有关,并且特定选择的核酸区域的等位基因标志的总和可以与含有选择的核酸区域的特定染色体上该选择的核酸区域的拷贝的相对数目有关。在多态性特异寡核苷酸组和多态性或snp不可知论的寡核苷酸组二者被用于单个测定中的实施方案中,等位基因标志和基因座标志二者可以被使用。
在又另一个实例中,用于选择的核酸区域的扩增的引物可被设计为在与选择的核酸区域互补的区域和通用扩增引物位点之间提供鉴定标志。在这样的方面中,展示足够数目的鉴定标志以独特地鉴定样品中的每一个扩增的分子。鉴定标志序列优选地长度是6个或更多个核苷酸。在优选的方面中,鉴定标志足够长以具有用单核酸区域独特地标记每一个分子的统计概率。例如,如果有3000个拷贝的特定单核酸区域,则大体上有多于3000个鉴定标志,使得每一个拷贝的特定单核酸区域有可能用独特的鉴定标志标记。如同其他标志,鉴定标志可以包含允许鉴定和修正测序错误的另外的核苷酸,所述鉴定和修正包括检测在测序期间一个或更多个碱基的缺失、取代或插入以及可以在测序之外诸如寡核苷酸合成、扩增、和分析的任何其它方面发生的核苷酸改变。
鉴定标志可以与任何其它标志联合以生成为两种特性提供信息的一个标志。鉴定标志还可被用以检测和定量在来自样品的选择的核酸区域的初始分离的下游出现的扩增偏倚并且此数据可以被用于归一化样品数据。
除了本文描述的其他标志,可以使用修正标志。修正标志是允许修正扩增、测序或其他实验错误的短核苷酸序列,所述修正包括检测在测序期间一个或更多个碱基的缺失、取代或插入以及可在测序之外诸如寡核苷酸合成、扩增、或测定的其它方面发生的核苷酸改变。修正标志可以是为单独的序列的独立标志,或其可以被嵌入其它标志内以帮助证实所使用的实验技术的准确性,例如,修正标志可以是基因座标志或鉴定标志的序列的亚组。
在一些方面中,指示选择的核酸区域从其分离的样品的标志被用于鉴定多路测定系统中选择的核酸区域的来源。在这样的方面中,来自一个个体的选择的核酸区域将被指定并且与特定独特的样品标志有关。样品标志因此可以被用于帮助用于单反应容器(即,在混合样品的情况下)中的不同样品的多路的核酸区域鉴定,使得每一个样品可以基于其样品标志而被鉴定。在优选的方面中,对于一组样品中的每个样品有独特的样品标志,并且在测序期间样品被混合。例如,如果12个样品被混合进入单个测序反应,有至少12个独特的样品标志使得每个样品被独特地标记。在执行测序步骤之后,在确定每一个样品的每一个选择的核酸区域的频率之前,且在确定每一个样品是否有染色体异常之前,测序数据优选地首先被样品标志分开。
样品内变异最小化
检测混合的样品中染色体异常的一个挑战是,来自具有染色体异常的细胞类型的dna(即,胎儿dna)经常以比来自整倍性细胞类型的dna(即,母体dna)低得多的丰度存在。在含有胎儿和母体的无细胞dna的母体样品的情况下,作为总的无细胞dna的百分比的无细胞胎儿dna可以从小于百分之一到百分之四十变化,并且最通常地以百分之二十或小于百分之二十并且经常地以百分之十或小于百分之十存在。例如,在检测这样的混合的母体样品的胎儿dna中y染色体非整倍性时,如果胎儿是正常的男性,y染色体序列的相对增加是y序列的预计百分比的倍数,并且因此作为混合的样品中总dna的百分比,当作为一个实例,胎儿dna是总的5%时,y染色体的贡献的增加作为总的百分比是5%的1/47th(样品中总dna百分比的0.11%)。如果人们将通过本文描述的方法稳健地检测该区别,y染色体的测量中的变化必须比y染色体的增加百分比小的多。
在一些方面中,染色体上一个或更多个选择的核酸区域的测量的定量被归一化以解释来自来源的已知变化,例如测定系统(比如,温度、试剂批次差异)、样品的潜在生物学(比如,核酸含量)、操作者差异,或任何其他变量。进一步,用于确定选择的核酸区域的频率的数据可以排除可能由于实验误差出现的离群数据,或具有基于特定样品内先天遗传偏差的提高的或降低的水平的离群数据。在一个实例中,用于总和的数据可以排除一个或更多个样品中具有特定地升高的频率的核酸区域。在另一个实例中,用于总和的数据可以排除一个或更多个样品以特别低的丰度发现的选择的核酸区域。
利用分析方法的组合可以最小化样品和/或样品内选择的核酸区域之间的变化。例如,通过在分析中使用内部参考减少变化。内部参考的实例是利用以“正常”丰度(比如,常染色体的二体性)存在的染色体与相同样品中的以异常的丰度,即非整倍性或痕量污染物存在的x和y染色体比较。虽然使用单一这样的“正常”染色体作为参考染色体可以是足够的,但是优选使用两个至若干个常染色体作为内部参考染色体以增加定量的统计功效。
内部参考的一个使用是计算样品中的假定异常x和/或y染色体频率的丰度与常染色体的丰度的比,称为染色体比。在计算染色体比时,对于每一个染色体的每一个选择的核酸区域的丰度或计数被总和在一起以计算每一个染色体的总计数。然后一个染色体的总计数除以不同的染色体的总计数以生成那两个染色体的染色体比。
可选地,每一个染色体的染色体比可以通过首先将每一个染色体的每一个选择的核酸区域的计数求和并且然后将一个染色体的总和除以两个或更多个染色体的总和来计算。计算后,然后将染色体比与来自整倍性群体的平均染色体比比较。
平均值可以是带有或不带有归一化或离群数据的排除的平均值(mean)、中值、众数或其它平均数(average)。在优选的方面中,使用平均值。在从整倍性群体建立染色体比的数据集时,计算测量的染色体的标准变化。此变化可以很多方式表示,最通常地作为变异系数,或cv。当来自样品的x染色体比与来自整倍性群体的平均染色体比比较时,如果样品的x染色体比统计上落在整倍性群体的平均染色体比之外,则样品含有指示,比如x非整倍性和/或x染色体镶嵌现象的x染色体异常。类似地,当来自样品的y染色体比与来自整倍性群体的平均染色体比比较时,如果样品的y染色体比统计上落在整倍性群体的平均染色体比之外,则样品含有指示,比如y非整倍性和/或y染色体镶嵌现象的y染色体异常。
用于设定表明非整倍性的统计阈值的标准取决于在染色体比的测量上的变化以及对于期望的方法的可接受的假阳性和假阴性率。通常,此阈值可以是在染色体比中观察到的多倍的变化。在一个实例中,该阈值为染色体比率的变化的三倍或更多倍。在另一个实例中,它是染色体比的变化的四倍或更多倍。在另一个实例中,它是染色体比的变化的五倍或更多倍。在另一个实例中,它是染色体比的变化的六倍或更多倍。在以上实例中,通过对每染色体的选择的核酸区域的计数求和来确定染色体比。通常地,对每一个染色体使用相同数目的选择的核酸区域。
用于生成染色体比的可选方法将是计算每一个染色体或染色体区域的选择的核酸区域的平均计数。虽然通常地使用平均值,但是平均值可以是平均值、中值或众数的任何估算。平均值可以是所有计数的平均值或一些变化,诸如微调的或加权的平均值。已计算每一个染色体的平均计数之后,可将每一个染色体的平均计数除以另一个染色体的平均计数以获得两个染色体之间的染色体比,可将每一个染色体的平均计数除以所有测量的染色体的平均值的总和以获得如以上描述的每一个染色体的染色体比。如以上强调的,在测定中,检测母体样品中x染色体、x染色体频率、y染色体或y染色体频率的能力极大地取决于不同的选择的核酸区域的测量中的变化,在所述母体样品中胎儿dna是以低的相对丰度。很多分析方法可被使用,所述很多分析方法减少此变化并且因此提高此方法检测非整倍性的灵敏度。
用于减少分析的变化性的一种方法是增加用以计算染色体的丰度的选择的核酸区域的数目。通常地,如果染色体的单个选择的核酸区域的测量的变化是b%并且在相同的染色体上测量c个不同的选择的核酸区域,则通过对该染色体上的每一个选择的核酸区域的丰度求和或求平均值计算的染色体丰度的测量的变化将是大约b%除以c1/2。换句话说,染色体丰度的测量的变化将大约是每一个选择的核酸区域的丰度的测量的平均变化除以选择的核酸区域的数目的平方根。
在本发明的优选的方面中,对于每一个染色体(x染色体、y染色体和一个或更多个常染色体)测量的选择的核酸区域的数目是至少8。在本发明的另一个优选的方面中,对于每一个染色体测量的选择的核酸区域的数目是至少24。在本发明的又另一个优选的方面中,对于每一个染色体测量的选择的核酸区域的数目是至少32。在本发明另一个优选的方面中,对于每一个染色体测量的选择的核酸区域的数目是至少100。在本发明的另一个优选的方面中,对于每一个染色体测量的选择的核酸区域的数目是至少200。测量每一个选择的核酸区域有增加的成本并且因此最小化选择的核酸区域的数目,同时仍旧生成在统计上强健的数据是重要的。在本发明的优选的方面中,对于每一个染色体测量的选择的核酸区域的数目是少于2000。在本发明的优选的方面中,对于每一个染色体测量的选择的核酸区域的数目是少于1000。在本发明的最优选的方面,对于每一个染色体测量的选择的核酸区域的数目是至少32且少于1000。
在一个方面中,测量每一个选择的核酸区域的丰度之后,选择的核酸区域的亚组可被用于确定x或y染色体异常的存在或不存在。有用于选择选择的核酸区域的亚组的许多标准方法,包括排除,其中选择的核酸区域以某百分位数以下或以上的检测的水平从分析中被丢弃。在一个方面中,百分位数可以是最低和最高的5%,如通过频率测量的。在另一个方面中,将被丢弃的百分位数可以是最低和最高的10%,如通过频率测量的。在另一个方面中,将被丢弃的百分位数可以是最低和最高的25%,如通过频率测量的。
用于选择选择的核酸区域的亚组的另一种方法包括消除落在一些统计限值之外的区域。例如,落在平均丰度的一个或更多个标准偏差之外的区域可被从分析移除。例如,用于选择选择的核酸区域的亚组的另一种方法可以是将选择的核酸区域的相对丰度与健康群体中相同的选择的核酸区域的预期丰度比较,并丢弃期望值测试不合格的任何选择的核酸区域。为了进一步使分析中的变化最小化,可以增加每一个选择的核酸区域被测量的次数。如所讨论的,与其中基因组被测量平均少于一次的检测x和y染色体频率异常的随机方法相比,本发明的方法有意地测量每一个选择的核酸区域多次。通常地,当计数事件时,通过poisson统计来确定计数中的变化,并且计数变化通常地等于一除以计数的数字的平方根。在本发明的优选的方面中,选择的核酸区域每一个被测量平均至少5次。在本发明的某些方面中,选择的核酸区域每一个被测量平均至少10、50或100次。在本发明的某些方面中,选择的核酸区域每一个被测量平均至少250次。在本发明的某些方面中,选择的核酸区域每一个被测量平均至少500次。在本发明的某些方面中,选择的核酸区域每一个被测量平均至少1000次或至少5,000次或至少10,000次。
在另一个方面中,在确定染色体异常是否存在时,利用足够的数目,选择的核酸区域的亚组能被随机地选择以获得统计上显著的结果。在母体样品内可以执行选择的核酸区域的不同亚组的多重分析以获得更大的统计功效。在此实例中,在随机分析之前,可以需要或可以不需要移除或消除任何的选择的核酸区域。例如,如果对于y染色体有100个选择的核酸区域且对于比如染色体2有100个选择的核酸区域,则对染色体中的每一个可以进行评价少于100个区域的一系列分析。
可以通过系统地移除样品来归一化序列计数并且通过利用关于对数变换计数的中位数平滑(medianpolish)来归一化分析偏差。对于每一个样品,度量可以被计算为选择的核酸区域的计数的平均值除以特定的染色体上选择的核酸区域的计数的平均值和不同染色体上选择的核酸区域的计数的平均值的总和。比例的标准z检验可以被用于计算z统计:
其中pj是给定的样品j中感兴趣的给定的染色体的观察到的比例,p0是计算为中值pj的给定测试染色体的期望的比例,并且nj是比例度量的分母。可以利用迭代审查进行z统计标准化。在每一个迭代时,移除落在比如三个中值绝对偏差以外的样品。在十次迭代之后,仅利用未经审查的样品计算平均值和标准偏差。然后将所有样品针对此平均值和标准偏差标准化。可以使用kolmogorov-smirnov检验(参见conover,practicalnonparametricstatistics,295-301页(johnwiley&sons,newyork,ny,1971))和shapiro-wilk's检验(参见royston,appliedstatistics,31:115–124(1982))检验整倍性样品的z统计的正规性。
除了用于减小分析中的变化的以上方法之外,其它分析技术可被组合使用,在该申请中所述其它分析技术中的很多在前面被描述。例如,当每一个样品的所有选择的核酸区域在单个容器中的单个反应中被询问时,分析中的变化可被减少。相似地,当使用通用扩增系统时,分析中的变化可以被减少。此外,当扩增的循环数是有限的时,分析的变化可以被减少。
母体样品中胎儿dna含量的确定
确定母体样品中胎儿dna的百分比提高对选择的核酸区域计算的频率的准确性,因为胎儿贡献的知识提供关于来自x和y染色体的选择的核酸区域的预期的统计存在的重要信息。在环境中考虑胎儿百分比是尤其重要的,所述环境中母体样品中胎儿dna的水平是低的,因为胎儿贡献百分比被用于确定样品中x和y染色体序列的定量统计显著性。当评估x染色体非整倍性、y染色体非整倍性或性染色体镶嵌现象的存在和/或确定是否有样品污染时,考虑胎儿百分比是重要的。
在感兴趣的等位基因处的母体dna的相对母体贡献可以与在该等位基因处的非母体贡献比较,以确定样品中的近似胎儿dna浓度。在优选的方面中,单独父系衍生序列,比如常染色体上父系特异多态性的相对定量被用于确定母体样品中胎儿dna的相对浓度。确定母体样品中胎儿贡献百分比的另一种示例性方法是通过分析具有胎儿和母体dna之间的dna甲基化的不同模式的dna片段。
因为x和y染色体通常不用于计算本发明方法中胎儿百分比,确定胎儿多态性需要靶标的snp和/或突变分析以鉴定母体样品中胎儿dna的存在。在每一个母体衍生的样品中,来自胎儿的dna将具有约50%的遗传自母亲的其基因座和50%的遗传自父亲的基因座。确定来自父亲来源的对胎儿贡献的基因座允许估计母体样品中的胎儿dna,并且因此提供用来计算感兴趣的染色体的染色体频率的统计上的显著差异的信息。在一些方面中,可以进行使用父本和母本的先前的基因分型。例如,亲本可以已经历用于鉴定疾病标志物的基因型确定,例如,用于诸如囊性纤维化、肌肉萎缩症、脊髓性肌萎缩或甚至rhd基因的状态的紊乱的基因型的确定可被确定。如果这样,在多态性、拷贝数变化或突变中的差异可以被用于确定母体样品中的胎儿贡献百分比。
在可选的优选的方面中,母体样品中胎儿无细胞dna百分比可以利用多路的snp检测来定量,而无母体或亲本基因型的先前知识。在此方面中,使用每一个区域中具有一个或更多个已知snp的选择的多态性核酸区域。在优选的方面中,选择的多态性核酸区域位于不可能是非整倍性的常染色体例如6号染色体上。又,在优选的方面中,在一个容器中的一个反应中扩增选择的多态性核酸区域。利用,比如高通量测序确定并定量母体样品中选择的多态性核酸区域的每一个等位基因。序列确定之后,鉴定其中母体和胎儿基因型不同的基因座,比如,母体基因型是纯合的并且胎儿基因型是杂合的。父本遗传序列可以通过以低的但统计上相关频率发生的检测的多态性来鉴定。通过针对特定的选择的核酸区域观察一个等位基因的高相对频率(>60%)和其他等位基因的低相对频率(<20%且>0.15%)来完成鉴定。由于多个基因座在等位基因的丰度的测量中减少变化的量,其的使用是特别地有优势的。满足该要求的所有的基因座或其亚组被用来通过统计分析确定胎儿浓度。
在一个方面中,通过将来自两个或更多个基因座的低频率等位基因加和在一起,除以高和低频率等位基因的和并且乘以2来确定胎儿浓度。在另一个方面中,通过对来自两个或更多个基因座的低频率等位基因求平均值,除以高和低频率等位基因的平均值并乘以2来确定胎儿无细胞dna百分比。
对于许多等位基因,母体和胎儿序列可以是纯合且是同一的,并且由于此信息在母体和胎儿dna之间无区别,其在确定母体样品中的胎儿dna百分比中是没有用的。在胎儿百分比的计算中,本方法利用其中胎儿和母体dna(比如,含有与母体等位基因不同的至少一个等位基因的胎儿等位基因)之间有差异的等位基因的信息。因此与对于母体和胎儿dna来讲相同的等位基因区域有关的数据不被选择用于分析,或在确定胎儿dna百分比之前被从相关数据移除,以不使有用的数据无效。用于定量母体血浆中胎儿dna的示例性方法可以发现,在例如chu等人,prenatdiagn30:1226-29(2010)中,其通过引用被并入本文。
在一个方面中,如果由于实验性错误、或来自特定样品内的先天的遗传倾向使得区域的量或频率表现是异常值,选择的核酸区域可被排除。在另一个方面中,选择的核酸可以在求和或求平均值,比如如本领域中已知的或以上描述的之前,经历统计的或数学的调整,诸如归一化、标准化、聚类或转化。在另一个方面中,选择的核酸可以在求和或求平均值之前经历归一化和数据实验性错误排除两者。在优选的方面中,12个或更多个基因座被用于分析。在另一个优选的方面中,24个或更多个基因座被用于分析。在另一个优选的方面中,32个或更多个基因座,48个或更多个基因座,72个或更多个基因座,96个或更多个基因座,100个或更多个基因座,或200个或更多个基因座被用于分析。
在一个优选的方面,可利用母体和胎儿等位基因中的串联snp检测来定量母体样品中的胎儿贡献百分比。用于鉴定提取自母体样品的dna中串联snp的技术被公开在mitchell等人,美国专利号7,799,531和ussn12/581,070;12/581,083;12/689,924和12/850,588中。这些参考文献通过检测在胎儿和母体基因组之间具有不同的单倍型的母体样品中的至少一个串联单核苷酸多态性(snp)来描述了胎儿和母体基因座的区别。这些单倍型的鉴定和定量可以对母体样品直接地进行,如在mitchell等人的公开内容中描述的,并被用于确定母体样品中的胎儿贡献百分比。
在又另一个可选地替代选择中,某些基因已经被鉴定为具有母体和胎儿基因拷贝之间的表观遗传学区别,并且这样的基因是母体样品中胎儿dna标志物的候选基因座。参见,比如,chim等人,pnasusa,102:14753-58(2005)。可以通过利用甲基化特异pcr(msp)以高特异性容易地检测这些基因座,甚至当这样的胎儿dna分子存在于母体来源的过量的背景血浆dna之中时,所述这些基因座可以是在胎儿dna中甲基化但在母体dna中未甲基化的(或反之亦然)。比较母体样品中甲基化的和未甲基化的扩增产物可以被用于通过计算这样的已知序列的一个或更多个的表观遗传的等位基因比来定量胎儿dna对母体样品的贡献百分比,已知这样的序列相比于母体dna被胎儿dna中的甲基化区别地调节。
为了确定母体样品中核酸的甲基化状态,样品的核酸经历亚硫酸氢盐转化样品并且然后经历msp,随后是等位基因特异性引物延伸。用于这样的硫酸氢盐转化的常规方法包括但不限于使用商业上可得的试剂盒,诸如methylamptmdna修饰试剂盒(epigentek,brooklyn,ny)。等位基因的频率和比率可以直接被计算并且从数据中输出以确定母体样品中胎儿dna的相对百分比。
x和y染色体频率分析中胎儿无细胞dna百分比的用途
胎儿无细胞dna百分比已经被计算之后,将此数据与用于检测和定量x和y染色体序列的方法联合以确定胎儿可能是女性、男性、x染色体的非整倍性、y染色体的非整倍性、x染色体镶嵌现象、y染色体镶嵌现象的似然。其在确定母体非整倍性包括镶嵌现象中也可以使用,或以鉴定被测试的母体样品是否被污染。
例如,在是10%胎儿dna的母体样品中,每一个染色体将贡献整倍性胎儿中的10%的1/46th(或约0.22%)。在整倍性男性胎儿中,因此y染色体将贡献10%的1/46th(0.22%),x染色体将贡献10%的1/46th(0.22%),并且常染色体对将贡献10%的2/46th(0.44%,因为每一个常染色体有两个)。因此,在确定胎儿是否是整倍性男性胎儿时,样品中是10%胎儿的y染色体特异性序列的频率应该是0.22%并且例如染色体3特异性序列的频率应该是0.44%,因为男性胎儿具有两个染色体3。在确定是否存在y染色体非整倍性时(即,两个或更多个y染色体),对于两个y染色体,y染色体特异性序列的频率将是约0.44%并且对于三个y染色体是约0.66%。在确定胎儿是否可以是y染色体镶嵌现象时,y染色体特异性序列的频率应该是较小的并且对于xx/xy镶嵌现象可以是基本上小于0.22%,并且对于评估具有来自女性胎儿的核酸的母体样品被具有来自男性胎儿的核酸的母体样品污染的可能性也是如此。在xy/xyy镶嵌现象中,y染色体特异性序列的频率应该是在0.22%和0.44%之间。在另一个实例中,在是5%胎儿dna的母体样品中每一个染色体将贡献整倍性胎儿中的5%的1/46th(或约0.11%)。在整倍性男性胎儿中,y染色体将因此贡献5%的1/46th(0.11%)并且常染色体对将贡献5%的2/46th或1/23rd(0.22%,因为每一个常染色体有两个)。
在另一个实例中,整倍性女性胎儿中x染色体将贡献10%的2/46th(即,1/23rd)(0.44%,因为在整倍性女性胎儿中有两个x染色体)并且常染色体对将贡献10%的2/46th或1/23rd(0.44%,因为每一个常染色体有两个)。因此,在确定胎儿是否是整倍性女性胎儿时,样品中是10%胎儿的x染色体特异性序列的频率应该是0.44%并且例如染色体3特异性序列的频率应该是0.44%,因为女性胎儿具有两个染色体3。在确定是否存在x染色体非整倍性时(即,一个、三个或多于三个x染色体),对于一个x染色体,x染色体特异性序列的频率将是约0.22%,对于三个x染色体是0.66%并且对于四个x染色体是约0.88%。在确定胎儿是否可以是x染色体镶嵌现象时,x染色体特异性序列的频率应该是较小的并且对于xx/xo镶嵌现象可以是基本上小于0.44%或对于xx/xxx镶嵌现象在0.44%和0.66%之间。在另一个实例中,是5%胎儿dna的母体样品中每一个染色体将贡献整倍性胎儿中的5%的1/46th(或约0.11%)。在整倍性女性胎儿中,x染色体将因此贡献5%的2/46th或1/23rd(0.22%)并且常染色体对将贡献5%的2/46th或1/23rd(0.22%,因为每一个常染色体有两个)。
图8是用于执行根据本发明的统计分析的示例性方法800的简化的流程图。在方法800的步骤801中,x和y染色体上以及至少一个常染色体上的基因座被询问。在步骤803中,在步骤801中询问的染色体的每一个的染色体频率被估计。在步骤805中,计算y染色体以无拷贝、一个拷贝或两个或更多个拷贝存在的似然值,并且在步骤807中,通过比较计算的似然值与假设y染色体以0、1、或2+个拷贝存在的数学模型来计算y非整倍性的风险。类似地,在步骤809中计算x染色体以一个拷贝、两个拷贝或三个或更多个拷贝存在的似然值,并且在步骤811中,通过比较计算的似然值与假设x染色体以1、2、或3+个拷贝存在的数学模型来计算x非整倍性的风险。
图9是用于执行根据本发明的分析的示例性方法900的另一个实施方案的简化的流程图。在方法900中,确定母体样品的胎儿比例并且计算胎儿x和y非整倍性的风险。在步骤901中,至少一个常染色体上的至少一个多态性基因座被询问。在本发明的大部分实施方案中,至少两个并且通常多于两个常染色体上的若干至许多多态性基因座将被询问。在步骤903中,利用关于多态性基因座的信息计算母体样品的胎儿核酸比例(胎儿百分比)。在步骤905中,进行确定母体样品的胎儿核酸比例是否适合执行进一步分析。在步骤907中,y染色体上的基因座(多态性或非多态性)被询问。在大部分实施方案中,将同时执行询问y染色体和x染色体上的基因座,并且优选地在询问至少一个常染色体上的基因座的相同容器中。在步骤909中,利用计算的胎儿核酸比例计算y染色体以无拷贝、一个拷贝或两个或更多个拷贝存在的似然值,并且在步骤911中,通过比较计算的似然值与假设0、1、或2+个拷贝的y染色体的数学模型来计算y胎儿非整倍性的风险。在步骤913中,x染色体上的基因座(多态性或非多态性)被询问,如之前注明的,将同时执行询问y染色体和x染色体上的基因座,并且优选地在询问至少一个常染色体上的基因座的相同容器中。在步骤915中,利用计算的胎儿核酸比例计算x染色体以一个拷贝、两个拷贝或三个或更多个拷贝存在的似然值,并且在步骤917中,通过比较计算的似然值与假设的1、2、或3+个拷贝的x染色体数学模型来计算x胎儿非整倍性的风险。
如以上提及的,在优选的方面中在单个反应中(即,在单个容器中)执行询问用于确定样品中胎儿dna百分比的选择的多态性核酸区域和来自x和y染色体的选择的核酸区域二者的反应。当利用胎儿dna含量以帮助确定染色体异常的存在或不存在时,单个反应有助于使在分析系统中的多个步骤期间可能被引入的污染或偏倚的风险最小化,所述污染或偏倚否则可使结果出现偏差。因此,如提及的当描述测定时,对于测量胎儿分数用于询问选择的核酸区域的寡核苷酸的一些组将是多态性特异的并且对于确定胎儿性别或x和y染色体的非整倍性用于询问选择的核酸区域的寡核苷酸的一些组将是多态性的或snp不可知论的。
在其它的方面中,可以利用一个或更多个选择的核酸区域用于确定胎儿dna含量百分比以及检测x和y染色体异常两者。选择的核酸区域的等位基因可以被用于确定胎儿dna含量,并且然后这些相同的选择的核酸区域可以被用于检测胎儿染色体异常,忽略等位基因或snp特异性信息。对于胎儿dna含量和检测染色体异常两者利用相同的选择的核酸区域还帮助使由实验性错误或污染造成的任何偏倚最小化。
在一个实施方案中,不考虑胎儿性别,使用常染色体snp测量母体样品中胎儿来源贡献(参见,sparks等人,am.j.obstet&gyn.,206:319.e1-9(2012))。使用的过程不需要父系基因型的先前知识,因为在该方法期间鉴定非母体等位基因而不考虑父系遗传的知识。利用二项分布的最大似然估计可以被用于计算跨每一个母体样品中若干提供信息的基因座的估计的胎儿核酸分布。使用的用于计算胎儿核酸贡献的过程被描述在,例如2012年7月19日提交的ussn13/553,012中,其通过引用被并入。用于确定胎儿贡献的多态性区域可以是来自染色体1-12并且优选地不以血型抗原为靶标。
在某些方面中,确定胎儿dna中y染色体的数目可以不依赖于确定胎儿dna中x染色体的数目来执行。
在某些方面中,来自多态性测定的胎儿贡献的估值被用于确定y染色体胎儿频率(yff)值。例如,在某些方面中胎儿频率可以被定义为
其中pf_poly是来自多态性测定的胎儿贡献百分比的估值并且pf_chry是染色体y计数的归一化比例,其中pf_chry可以通过,比如计算每一个染色体y测定的中值计数与每一个常染色体或参考染色体测定的中值计数的比来确定。当yff值接近于零时,胎儿dna不可能包含y染色体。当yff值接近于一时,胎儿dna可能包含单一y染色体。如果yff值接近于二,胎儿dna可能包含两个拷贝的y染色体,并且对于另外的拷贝的y依此类推。在某些方面中,yff值被用于确定样品的胎儿dna中多于两个拷贝,例如三个、四个或五个拷贝,的y染色体的存在。
在某些方面中,pf_poly的估值被用于确定x染色体胎儿频率(xff)值。在某些方面中,可以利用样品中每一个潜在x染色体组成的不同定义来定义xff。例如,在某些方面中,对于包含单个x染色体的胎儿dna,样品j的x胎儿概率
其中
在某些方面中,对于包含两个x染色体的胎儿dna,x胎儿概率
其中
在某些方面中,对于包含三个x染色体的胎儿dna,样品j的x胎儿概率
其中
在某些方面中,胎儿dna中x染色体的数目可以通过比较
在某些实施方案中,最高归一化的概率与胎儿dna中x染色体的估计的数目对应。例如,如果p2比p1和p3更高,那么胎儿dna可能包含两个x染色体。在某些方面中,x归一化概率值计算被用于确定样品的胎儿dna中多于三个拷贝,诸如四个、五个或六个拷贝,的x染色体的存在。
测量的xff和yff值可以被在分析期间发生的变化影响,所述变化诸如来自测定系统的变化、操作者差异或其他变量。在某些方面中,为了报告结果的目的,胎儿差异值的特定范围可以被排除在确定性的基线水平外。在一些方面中,在零以下的yff值将被认为清楚地表明胎儿不具有y染色体。在一些方面中,在零和一之间的yff值不提供确定y染色体的存在或不存在的确定性的需要的水平。因此,在某些方面中,落在某种范围内,诸如0至1的yff值被认为在确定性范围之外,诸如在0.1至0.9的范围内的胎儿差异值,诸如0.2至0.8。这样的确定,无论是否是包含性的,可以被用于基于如以上展示的概率的值计算风险得分。这样的风险得分可以被用于,比如建议母亲和/或胎儿的临床护理。
本发明的过程的计算机实现
本发明的过程可以通过计算机或计算机系统来实现。例如,将来自方法的“读出(readout)”的原始数据-即高通量测序扩增产物或杂交至阵列-与计算机或处理器通讯,并且计算机可以执行软件,所述软件比如“计数”或“记录”感兴趣的多种序列的发生的频率、比较频率、归一化频率、执行质量控制和/或统计分析、计算母体样品的胎儿比例或百分比、鉴于胎儿核酸百分比计算基因组区域和/或染色体的量或频率、确定风险概率或执行其他计算以确定染色体异常。在一个实施方案中,计算机可以包括个人计算机,但计算机可以包括包含至少一个处理器和存储器的任何类型的机器。
软件组件的输出包括具有比如基因组区域和/或染色体(诸如,在此情况下,x和/或y染色体)具有量异常的概率的值的报告。在一些方面中,此报告是区域或染色体具有两个拷贝(比如,是二体)的似然值和区域或染色体具有更多个拷贝(比如,是三体)或更少(比如,是单体)拷贝的似然值。该报告可以是打印出来的纸或电子的,报告可以在显示器上来展示和/或通过电子邮件、ftp、手机短消息发送、发布在服务器上等电子通讯给使用者。尽管本发明的归一化过程被描述为作为软件实现,其也可以作为硬件和软件的组合实现。此外,用于归一化的软件可以作为在相同或不同计算机上操作的多组件实现。服务器(如果存在)和计算机二者可以包括典型计算设备的硬件组件(未显示),包括处理器、输入设备(比如,键盘、点击设备、用于语音命令的传声器、按钮、触摸屏等)和输出设备(比如,显示器、扬声器以及类似物)。当通过处理器来执行时,服务器和计算机可以包括计算机可读介质,比如包含实施公开的功能的计算机说明书的存储器和储存设备(比如,闪存、硬盘驱动器、光盘驱动器、磁盘驱动器以及类似物)。服务器和计算机还可以包括用于通讯的有线或无线网络通讯接口。
实施例
提出以下实施例以便为本领域中普通技术人员提供如何进行和利用本发明的完整的公开内容和说明,并且不意图限制发明人视为其发明的内容的范围,也不意图表示或暗示以下实验是进行的全部实验或仅有的实验。本领域技术人员将领会,可对本发明进行很多变化和/或修该,如在特定的方面显示的,而不偏离如广泛地描述的本发明的精神或范围。因此本方面将被认为在所有的方面为例证性的且非限制性的。
已经做出努力以确保关于使用的数字(比如,量、温度等)的准确性,但应该考虑某些实验误差和偏差。除非另外指示,否则份数按重量份数计,分子量是重量平均分子量,温度是以摄氏度,并且压力是在大气压或接近大气压。
实施例1:制备用于串联连接程序的dna
从coriellcellrepositories(camden,newjersey)获得来自受试者的基因组dna并且通过声学剪切(covaris,woburn,ma)将基因组dna片段化成约200bp的平均片段大小。
使用标准程序将dna生物素化。简略地,通过在1.5ml微管中生成以下反应来将covaris片段化的dna末端修复。5ugdna、12μl10xt4连接酶缓冲液(enzymatics,beverlyma)、50ut4多核苷酸激酶(enzymatics,beverlyma)和h20至120μl。将此微管在37℃孵育30分钟。将dna用10mmtris1mmedtaph8.5稀释至~0.5ng/μl的期望的最终浓度。
将5μldna置于96孔板的每一个孔中,并且板用粘合性板封口膜密封并且在250xg旋转10秒钟。然后将板在95℃孵育3分钟,并且冷却至25℃,并且再次在250xg旋转10秒钟。将生物素化主混合物(mastermix)在1.5ml微管中制备成以下的最终浓度:1xtdt缓冲液(enzymatics,beverlyma),8utdt(enzymatics,beverlyma),250μmcocl2,0.01nmol/μl生物素-16-dutp(roche,nutleynj)和h20至1.5ml。将15μl的主混合物等分入96孔板的每一个孔中,并且板用粘合性板封口膜密封。将板在250xg旋转10秒钟并且在37℃孵育60分钟。孵育之后,将板在250xg再次旋转10秒钟并且将7.5μl沉淀混合物(1ng/μl葡聚糖蓝,3mmnaoac)加至每一个孔。
将板用粘合性板封口膜密封并且用ika板涡旋仪在3000rpm混合2分钟。将27.5μl的异丙醇加入每一个孔中,将板用粘合性板封口膜密封并且在3000rpm涡旋5分钟。将板在3000xg旋转20分钟,倾析上清液,并且将板倒置并且在吸收剂擦拭物上以10xg离心1分钟。将板风干5分钟,并且将小团重悬在10μl10mmtrisph8.0,1mmedta中。从如以上列出的制备的寡核苷酸产生第一和第二基因座特异性固定寡核苷酸组的等摩尔的池(各40nm)。基于选择的基因组基因座的序列对测定过程同样地产生桥接的寡核苷酸的分离的等摩尔的池(各20nm)。
将10μg的链霉亲和素珠转移进入96孔板的孔中,并且将上清液移除。将60μl结合缓冲液(100mmtrisph8.0、10mmedta、500mmnacl2、58%甲酰胺、0.17%tween-80),10μl40nm固定序列寡核苷酸池和30μl的实施例2中制备的生物素化模板dna加至珠。将板用粘合性板封口膜密封并且在3000rpm涡旋直到将珠重悬。通过在70℃孵育5分钟将寡核苷酸退火至模板dna,随后慢慢冷却至30℃。
将板置于凸起的条形磁板上2分钟以将磁珠拉出并且将dna与孔的侧面关联。通过移液器将上清液移除并且用50μl的60%结合缓冲液(水中v/v)替换。通过涡旋将珠重悬,再次置于磁体上并且将上清液移除。使用50ul60%结合缓冲液重复此珠洗涤程序一次,并且使用50μl洗涤缓冲液(10mmtrisph8.0,1mmedta,50mmnacl2)再重复两次。
将珠重悬在由1xtaq连接酶缓冲液(enzymatics,beverlyma)、10utaq连接酶、和2um桥接寡核苷酸池(取决于测定格式)组成的37μl连接反应混合物中,并且在37℃孵育一小时。在适当时并且取决于测定格式,将非校正热稳定聚合酶加200nm每一种dntp包括在此混合物中。将板置于凸起的条形磁板上2分钟以将磁珠拉出并且将dna与孔的侧面关联。通过移液器将上清液移除,并且用50μl洗涤缓冲液来替代。通过涡旋将珠悬浮,再次置于磁铁上并且将上清液移除。重复洗涤程序一次。
为从链霉亲和素珠中洗脱产物,将30μl的10mmtris1mmedta,ph8.0加至96孔板中的每一个孔中。将板密封并且使用ika涡旋仪在3000rpm混合2分钟以重悬珠。将板在95℃孵育1分钟,并且使用8通道移液器将上清液吸出。将来自每一个孔的25μl的上清液转移进入新的96孔板用于通用扩增。
实施例2:连接的产物的通用扩增
使用与存在于杂交至感兴趣的核酸区域的第一和第二固定序列寡核苷酸中的通用序列互补的通用pcr引物扩增聚合的和/连接的核酸。在每一个扩增反应中使用实施例3中的反应混合物的每一种的25μl。由25μl洗脱的连接产物加1xpfusion缓冲液(finnzymes,finland)、1m甜菜碱、400nm每一种dntp、1upfusion误差校正热稳定dna聚合酶和带有样品标签的引物对组成的50μl通用pcr反应用于在混合和测序之前独特地鉴定个体样品。使用bioradtetradtm热循环仪在严格的条件下实施pcr。
将来自样品的每一种的10μl的通用pcr产物混合并且使用quant-ittmpicogreen,(invitrogen,carlsbad,ca)将混合的pcr产物纯化并且定量。将纯化的pcr产物在illuminahiseqtm2000上的载玻片的单泳道上测序。测序运行通常产生~100m原始读数,其中~85m(85%)映射至预期的测定结构。这转化为跨实验的平均~885k读数/样品,和跨96个选择的核酸区域的(在使用96基因座的实验的实例中)9.2k读数/复制物/基因座。
实施例3:分析多态性基因座以评估胎儿贡献百分比
为了评估母体样品中胎儿核酸比例,针对染色体1至12上的一组含snp基因座设计测定,其中一个碱基不同的两个桥接寡核苷酸被用于询问每一个snp(参见,比如图3)。在hapmap3数据集中针对次要等位基因频率优化snp。duan,等人,bioinformation,3(3):139-41(2008);epub2008nov9。
通过idt(coralville,iowa)来合成寡核苷酸并且汇集在一起以生成单个多路的测定池。如之前描述的从每一个受试者样品产生pcr产物。提供信息的多态性基因座被定义为其中胎儿等位基因不同于母体等位基因的基因座。因为测定表现出超过99%的等位基因特异性,所以当基因座的胎儿等位基因比例被测量是在1%和20%之间时,提供信息的基因座容易被鉴定。诸如在2012年7月19日提交的共同待审的申请ussn13/553,012中描述的,利用二项分布来估计最大似然,以基于来自几个提供信息的基因座的测量结果来确定最可能的胎儿比例。结果与由chu和合作者(参见,chu,等人,prenat.diagn.,30:1226-29(2010))提出的加权平均法良好地相关(r2>0.99)。
实施例4:利用染色体特异性基因组区域中非多态性位点检测y染色体频率异常
在第一实施方案中,使用针对y染色体上特异性基因组区域的测定鉴定y染色体频率异常的存在或不存在。本测定系统允许使用高多路系统鉴定多个个体的dna中这样的异常的存在或不存在。
使用与y染色体(chry)和染色体13、18和21(chr13、chr18和chr21)互补或来源于y染色体(chry)和染色体13、18和21(chr13、chr18和chr21)的寡核苷酸制备多重询问。使用常规固相化学反应合成串联连接格式中使用的所有寡核苷酸。第一固定组的寡核苷酸和桥接寡核苷酸合成为带有能够连接至毗邻寡核苷酸的3’羟基末端的5'磷酸部分。在chry开发了三十二种非多态性测定并且与对chr13、chr18和chr21开发的测定比较(参见,比如sparks等人,prenat.diagn.,32(1):3-9(2012)和sparks等人,amj.obstet.gynecol.(2012),doi:10.1016/j.ajog.2012.01.030)。使用染色体1至12或chr13、chr18和chr21上一组含有snp的基因座测量胎儿分数。使用多态性检测和概率算法计算来自这些染色体的胎儿分数估计,如2011年12月9日提交的ussn13/316,154和2011年12月28日提交的13/338,963中描述的。
检测并且计算y染色体计数的分数以确定pf_chry。将pf_chry除以计算的pf_poly。从一减去结果以提供yff。
通过y测定计数、常染色体测定计数和计算的pf_poly的bootstrap抽样计算此实例中yff的方差。使用0个拷贝的y、1个拷贝的y和2+个拷贝的y的模型执行贝叶斯分析以估计具有0个、1个或2+个拷贝的概率。使用在前述步骤中执行的标准偏差,一个实现使用对0个拷贝的截取正态模型和对1或2+个拷贝的正态分布模型。当差异接近0时,胎儿可能是女性并且当差异接近1时,胎儿可能是男性。在本实施例中,当yff的结果是在0.25和0.65之间或超过3.5时,结果被认为是在可报告的范围之外;但是来源于观察结果的其他阈值可能被用于建立什么是可报告的。
利用bootstrap抽样计算此胎儿差异值的分布,并且计算log10比值比,比较差异的似然是来自拟合0、1或2+个chry拷贝的模型的样品。
获得的结果显示用于胎儿性别的测试的准确性是100%,正确鉴定了745名女性和正确鉴定了797名男性。对于胎儿性别确定和y染色体非整倍性的确定实现了核型分析和使用如本文描述的chry分析之间的99.8%的一致性。表1显示确证组的胎儿y染色体状态的一致性。
实施例5:确定胎儿xo、xx、xy、xxx、xxy、xyy和xxyy基因型
为允许分析胎儿x染色体,除了用于询问y染色体和染色体13、18和21的寡核苷酸的组,固定序列和桥接寡核苷酸的组如所述的被用于询问染色体x基因座。将计算存在于胎儿的x染色体的数目的似然的计算加至计算chry、chr13、chr18和chr21的数目的似然以及确定pf_poly的计算。
在本研究中,处理432个血浆样品的组。全部受试者是不知情的、病例对照研究的部分,其中全部妊娠受试者已经经历过入侵性测试。将获得的结果与从入侵性测试获得的核型比较。将来自核型分析的妊娠个体的全部血浆样品根据在以上实施例1-4中详细描述的方案处理。用于选择样品的样品接受标准是与母体年龄等于或大于18岁,妊娠期等于或大于10周,并且单胎妊娠。允许自我和遗传不相关的卵子供体二者的卵子供体妊娠。
首先计算每一个样品的t21、t18和t13的风险(参见,比如2011年12月9日提交的ussn13/316,154和2011年12月28日提交的ussn13/338,963)。风险得分被限制在上端99%和下端0.01%。超过1%的风险将受试者分类为t21、t18或t13的高风险。利用测定确定的t21、t18和t13的基于风险得分的分类和如由核型确定的遗传状态的一致性被鉴定。
y染色体分析如在实施例4中描述的被执行。对于x染色体,使用chrx对chr13、chr1和chr21计数的比例。利用样品中基于测量的pf_poly的预期的损失或增加衍生用于1、2或3+个胎儿拷贝的chrx的模型。
对于包含单个x染色体的胎儿dna,样品j的x胎儿概率
其中
其中
其中
最高归一化的概率与胎儿dna中x染色体的估计的数目对应。
用于确定存在于胎儿的x染色体的数目的似然的数据接受标准实质上与用于确定存在于胎儿的y染色体的数目的似然的数据接收标准是一样的,除了三种计算被用于计算各自匹配1、2或3+个胎儿染色体x拷贝的模型的患者样品的log10比率。与对胎儿y染色体的数目执行的计算联合,计算存在于胎儿的x染色体的数目的似然的测试使用贝叶斯模型评估来自x和y染色体的数据并且比较对基因型xo、xx、xy、xxx、xxy、xyy和xxyy的假定。如果胎儿性别(男性对女性)的概率是<99%,那么生成胎儿x和y染色体状态的“无结果”。xff粗略代表损失的或增加的胎儿x染色体的数目。
432种血浆样品的414种通过qc度量,样品通过率为95.8%。t21、t18和t13的结果给出与核型分析的>99%一致性并且报告胎儿染色体x和y状态的“无结果”率<1%。胎儿x和y染色体分析给出每一个性染色体非整倍性(xo、xx、xy、xxx、xxy、xyy和xxyy)的≥99%的特异性;对于样品,给出≥80%的染色体非整倍性xo(其与特纳综合征有关)的灵敏度;并且胎儿x和y染色体分析给出胎儿性别(男/女)>99%的精确度。
使用1%的风险得分作为t21对非t21分类的边界条件,观察到似然的计算和核型分析之间的100%一致性;使用1%的风险得分作为t18对非t18分类的边界条件,观察到似然的计算和核型分析之间的100%一致性;以及使用1%的风险得分作为t13对非t13分类的边界条件,观察到似然的计算和核型分析之间的100%一致性。用于确定胎儿性别和非整倍性的结果被概括在表2中:
xff+yff测试结果给出每一个xo和xxx非整倍性样品的99.5%的特异性(95%wilson置信区间98.1-99.9)以及每一个xxy、xyy和xxyy非整倍性样品的100%的特异性(95%wilson置信区间99-100)。xff+yff测试结果给出xo非整倍性样品的96.3%的灵敏度(95%wilson置信区间82-99.8),其通过≥80%的接受标准。xff+yff测试结果给出胎儿性别的100%的精确度(95%wilson置信区间99-100)。
虽然本发明由以很多不同形式的方面满足,如结合本发明优选的方面详细描述的,但是应理解本公开内容应被视为是本发明的原则的示范且不意图将本发明限制为本文例证和描述的特定方面。本领域技术人员可做出很多变化而不偏离本发明的精神。本发明的范围将通过所附的权利要求书和其等价物来判断。摘要和题目不被解释为限制本发明的范围,因为它们的目的是使有关当局以及一般公众能够快速地确定本发明的一般性质。在以下的权利要求书中,除非使用术语“手段(means)”,否则本文提及的特征或要素不应解释为根据35u.s.c.§112,
- 还没有人留言评论。精彩留言会获得点赞!