环己基吡啶衍生物的制作方法
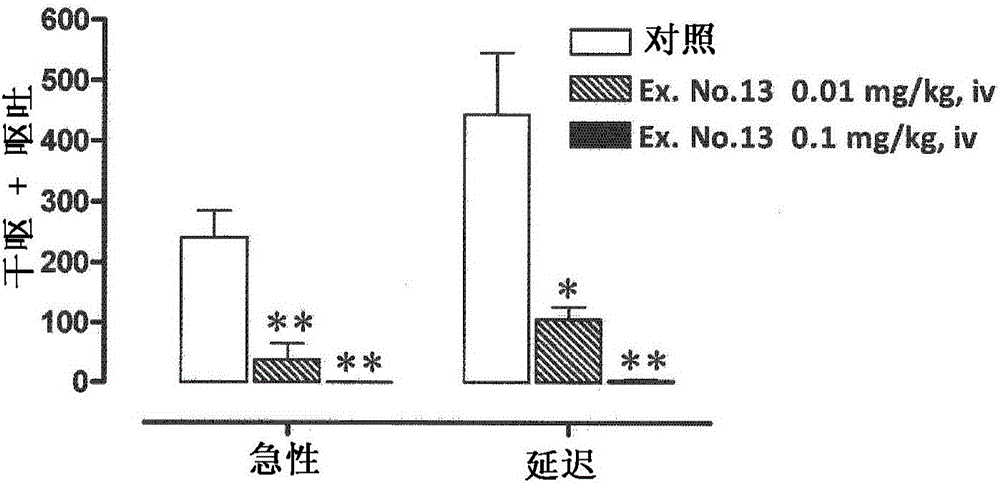
本发明涉及可用作药物的环己基吡啶衍生物。更具体地,本发明涉及环己基吡啶衍生物或其可药用盐,其具有P物质/神经激肽1(NK1)受体拮抗剂活性,并且其可用作预防或治疗癌症化疗所致恶心和呕吐(CINV)等的药剂。
背景技术:
:CINV在位于延髓外侧网状结构中的呕吐中枢受到刺激时发生。延髓的最后区和孤束核含有NK1受体,并且所述NK1受体被认为与呕吐密切相关。施用抗肿瘤剂促进消化道中肠嗜铬(EC)细胞的血清素分泌,而血清素通过消化道中的5-羟色胺3(5-HT3)受体直接刺激呕吐中枢。还有,当血清素通过位于第四脑室的最后区中的化学感受器触发区(CTZ)刺激呕吐中枢时,发生恶心和呕吐。P物质,如血清素,存在于消化道的EC细胞中,并且施用抗肿瘤剂促进了它的分泌。近年来,已经揭示,P物质通过CTZ中的NK1受体或通过与中枢神经系统中的NK1受体结合诱发呕吐,因此NK1受体作为开发止吐剂的靶标已经引起了关注(非专利文献1)。阿瑞吡坦(Aprepitant)是世界上第一个选择性NK1受体拮抗剂,它被批准作为与施用抗肿瘤剂相关的恶心和呕吐的预防药。关于阿瑞吡坦的作用机制,认为阿瑞吡坦选择性抑制中枢神经系统中P物质和NK1受体的结合,这是CINV的诱发途径之一,并由此预防CINV。阿瑞吡坦已经作为CINV的预防药投放市场(非专利文献2)。已知阿瑞吡坦通过细胞色素P450(CYP)3A4代谢。还有,已知阿瑞吡坦具有剂量依赖性的CYP3A4抑制作用、CYP3A4诱导作用和CYP2C9诱导作用。因此,阿瑞吡坦可能引起与抑制或诱导CYP3A4的药物或者与通过CYP3A4或CYP2C9代谢的药物的药物间相互作用。例如,据报告,阿瑞吡坦对CYP3A4的抑制作用有时抑制地塞米松的代谢,因此当地塞米松与阿瑞吡坦联用时,应该调节剂量(非专利文献3)。因此,当使用阿瑞吡坦时,应该对于基于阿瑞吡坦的CYP3A4抑制作用的药物间相互作用有充分的注意。出于以上原因,在预防或治疗CINV中需要药物间相互作用较少的新的NK1受体拮抗剂。有NK1受体拮抗剂活性的化合物例如卡索匹坦、奈妥匹坦、依洛匹坦、罗拉吡坦、维替匹坦、伏氟匹坦等等,是已知的。然而,卡索匹坦据报告具有CYP3A4抑制作用并且由于所述作用而引起药物间相互作用(非专利文献4)。对卡索匹坦作为癌症化疗所致恶心和呕吐的预防药的临床试验已经在美国和欧洲进行;然而,它的开发在申请后中止。奈妥匹坦目前作为癌症化疗所致恶心和呕吐的预防药正在开发中;然而,奈妥匹坦据报告具有CYP3A4抑制作用并引起由于所述作用的药物间相互作用(非专利文献5)。对依洛匹坦作为癌症化疗所致恶心和呕吐的预防药的临床试验已经在美国进行;然而,它的开发中止。对伏氟匹坦作为癌症化疗所致恶心和呕吐的预防药的临床试验已经在欧洲进行;然而,它的开发中止。以上化合物许多以中止为结果。所有以上化合物均未上市。专利文献1至16中描述了宣称具有NK1受体拮抗剂活性的吡啶衍生物。并且,专利文献17和18中描述了吡啶衍生物的前体药物。然而,以上文献中均未描述本发明的环己基吡啶衍生物。现有技术文献专利文献专利文献1:美国专利第6,479,483号说明书专利文献2:美国专利第6,770,637号说明书专利文献3:美国专利第7,939,533号说明书专利文献4:欧洲专利第1,103,545号说明书专利文献5:美国专利第7,211,579号说明书专利文献6:美国专利公布第2006/0030600号说明书专利文献7:美国专利第6,576,762号说明书专利文献8:美国专利第6,225,316号说明书专利文献9:美国专利第7,683,056号说明书专利文献10:美国专利第8,344,005号说明书专利文献11:国际公布第WO2011/054773号说明书专利文献12:美国专利公布第2007/0071813号说明书专利文献13:美国专利公布第2003/0083345号说明书专利文献14:美国专利公布第2003/0004157号说明书专利文献15:美国专利第6,849,624号说明书专利文献16:美国专利第6,297,375号说明书专利文献17:美国专利第6,593,472号说明书专利文献18:美国专利第8,426,450号说明书非专利文献非专利文献1:P.J.Hesketh等,EuropeanJournalofCancer,2003,第39卷,1074-1080页非专利文献2:ToniM.Dando等,Drugs,2004,第64卷,第7号,777-794页非专利文献3:JacquelineB.McCrea等,CLINICALPHARMACOLOGY&THERAPEUTICS,2003,第74卷,第1号,17-24页非专利文献4:StefanoZamuner等,BritishJournalofClinicalPharmacology,2010,第70卷,第4号,537-546页非专利文献5:CorinnaLanzarotti等,SupportCareCancer,2013,第21卷,第10号,2783-2791页技术实现要素:发明要解决的课题本发明的课题是提供具有NK1受体拮抗剂活性的新化合物,它的CYP3A4抑制活性与阿瑞吡坦相比降低,并且它可用于预防或治疗癌症化疗所致恶心和呕吐。本发明的课题优选是提供中枢迁移性和药效持续性优异的上述化合物。课题解决手段本发明涉及由下面式(I)表示的化合物或其可药用盐。亦即,本发明与以下[1]至[12]等相关。[1]式(I)表示的化合物,或其可药用盐:[化学式1]其中环A是下式表示的基团:[化学式2]X是氢原子、氰基、卤素、C1-6烷基或羟甲基;R1是下式表示的基团:[化学式3]其中R1a和R1b各自独立地是氢原子、氟原子或C1-6烷基中的任意一种;m是0、1或2;当m是2时,这些R1a和R1b任选彼此不同;R2是C1-6烷基、羟基或C1-6烷氧基;U是下式表示的基团:[化学式4]其中R3a和R3b各自独立地是氢原子、C1-6烷基、羟基C1-6烷基或C1-6烷氧基C1-6烷基;Y是0、1或2;当Y是2时,两个R2任选彼此不同。[2]根据上述[1]的式(Ia)表示的化合物,或其可药用盐:[化学式5]其中环A和X具有与上述[1]中所述的相同的含义;R1c和R1d各自独立地是氢原子或甲基;U1是下式表示的基团:[化学式6]其中R3c和R3d各自独立地是氢原子、甲基或羟甲基;n是0、1或2;当n是2时,这些R1c和R1d任选彼此不同。[3]根据上述[2]的式(Ib)表示的化合物,或其可药用盐:[化学式7]其中R1c和R1d具有与上述[2]中所述的相同的含义。[4]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式8][5]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式9][6]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式10][7]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式11][8]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式12][9]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式13][10]根据上述[1]的下式表示的化合物,或其可药用盐:[化学式14][11]药物组合物,其包含根据上述[1]至[10]任一项的化合物或其可药用盐作为活性成分。[12]根据上述[11]的药物组合物,其用于预防癌症化疗所致恶心和呕吐。发明的效果本发明的化合物具有优异的NK1受体拮抗剂活性。并且,本发明的化合物的CYP3A4抑制活性与阿瑞吡坦相比降低。本发明的优选化合物在中枢迁移性上优异。本发明的更优选的化合物在中枢迁移性和药效持续性上优异。因此,本发明的化合物或其可药用盐可用作预防或治疗癌症化疗所致恶心和呕吐的药剂。附图说明[图1]图1显示了试验例6中对顺铂诱发急性和延迟性呕吐反应的作用。图中,从左起的每个柱状图块分别显示了急性期中对照组(对照)、静脉内施用0.01mg/kg的实施例13化合物(Ex.No13,0.01mg/kg,iv)的组和静脉内施用0.1mg/kg的实施例13化合物(Ex.No13,0.1mg/kg,iv)的组的值,以及在延迟期中对照组、静脉内施用0.01mg/kg的实施例13化合物(Ex.No13,0.01mg/kg,iv)的组和静脉内施用0.1mg/kg的实施例13化合物(Ex.No13,0.1mg/kg,iv)的组的值。纵轴显示干呕和呕吐的次数(干呕+呕吐)(对照组中3例的平均值+标准误差,静脉内施用0.01mg/kg的组中3例的平均值+标准误差,和静脉内施用0.1mg/kg的组中3例的平均值+标准误差)。具体实施方式以下,将进一步详细描述本发明的实施方式。在本发明中,除非另作说明,各术语具有以下含义。术语“C1-6烷基”是指具有1至6个碳原子的直链或支链烷基,例如,甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基等可以作为举例说明。术语“C1-6烷氧基”是指具有1至6个碳原子的直链或支链烷氧基,例如,甲氧基、乙氧基、丙氧基、异丙氧基等可以作为举例说明。术语“羟基C1-6烷基”是指被羟基取代的C1-6烷基,例如羟甲基、1-羟基乙基、1-羟基-1,1-二甲基甲基、2-羟基乙基、2-羟基-2-甲基丙基、3-羟基丙基等。术语“C1-6烷氧基C1-6烷基”是指被上述C1-6烷氧基取代的上述C1-6烷基。在本发明的式(I)表示的化合物含有一个或多个不对称碳原子的情况下,每个不对称碳处的R或S构型的所有立体异构体和它们的混合物包括在本发明中。在这样的情况下,消旋化合物、消旋混合物、单个对映体以及非对映体的混合物包括在本发明的范围内。在本发明的式(I)表示的化合物具有顺-反异构体的情况下,所有顺-反异构体都包括在本发明中。在本发明中,立体化学确定也可以根据本领域公知的方法确定。例如,也参照"特论NMR立体化学"(讲谈社),(2012年发行),59页。本发明的式(I)表示的化合物还可以根据常规方法转变为其可药用盐。作为这样的盐,酸加成盐和与碱的盐可以作为举例说明。作为酸加成盐,与无机酸例如盐酸、氢溴酸、氢碘酸、硫酸、硝酸、磷酸等的酸加成盐,与有机酸例如甲酸、乙酸、三氟乙酸、甲磺酸、苯磺酸、对甲苯磺酸、丙酸、柠檬酸、琥珀酸、酒石酸、富马酸、丁酸、草酸、丙二酸、马来酸、乳酸、苹果酸、碳酸、苯甲酸、谷氨酸、天冬氨酸等的酸加成盐,可以作为举例说明。作为与碱的盐,与无机碱形成的盐例如锂盐、钠盐、钾盐、钙盐、镁盐等,和与有机碱例如N-甲基-D-葡糖胺、N,N'-二苄基乙二胺、三乙胺、哌啶、吗啉、吡咯烷、精氨酸、赖氨酸、胆碱等形成的盐可以作为举例说明。在本发明中,可药用盐还包括其与可药用溶剂例如水、乙醇等的溶剂合物。在本发明的式(I)表示的化合物中,符号R1和R2是指环己烷环的取代基。在本发明的式(I)表示的化合物的实施方式中,作为在环上具有取代基的环己烷环,下式表示的基团可以作为举例说明。[化学式15]其中,带有(*)的键是与吡啶环的键合部位,并且R1和R2具有与上述[1]中所述的相同的含义。本发明的式(I)表示的化合物还可以,例如,通过下面描述的方法或与其相似的方法、或者在文献中描述的方法或或与其相似的方法制备。方案1[化学式16]式中,L1和L2各自独立地是离去基团例如氯原子、溴原子、碘原子、三氟甲磺酰氧基等,并且环A、X、R1、R2、R3a、R3b和Y具有与上文规定的相同的含义。工序1化合物(4)也可以通过化合物(2)与化合物(3)在惰性溶剂中在碱和钯催化剂存在下进行偶联反应而制备。工序2化合物(6)也可以通过化合物(4)与化合物(5)在惰性溶剂中在碱存在下进行缩合反应而制备。工序3化合物(8)也可以通过化合物(6)与化合物(7)在惰性溶剂中在碱和钯催化剂存在下进行偶联反应而制备。作为惰性溶剂,例如,N,N-二甲基甲酰胺、N-甲基吡咯烷酮、二甲亚砜、二乙醚、四氢呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯、乙醇、水及其混合溶剂可以作为举例说明。作为碱,例如,碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢氧化锂、氟化钾、氟化铯、三乙胺、吡啶、N,N-二异丙基乙胺、2,6-卢剔啶、和1,8-二氮杂双环[5,4,0]-7-十一烯可以作为举例说明。作为钯催化剂,[1,1'-双(二苯基膦)二茂铁]-二氯化钯(II)-二氯甲烷复合物(1:1)、四(三苯基膦)钯(0)等可以作为举例说明。反应温度通常在0℃至回流温度。反应时间基于所使用的起始材料、溶剂和反应温度等而异,通常从30分钟至7天。上述偶联反应还可以使用微波反应器(Biotage)进行。当使用微波反应器时,基于所使用的起始材料、溶剂和机种而异,所述反应在压力范围:1至30巴、功率范围:1至400W、反应温度:室温至300℃、和反应时间:1分钟至1天下进行。另外,当R1或R2的官能团需要保护基团时,上述偶联反应也可以在引入保护基团后进行。工序4式(I)表示的化合物也可以通过进行化合物(8)的烯烃的还原例如催化还原法而制备。所述催化还原法可以,例如,通过在惰性溶剂中在氢气气氛下使用催化剂以让化合物(8)反应来进行。作为所述惰性溶剂,例如,甲醇、乙醇、乙酸乙酯、四氢呋喃和乙酸可以作为举例说明。作为所述催化剂,例如,钯-碳粉末、铑-碳粉末、铂-碳粉末、掺杂钒的铂-碳粉末可以作为举例说明。所述反应温度通常在室温至回流温度。所述反应时间基于所使用的起始材料、溶剂和反应温度等而异,通常从30分钟至7天。另外,当在上述工序3中保护基团被引入官能团时,式(I)表示的化合物也可以通过在上述还原反应后进行去保护反应而制备。方案2[化学式17]式中,R1、R2和Y具有与上文规定的相同的含义。工序5化合物(10)也可以通过化合物(9)在惰性溶剂中在碱存在下与三氟甲烷磺酸酐进行反应而制备。作为惰性溶剂,例如,N,N-二甲基甲酰胺、N-甲基吡咯烷酮、二甲亚砜、二乙醚、四氢呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯、及它们的混合溶剂可以作为举例说明。作为碱,例如,碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢氧化锂、氟化钾、氟化铯、三乙胺、吡啶、N,N-二异丙基乙胺、2,6-卢剔啶、2,6-二叔丁基-4-甲基吡啶和1,8-二氮杂双环[5,4,0]-7-十一烯可以作为举例说明。反应温度通常在0℃至回流温度。反应时间基于所使用的起始材料、溶剂和反应温度等而异,通常从30分钟到7天。工序6化合物(7)也可以通过化合物(10)与化合物(11)在惰性溶剂中在碱和钯催化剂存在下进行偶联反应而制备。作为惰性溶剂,例如,N,N-二甲基甲酰胺、N-甲基吡咯烷酮、二甲亚砜、二乙醚、四氢呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯、及它们的混合溶剂可以作为举例说明。作为碱,例如,碳酸钾、乙酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢氧化锂、氟化钾、氟化铯、三乙胺、吡啶、N,N-二异丙基乙胺、2,6-卢剔啶、和1,8-二氮杂双环[5,4,0]-7-十一烯可以作为举例说明。作为钯催化剂,例如,[1,1'-双(二苯基膦)二茂铁]-二氯化钯(II)-二氯甲烷复合物(1:1)、二(三苯基膦)钯(II)等可以作为举例说明。反应温度通常在室温至回流温度。反应时间基于所使用的起始材料、溶剂和反应温度等而异,通常从30分钟至7天。上述方案对于制备本发明的式(I)表示的化合物及其合成中间体是示例性的。上述方案可以改变或修改为本领域普通技术人员可以易于理解的方案。在上述方案中,当基于官能团的种类,保护基团是必要的时,也可以根据常规方法任选进行引入和除去操作的组合。如果必要,本发明的式(I)表示的化合物及其中间体也可以根据相关领域普通技术人员公知的常规分离和提纯技术,例如溶剂提取、结晶、再结晶、层析、制备型高效液相色谱等,进行分离和提纯。本发明的化合物具有优异的NK1受体拮抗剂活性,因此也可以用作预防或治疗由NK1受体介导的各种疾病的药剂。例如,本发明的化合物可用作止吐剂,尤其可用作癌症化疗(例如顺铂)所致胃肠道症状(例如恶心和呕吐)的预防药。本发明的优选化合物不仅可用于急性期癌症化疗所致恶心和呕吐,而且可用于延迟期癌症化疗所致恶心和呕吐。在一种实施方式中,本发明的化合物具有优异的NK1受体拮抗剂活性,因此也可以用作术后恶心和呕吐(PONV)、放疗伴发的恶心和呕吐、吗啡诱发的呕吐或运动病的预防药,和精神分裂症、社交恐惧症、焦虑和抑郁、酒精中毒、过敏性肠综合征、溃疡性结肠炎、咳嗽、哮喘、特应性皮炎、银屑癣、搔痒症、疼痛、偏头痛、耳鸣、良性前列腺肥大、膀胱过度活动症或尿失禁的治疗药。本发明的药物组合物可以取决于它们的用法以各种剂型施用。作为这样的剂型,例如,散剂、颗粒剂、细粒剂、干糖浆剂、片剂、胶囊剂、注射剂、液剂、软膏剂、栓剂和贴敷剂可以作为举例说明,其口服或肠胃外施用。本发明的药物组合物可以通过使用式(I)表示的化合物或其可药用盐和至少一种药物添加剂而制备。这些药物组合物可以取决于它们的剂型,按照常规配制程序,通过用适当的药物添加剂例如赋形剂、崩解剂、粘合剂、润滑剂、稀释剂、缓冲剂、等张化剂、防腐剂、润湿剂、乳化剂、分散剂、稳定剂、增溶剂等混合、稀释或溶解而配制。当本发明的药物组合物用于预防或治疗时,式(I)表示的化合物或其可药用盐作为活性成分的剂量取决于各患者的年龄、性别、体重、疾患和治疗的程度等适当决定。成人的剂量在口服施用的情况下可以在例如0.1至1000mg/日、0.1至500mg/日、0.1至100mg/日或0.1至50mg/日的范围内决定,并且所述日剂量可以分成每日一、二、三或四次施用。并且,成人的剂量在肠胃外给药的情况下可以在例如0.1至1000mg/日、0.1至500mg/日、0.1至100mg/日或0.1至50mg/日的范围内决定,并且所述日剂量可以分成每日一、二、三或四次施用。当本发明的药物组合物用于预防癌症化疗所致恶心和呕吐时,本药物也可以在抗肿瘤剂给药之前施用。例如,所述药物可以在化疗中给药之前立即至给药的一个半小时之前施用,并且在第二日之后,还可以在上午施用所述药物。在一种实施方式中,本发明的式(I)表示的化合物或其可药用盐也可以与NK1受体拮抗体以外的任何其他药物组合使用。作为组合使用的这类其他药物,例如,皮质类固醇和5-HT3受体拮抗剂型止吐剂可以作为举例说明。当本发明的式(I)表示的化合物或其可药用盐与其他药剂组合使用时,它可以作为与它们的活性成分包含在一起的制剂或作为每种活性成分分别配制的各制剂施用。当个别配制时,这些制剂可以分别或同时施用。此外,本发明的式(I)表示的化合物的剂量或其可药用盐可以根据所述组合使用的其他药剂的剂量而适度减少。实施例本发明通过以下参考例、实施例和试验例进一步更详细地说明。然而,本发明不限于这些内容。参考例14-(4,4,5,5-四甲基-[1,3,2]-二氧杂硼戊环-2-基)环己-3-烯羧酸乙酯在室温下向4-氧环己烷羧酸乙酯(1.00g)和2,6-二叔丁基-4-甲基吡啶(1.39g)在二氯甲烷(40mL)中的溶液添加三氟甲磺酸酐(1.74g),并将所述混合物在室温下搅拌16小时。通过过滤除去不溶性物质,然后用二氯甲烷(5mL)洗涤。滤液在减压下浓缩,并向残渣添加二氯甲烷(5mL)。通过过滤除去不溶性物质,然后用二氯甲烷(3mL)洗涤。滤液在减压下浓缩,并向残渣添加二氯甲烷(3mL)。通过过滤除去不溶性物质,然后用二氯甲烷(2mL)洗涤。滤液在减压下浓缩,产生4-三氟甲磺酰氧基-环己-3-烯羧酸乙酯(1.57g)。在氩气气氛下,将4-三氟甲磺酰氧基-环己-3-烯羧酸乙酯(1.57g)、双(频那醇合)二硼(1.39g)、[1,1'-双(二苯基膦)二茂铁]-二氯化钯(II)二氯甲烷复合物(1:1)(0.13g)和乙酸钾(1.53g)在二甲亚砜(26mL)中的悬液在50℃搅拌4.5小时。所述反应混合物冷却到室温并添加水。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-85/15),获得标题化合物(0.84g)。参考例2至7参考例2至7的化合物以与参考例1所描述的相似的方法,使用相应的起始材料制备。参考例8(6-氯-4-碘吡啶-3-基)氨基甲酸叔丁酯在氩气气氛下,在-78℃向(6-氯吡啶-3-基)氨基甲酸叔丁酯(5.0g)和N,N,N',N'-四甲基乙-1,2-二胺(7.7g)在二乙醚(120mL)中的溶液逐滴添加正丁基锂(2.65mol/L四氢呋喃溶液,25mL)。所述混合物在-10℃搅拌2小时后,在-78℃下向所述混合物逐滴添加碘(11.4g)在二乙醚(40mL)中的溶液,并将所生成的混合物在室温下搅拌1天。向所述反应混合物添加饱和氯化铵水溶液,并用二乙醚提取所生成的混合物。有机层用10%焦亚硫酸钠水溶液和饱和盐水洗涤,经过无水硫酸镁干燥,和在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-60/40),获得标题化合物(2.59g)。参考例9(6-氯-4-碘吡啶-3-基)甲基氨基甲酸叔丁酯在冰冷却下向(6-氯-4-碘吡啶-3-基)氨基甲酸叔丁酯(2.59g)在N,N-二甲基甲酰胺(30mL)中的溶液添加氢化钠(60%,0.32g),并将所述混合物在室温下搅拌30分钟。在冰冷却下向所述混合物添加碘甲烷(2.60g)并将所述混合物在室温下搅拌过夜。向所述反应混合物添加饱和碳酸氢钠水溶液,并用乙酸乙酯提取所生成的混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-70/30),获得标题化合物(2.66g)。参考例10(6-氯-4-碘吡啶-3-基)甲胺在冰冷却下向(6-氯-4-碘吡啶-3-基)甲基氨基甲酸叔丁酯(2.66g)在二氯甲烷(10mL)中的溶液添加三氟乙酸(8.23g),并且所述混合物在室温下搅拌2小时。所述反应混合物在减压下浓缩,向残渣添加饱和碳酸钠水溶液,并用乙酸乙酯提取所述混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(1.89g)。参考例11[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]甲胺在室温下向(6-氯-4-碘吡啶-3-基)甲胺(1.89g),4-氟-2-甲基苯基硼酸(1.30g)、1,2-二甲氧基乙烷(20mL)和水(20mL)的混合物添加乙酸钯(II)(0.16g)、三苯基膦(0.37g)和碳酸钠(3.73g),并将所述混合物在90℃搅拌过夜。所述反应混合物冷却到室温并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-50/50),获得标题化合物(1.56g)。参考例12(6-氯-4-邻甲基苯基吡啶-3-基)甲胺在室温下向(6-氯-4-碘吡啶-3-基)甲胺(0.70g)、2-甲基苯基硼酸(0.42g)、1,2-二甲氧基乙烷(10mL)和水(10mL)的混合物添加乙酸钯(II)(0.058g)、三苯基膦(0.14g)和碳酸钠(1.38g),并将所述混合物在90℃搅拌过夜。所述反应混合物冷却到室温并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-70/30),获得标题化合物(0.54g)。参考例136-氯-3-硝基吡啶-2-甲腈在室温下向2,6-二氯-3-硝基吡啶(2.50g)在N-甲基吡咯烷酮(25mL)中的溶液添加氰化铜(I)(2.32g),并将所述混合物在180℃搅拌1小时。所述反应混合物冷却到室温,并向所述混合物添加乙酸乙酯和水。过滤除去不溶性物质。滤液用饱和盐水洗涤,分离水层用乙酸乙酯再提取。合并的有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-70/30),获得标题化合物(0.90g)。参考例143-氨基-6-氯吡啶-2-甲腈在室温下向6-氯-3-硝基吡啶-2-甲腈(0.32g)和浓盐酸(1.2mL)在乙醇(3.6mL)中的溶液添加铁粉(0.34g),和将所述混合物在回流下加热30分钟。所述反应混合物冷却到室温,并通过添加饱和碳酸氢钠水溶液碱化。向所述反应混合物添加乙酸乙酯,所生成的混合物通过Celite(注册商标)垫过滤。滤液用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.24g)。参考例153-氨基-4-溴-6-氯吡啶-2-甲腈在室温下向3-氨基-6-氯吡啶-2-甲腈(0.24g)在N,N-二甲基甲酰胺(8mL)中的溶液添加N-溴代琥珀酰亚胺(0.37g),并将所述混合物在同样的温度下搅拌过夜。向所述反应混合物添加饱和硫代硫酸钠水溶液,并用乙酸乙酯提取所生成的混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=75/25-50/50),获得标题化合物(0.30g)。参考例163-氨基-6-氯-4-(4-氟-2-甲基苯基)吡啶-2-甲腈3-氨基-4-溴-6-氯吡啶-2-甲腈(0.15g)、4-氟-2-甲基苯基硼酸(0.08g)、四(三苯基膦)钯(0)(0.07g)、碳酸钠(0.20g),1,2-二甲氧基乙烷(3.2mL)和水(0.8mL)的混合物在微波照射下在100℃搅拌1小时。所述反应混合物冷却到室温并添加水。所生成的混合物用乙酸乙酯提取。有机层用饱和碳酸氢钠水溶液和饱和盐水洗涤,经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=50/50-0/100),获得标题化合物(0.14g)。参考例173-苄氧基-2-(3,5-双三氟甲基苯基)-2-甲基丙酸在氩气气氛下,在-78℃向2-(3,5-双三氟甲基苯基)丙酸甲酯(0.60g)在四氢呋喃(5mL)中的溶液逐滴添加二异丙基氨基锂(1.09mol/L四氢呋喃/正己烷溶液,2mL),并将所述混合物在同样的温度下搅拌15分钟。在-78℃向所述反应混合物添加苄基氯甲基醚(0.34g)在四氢呋喃(2mL)中的溶液,并将所述混合物在室温下搅拌1小时。向所述反应混合物添加饱和氯化铵水溶液,并用乙酸乙酯提取所生成的混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-70/30),获得3-苄氧基-2-(3,5-双三氟甲基苯基)-2-甲基丙酸甲酯(0.78g)。在室温下向所得到的化合物(0.78g)在乙醇(3mL)中的溶液添加5.0mol/L氢氧化钠水溶液(1mL),并将所述混合物在室温下搅拌3小时。向所述反应混合物添加2.0mol/L盐酸(3ml),并用乙酸乙酯提取所述混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下除去所述溶剂,获得标题化合物(0.77g)。参考例182-(3,5-双三氟甲基苯基)-N-[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺在室温下向2-(3,5-双三氟甲基苯基)-2-甲基丙酸(0.66g)在二氯甲烷(10mL)中的溶液添加草酰氯(0.56g)和N,N-二甲基甲酰胺(2滴),并将所述混合物在同样的温度下搅拌1小时。所述反应混合物在减压下浓缩,获得残渣。在氩气气氛下,在冰冷却下向[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]甲胺(0.50g)在四氢呋喃(10mL)中的溶液逐滴添加双(三甲基甲硅烷基)氨基钾(0.5mol/L甲苯溶液,5.0mL),并将所述混合物在室温下搅拌30分钟。在冰冷却下向所述反应混合物逐滴添加上述残渣在四氢呋喃(5mL)中的溶液,并将所述混合物在室温下搅拌2小时。向所述反应混合物添加1.0mol/L碳酸氢钠水溶液,并用乙酸乙酯提取所述混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-60/40),获得标题化合物(1.03g)。参考例19和20参考例19和20的化合物以与参考例18所描述的相似的方法,使用相应的起始材料制备。参考例212-(3,5-双三氟甲基苯基)-N-[6-氯-2-氰基-4-(4-氟-2-甲基苯基)吡啶-3-基]异丁酰胺在室温下向2-(3,5-双三氟甲基苯基)-2-甲基丙酸(0.31g)在二氯甲烷(2.6mL)中的溶液添加草酰氯(0.26g)和N,N-二甲基甲酰胺(2滴),并将所述混合物在同样的温度下搅拌1小时。所述反应混合物在减压下浓缩,获得残渣。在冰冷却下向3-氨基-6-氯-4-(4-氟-2-甲基苯基)吡啶-2-甲腈(0.14g)在四氢呋喃(5mL)中的溶液添加双(三甲基甲硅烷)氨基钠(1.0mol/L四氢呋喃溶液,1.1mL),并将所述混合物在同样的温度下搅拌30分钟。在冰冷却下向所述反应混合物逐滴添加上述残渣在四氢呋喃(2.0mL)中的溶液,并将所述混合物在室温下搅拌30分钟。向所述反应混合物添加饱和碳酸氢钠水溶液,并用乙酸乙酯提取所生成的混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=85/15-40/60),获得标题化合物(0.21g)。参考例222-(3,5-双三氟甲基苯基)-N-[6-氯-2-氰基-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺在冰冷却下向2-(3,5-双三氟甲基苯基)-N-[6-氯-2-氰基-4-(4-氟-2-甲基苯基)吡啶-3-基]异丁酰胺(0.21g)在N,N-二甲基甲酰胺(2.4mL)中的溶液添加氢化钠(60%,0.018g),并将所述混合物在同样的温度下搅拌5分钟。在冰冷却下向所述反应混合物添加碘甲烷(0.11g),并将所述混合物在室温下搅拌过夜。向所述反应混合物添加水,并将所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-50/50),获得标题化合物(0.09g)。参考例234-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯羧酸乙酯2-(3,5-双三氟甲基苯基)-N-[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺(0.08g)、4-(4,4,5,5-四甲基-[1,3,2]-二氧杂硼戊环-2-基)环己-3-烯羧酸乙酯(0.08g)、碳酸钠(0.05g)、四(三苯基膦)钯(0)(0.02g)、1,2-二甲氧基乙烷(1.0mL)、水(0.2mL)和乙醇(0.2mL)的混合物在微波照射下在120℃搅拌1小时。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-80/20),获得标题化合物(0.03g)。参考例24至26参考例24至26的化合物以与参考例23所描述的相似的方法,使用相应的起始材料制备。参考例272-(3,5-双三氟甲基苯基)-N-[6-(1,4-二氧杂螺[4.5]癸-7-烯-8-基)-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺2-(3,5-双三氟甲基苯基)-N-[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺(0.53g)、8-(4,4,5,5-四甲基-[1,3,2]-二氧杂硼戊环-2-基)-1,4-二氧杂螺[4.5]癸-7-烯(0.29g)、碳酸钠(0.32g)、四(三苯基膦)钯(0)(0.12g)、1,2-二甲氧基乙烷(7.5mL)、水(1.5mL)和乙醇(1.5mL)的混合物在微波照射下在120℃搅拌30分钟。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-10/90),获得标题化合物(0.52g)。参考例28至30参考例28至30的化合物以与参考例23同样的方法,使用相应的起始材料制备。参考例312-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}-2-甲基丙酸乙酯2-(3,5-双三氟甲基苯基)-N-[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺(0.11g)、2-甲基-2-[4-(4,4,5,5-四甲基[1,3,2]二氧杂硼戊环-2-基)环己-3-烯基]丙酸乙酯(0.12g)、碳酸钠(0.06g)、四(三苯基膦)钯(0)(0.02g)、1,2-二甲氧基乙烷(1.5mL)、水(0.3mL)和乙醇(0.3mL)的混合物在微波照射下在120℃搅拌30分钟。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-60/40),获得标题化合物(0.12g)。参考例32{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}乙酸甲酯2-(3,5-双三氟甲基苯基)-N-[6-氯-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺(2.00g)、[4-(4,4,5,5-四甲基[1,3,2]二氧杂硼戊环-2-基)环己-3-烯基]乙酸甲酯(1.26g)、四(三苯基膦)钯(0)(0.22g)、2.0mol/L碳酸钠水溶液(5.6mL)、1,2-二甲氧基乙烷(22.5mL)和乙醇(5.6mL)的混合物在微波照射下在120℃搅拌30分钟。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-50/50),获得标题化合物(2.14g)。参考例332-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(1-甲基-3-氧代环己基)吡啶-3-基]-N-甲基异丁酰胺在冰冷却下向碘化铜(I)(0.05g)在二乙醚(2mL)中的悬液添加甲基锂(1.13mol/L二乙醚溶液,0.45mL),并将所述混合物在同样的温度下搅拌15分钟。在冰冷却下向所述反应混合物逐滴添加在二乙醚(1mL)中的2-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(3-氧代环己-1-烯基)吡啶-3-基]-N-甲基异丁酰胺(0.10g),并将所述混合物在室温下搅拌1小时。向所述反应混合物添加饱和氯化铵水溶液,并用乙酸乙酯提取所生成的混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=85/15-50/50),获得标题化合物(0.50g)。参考例34{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]亚环-2-己烯基}乙酸乙酯在冰冷却下向氢化钠(60%,0.012g)在四氢呋喃(2mL)中的悬液添加膦酰乙酸二乙酯(0.08g),并将所述混合物在同样的温度下搅拌30分钟。在室温下向所述反应混合物添加在四氢呋喃(1mL)中的2-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(3-氧代环己-1-烯基)吡啶-3-基]-N-甲基异丁酰胺(0.10g),并将所述混合物在同样的温度下搅拌过夜和在50℃下搅拌24小时。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=85/15-40/60),获得标题化合物(0.05g)。参考例35参考例35的化合物以与参考例34同样的方法,使用相应的起始材料制备。参考例362-(3,5-双三氟甲基苯基)-N-[6-(1,4-二氧杂螺[4.5]癸-8-基)-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺在氢气气氛下,2-(3,5-双三氟甲基苯基)-N-[6-(1,4-二氧杂螺[4.5]癸-7-烯-8-基)-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺(0.52g)和10%钯碳(0.10g,湿)在甲醇(10mL)中的悬液在室温下搅拌过夜。所述反应混合物通过Celite(注册商标)垫过滤,并且滤液在减压下浓缩,获得标题化合物(0.50g)。参考例372-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(4-氧代环己基)吡啶-3-基]-N-甲基异丁酰胺在室温下向2-(3,5-双三氟甲基苯基)-N-[6-(1,4-二氧杂螺[4.5]癸-8-基)-4-(4-氟-2-甲基苯基)吡啶-3-基]-N-甲基异丁酰胺(0.50g)在丙酮中的溶液添加1.0mol/L盐酸(3.0mL),并将所述混合物在50℃下搅拌2小时。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-10/90),获得标题化合物(0.41g)。参考例38{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-1-羟基环己基}乙酸乙酯在氩气气氛下,在-78℃向乙酸乙酯(0.021g)在四氢呋喃(1mL)中的溶液逐滴添加二异丙基氨基锂溶液(1.09mol/L四氢呋喃/正己烷溶液,0.20mL),并将所述混合物在同样的温度下搅拌20分钟。在-78℃向所述反应混合物添加2-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(4-氧代环己基)吡啶-3-基]-N-甲基异丁酰胺(0.10g)在四氢呋喃(1mL)中的溶液,并将所述混合物在室温下搅拌2小时。向所述反应混合物添加饱和氯化铵水溶液,并用乙酸乙酯提取所生成的混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-10/90),获得标题化合物(0.10g)。参考例392-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]亚环己基}丙酸乙酯在冰冷却下向氢化钠(60%,0.017g)在四氢呋喃(2mL)中的悬液添加2-膦酰丙酸三乙酯(0.11g),并将所述混合物在同样的温度下搅拌30分钟。在室温下向所述反应混合物添加2-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(4-氧代环己基)吡啶-3-基]-N-甲基异丁酰胺(0.14g)在四氢呋喃(1mL)中的溶液,并将所述混合物在50℃搅拌过夜。所述反应混合物冷却到室温,并添加饱和氯化铵水溶液。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-50/50),获得标题化合物(0.13g)。参考例40{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸乙酯在氢气气氛下,{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]亚环-2-己烯基}乙酸乙酯(0.03g)和10%钯碳(0.01g,湿)在甲醇(1mL)中的悬液在室温下搅拌过夜。所述反应混合物通过Celite(注册商标)垫过滤,并将滤液在减压下浓缩。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=85/15-40/60),获得标题化合物(0.02g)。参考例41参考例41的化合物以与参考例40同样的方法,使用相应的起始材料制备。参考例422-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}丙酸乙酯在氢气气氛下,2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]亚环己基}丙酸乙酯(0.13g)和10%钯碳(0.025g,湿)在甲醇(5mL)中的悬液在室温下搅拌过夜。所述反应混合物通过Celite(注册商标)垫过滤,并将滤液在减压下浓缩,获得标题化合物(0.11g)。参考例434-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己烷羧酸乙酯在氢气气氛下,4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯羧酸乙酯(0.03g)和10%钯碳(0.010g,湿)在乙醇(1mL)中的悬液在室温下搅拌14小时。所述反应混合物通过Celite(注册商标)垫过滤,并将滤液在减压下浓缩。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=70/30-50/50),获得标题化合物(0.02g)。参考例44至47参考例44至47的化合物以与参考例43同样的方法,使用相应的起始材料制备。参考例48{4-[5-{[3-苄氧基-2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}乙酸在室温下向{4-[5-{[3-苄氧基-2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}乙酸乙酯(0.11g)、四氢呋喃(1mL)、甲醇(0.5mL)和水(0.5mL)的混合物添加氢氧化锂一水合物(0.03g),并将所述混合物在同样的温度下搅拌过夜。向所述反应混合物添加2.0mol/L盐酸(0.4ml),并在减压下除去溶剂。向残渣添加水,并用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:乙酸乙酯/甲醇=100/0-90/10),获得标题化合物(0.05g)。参考例49和50反式-2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-2-甲基丙酸乙酯(参考例49),和顺式-2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-2-甲基丙酸乙酯(参考例50)在氢气气氛下,2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}-2-甲基丙酸乙酯(0.11g)、10%钯碳(0.03g,湿)、甲醇(2mL)和四氢呋喃(1mL)的混合物在室温下搅拌过夜。所述反应混合物通过Celite(注册商标)垫过滤,并将滤液在减压下浓缩。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-60/40),获得参考例49(0.05g)和参考例50(0.04g)。在上述层析中,参考例49的化合物在高极性侧,而参考例50的化合物在低极性侧。参考例51{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸甲酯在氢气气氛下,{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}乙酸甲酯(2.14g)和10%钯碳(E101NE/W型(EVONIK))(0.21g)在甲醇(96mL)中的悬液在室温下搅拌过夜。所述反应混合物通过Celite(注册商标)垫过滤,并将滤液在减压下浓缩。得到的粗产物通过在氨丙基甲硅烷基化硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=100/0-40/60),获得标题化合物(2.13g)。参考例52{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-1-甲基环己基}乙酸甲酯在冰冷却下向氢化钠(60%,0.020g)在四氢呋喃(2mL)中的悬液添加二甲基膦酰乙酸甲酯(0.09g),并将所述混合物在室温下搅拌1小时。在室温下向所述反应混合物添加2-(3,5-双三氟甲基苯基)-N-[4-(4-氟-2-甲基苯基)-6-(4-氧代环己基)吡啶-3-基]-N-甲基异丁酰胺(0.15g)在四氢呋喃(1mL)中的溶液,并将所述混合物在50℃搅拌30分钟。所述反应混合物冷却到室温并添加饱和氯化铵水溶液。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-30/70),获得{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]亚环己基}乙酸甲酯(0.15g)。在冰冷却下向碘化铜(I)(0.05g)在二乙醚(0.40mL)中的悬液添加甲基锂(1.13mol/L二乙醚溶液,0.50mL),并将所述混合物在同样的温度下搅拌10分钟,并在减压下浓缩溶剂。在氩气气氛下,在冰冷却下向残渣添加二氯甲烷(0.40mL),将所述混合物搅拌5分钟,并在减压下浓缩溶剂。向残渣添加二氯甲烷(0.40mL),并冷却到-78℃。向所述混合物添加三甲基氯化甲硅烷(0.03g),并向所生成的混合物逐滴添加{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]亚环己基}乙酸甲酯(0.09g)在二氯甲烷(1.0mL)中的溶液。所生成的混合物在冰冷却下搅拌1小时和在室温下搅拌过夜。向所述反应混合物添加饱和氯化铵水溶液,并用乙酸乙酯提取所生成的混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-40/60),获得标题化合物(0.08g)。参考例53参考例53的化合物以与参考例16的同样的方法,使用相应的起始材料制备。参考例54参考例54的化合物以与参考例21的同样的方法,使用相应的起始材料制备。参考例55参考例55的化合物以与参考例22的同样的方法,使用相应的起始材料制备。参考例56和57参考例56和57的化合物以与参考例23同样的方式,使用相应的起始材料制备。参考例58和59参考例58和59的化合物以与参考例43同样的方式,使用相应的起始材料制备。参考例60{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-1-氧基吡啶-2-基]环己基}乙酸乙酯在冰冷却下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸乙酯(0.37g)在二氯甲烷中的溶液添加间氯过苯甲酸(纯度70%,0.55g),并将所述混合物在室温下搅拌过夜。所述反应混合物用冰冷却,并向所述混合物添加1.0mol/L氢氧化钠水溶液。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=40/60-0/100),获得标题化合物(0.35g)。参考例61{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氯-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸乙酯{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-1-氧基吡啶-2-基]环己基}乙酸乙酯(0.20g)和磷酰氯(0.60mL)的混合物在120℃下搅拌2小时。所述反应混合物用冰冷却,并通过添加水和氨水碱化。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-50/50),获得标题化合物(0.16g)。参考例62{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-甲基吡啶-2-基]环己基}乙酸乙酯{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氯-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸乙酯(0.04g)、2,4,6-三甲基环三硼氧烷(0.014g)、四(三苯基膦)钯(0)(0.007g)、碳酸钠(0.016g)和1,2-二甲氧基乙烷(2.9mL)的混合物在微波照射下在120℃搅拌1小时。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=80/20-20/80),获得标题化合物(0.019g)。参考例63{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-羟基甲基吡啶-2-基]环己基}乙酸乙酯在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-1-氧基吡啶-2-基]环己基}乙酸乙酯(0.15g)在二氯甲烷(2.0mL)中的溶液添加三甲基氧鎓四氟硼酸(0.04g),并将所述混合物在同样的温度下搅拌2小时。所述反应混合物在减压下浓缩,并向残渣添加甲醇(2.0mL)。在65℃向所述混合物添加过硫酸铵(0.01g)在水(0.02mL)中的溶液,并将所述混合物在同样的温度下搅拌1小时。在65℃向所述反应混合物添加过硫酸铵(0.01g)在水(0.02mL)中的溶液,并将所述混合物在同样的温度下搅拌13小时。所述反应混合物冷却到室温后,在减压下浓缩溶剂。向残渣添加碳酸钠的水溶液,并用乙酸乙酯提取所述混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物和{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-羟基甲基吡啶-2-基]环己基}乙酸甲酯的混合物(0.06g)。实施例14-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己烷羧酸在室温下向4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己烷羧酸乙酯(0.022g)、四氢呋喃(0.375mL)、甲醇(0.375mL)和水(0.150mL)的混合物添加氢氧化锂一水合物(0.014g),并将所述混合物在同样的温度下搅拌72小时。向所述反应混合物添加2.0mol/L盐酸(0.170mL)和水,并用乙酸乙酯提取所生成的混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸钠干燥。在减压下浓缩溶剂,获得标题化合物(0.018g)。实施例23-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}丙酸在室温下向3-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}丙酸乙酯(0.019g)、四氢呋喃(0.375mL)、甲醇(0.375mL)和水(0.150mL)的混合物添加氢氧化锂一水合物(0.012g),并将所述混合物在同样的温度下搅拌6小时。向所述反应混合物添加2.0mol/L盐酸(0.140ml)和水,并用乙酸乙酯提取所生成的混合物。有机层用水和饱和盐水洗涤,并经过无水硫酸钠干燥。在减压下浓缩溶剂,获得标题化合物(0.017g)。实施例3{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸在室温下向{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸乙酯(0.023g)、四氢呋喃(0.50mL)、甲醇(0.25mL)和水(0.25mL)的混合物添加氢氧化锂一水合物(0.007g),并将所述混合物在同样的温度下搅拌过夜。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯/甲醇=50/50/0-0/100/0-0/90/10),获得标题化合物(0.003g)。实施例4{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-3-甲基环己基}乙酸在室温下向{3-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-3-甲基环己基}乙酸乙酯(0.022g)、四氢呋喃(0.40mL)、甲醇(0.20mL)和水(0.20mL)的混合物添加氢氧化锂一水合物(0.006g),并将所述混合物在同样的温度下搅拌过夜。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯/甲醇=50/50/0-0/100/0-0/90/10),获得标题化合物(0.007g)。实施例5{4-[5-{[2-(3,5-双三氟甲基苯基)-3-羟基-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸在氢气气氛下,{4-[5-{[3-苄氧基-2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己-3-烯基}乙酸(0.045g)和10%钯碳(0.03g,湿)在甲醇(1.5mL)中的悬液在室温下搅拌5小时。向所述反应混合物添加10%钯碳(0.03g,湿)。在氢气气氛下,所生成的混合物在室温下搅拌过夜。所述反应混合物通过Celite(注册商标)垫过滤,并将滤液在减压下浓缩,获得标题化合物(0.04g)。实施例6{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-1-羟基环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-1-羟基环己基}乙酸乙酯(0.05g)、四氢呋喃(1.00mL)、甲醇(0.50mL)和水(0.50mL)的混合物添加氢氧化锂一水合物(0.015g),并将所述混合物在同样的温度下搅拌过夜。向所述反应混合物添加2.0mol/L盐酸(0.20ml),并在减压下除去溶剂。向残渣添加水,并用乙酸乙酯提取所生成的混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.046g)。实施例76-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]螺[2.5]辛烷-1-羧酸6-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]螺[2.5]辛烷-1-羧酸乙酯(0.020g)、1.0mol/L氢氧化钠水溶液(0.09mL)、四氢呋喃(0.60mL)和甲醇(0.30mL)的混合物在微波照射下在140℃搅拌1个半小时。所述反应混合物冷却到室温,并添加水。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯/甲醇=40/60/0-0/100/0-0/90/10),获得标题化合物(0.005g)。实施例8{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸甲酯(0.017g)、四氢呋喃(0.30mL)、甲醇(0.15mL)和水(0.15mL)的混合物添加氢氧化锂一水合物(0.005g),并将所述混合物在同样的温度下搅拌2天。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=50/50-0/100),获得标题化合物(0.008g)。实施例9{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-邻甲基苯基吡啶-2-基]-环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-邻甲基苯基吡啶-2-基]-环己基}乙酸甲酯(0.08g)、四氢呋喃(1.00mL)、甲醇(0.50mL)和水(0.50mL)的混合物添加氢氧化锂一水合物(0.021g),并将所述混合物在同样的温度下搅拌3小时。向所述反应混合物添加2.0mol/L盐酸(0.28ml),并在减压下除去溶剂。向残渣添加水,并用乙酸乙酯提取所生成的混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.071g)。实施例102-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}丙酸2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}丙酸乙酯(0.11g)、1.0mol/L氢氧化钠水溶液(0.50mL)、四氢呋喃(0.50mL)和甲醇(1.50mL)的混合物在微波照射下在140℃搅拌1个半小时。所述反应混合物冷却到室温,并添加1.0mol/L盐酸(0.60mL)。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.10g)。实施例11反式-2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-2-甲基丙酸反式-2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-2-甲基丙酸乙酯(0.054g)、1.0mol/L氢氧化钠水溶液(0.25mL)、四氢呋喃(0.25mL)和甲醇(0.75mL)的混合物在微波照射下在140℃搅拌1个半小时。所述反应混合物冷却到室温,并添加1.0mol/L氢氧化钠水溶液(0.25mL)。所生成的混合物在微波照射下在140℃搅拌1个半小时。所述反应混合物冷却到室温,并添加1.0mol/L盐酸(0.60mL)。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.022g)。实施例12顺式-2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-2-甲基丙酸顺式-2-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-2-甲基丙酸乙酯(0.044g)、1.0mol/L氢氧化钠水溶液(0.20mL)、四氢呋喃(0.20mL)和甲醇(0.60mL)的混合物在微波照射下在140℃搅拌1个半小时。所述反应混合物冷却到室温,并添加1.0mol/L氢氧化钠水溶液(0.20mL)。所生成的混合物在微波照射下在140℃搅拌1个半小时。所述反应混合物冷却到室温,并添加1.0mol/L盐酸(0.50mL)。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯=90/10-10/90),获得标题化合物(0.014g)。实施例13和14反式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸(实施例13),和顺式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸(实施例14)(1)反式和顺式异构体混合物的合成在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}-乙酸甲酯(2.13g)在四氢呋喃(32mL)-甲醇(16mL)-水(16mL)中的混合溶液添加氢氧化锂一水合物(0.41g),并将所述混合物在同样的温度下搅拌17小时。向所述反应混合物添加2.0mol/L盐酸(4.9ml),并在减压下除去溶剂。向残渣添加水,并用乙酸乙酯提取所生成的混合物。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得粗产物(反式-和顺式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸的混合物)(2.08g)。(2)反式和顺式异构体的分离反式-和顺式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸的混合物(36.6g)通过液相色谱在下列条件下分离,分别获得实施例13(17.0g)和实施例14(16.2g)。[液相色谱的分离条件](A)分离装置装置名:K-Prep(KYOTOCHROMATOCo.,Ltd.);(B)分离条件柱:CHIRALPAK(注册商标)IA;型号:5cmI.D.x25cmL.;粒径:5μm;流动相:正己烷/乙醇/乙酸=85/15/0.1<v/v/v>流速:35mL/min;温度:30℃;检测波长:254nm;注入方法:装液法注入;注入量:10-20mL(20g/L溶液)(C)保持时间实施例13:约21分钟,实施例14:约17分钟实施例13和14在下列分析条件下分析。[液相色谱的分析条件](A)分析系统泵:LC-20AD(岛津制作所);检测器:SPD-20A(岛津制作所);自动进样器:SIL-20A(岛津制作所)(B)分析条件柱:CHIRALPAK(注册商标)IA;型号:0.46cmI.D.x25cmL.;流动相:正己烷/乙醇/乙酸=85/15/0.1<v/v/v>流速:1.0mL/min;温度:40℃;检测波长:254nm;注入量:10μL(C)保持时间实施例13:6.164分钟,实施例14:5.016分钟实施例15{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-1-甲基环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)吡啶-2-基]-1-甲基环己基}乙酸甲酯(0.078g)、四氢呋喃(0.60mL)、甲醇(0.30mL)和水(0.30mL)的混合物添加氢氧化锂一水合物(0.022g),并将所述混合物在同样的温度下搅拌1小时和在50℃下搅拌3小时。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用饱和盐水洗涤,并经过无水硫酸镁干燥,并在减压下除去溶剂。得到的粗产物通过在硅胶上柱层析提纯(洗脱液:正己烷/乙酸乙酯/甲醇=20/80/0-0/100/0-0/90/10),获得标题化合物(0.069g)。实施例16[4-(5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-邻甲基苯基吡啶-2-基)环己基]乙酸在室温下向[4-(5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-邻甲基苯基吡啶-2-基)环己基]乙酸乙酯(0.018g)、四氢呋喃(0.40mL)、甲醇(0.20mL)和水(0.20mL)的混合物添加氢氧化锂一水合物(0.005g),并将所述混合物在同样的温度下搅拌过夜。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.016g)。实施例17{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氯-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氯-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸乙酯(0.020g)、四氢呋喃(0.50mL)、甲醇(0.25mL)和水(0.25mL)的混合物添加氢氧化锂一水合物(0.005g),并将所述混合物在同样的温度下搅拌2小时。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.018g)。实施例18{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-甲基吡啶-2-基]环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-甲基吡啶-2-基]环己基}乙酸乙酯(0.019g)、四氢呋喃(0.50mL)、甲醇(0.25mL)和水(0.25mL)的混合物添加氢氧化锂一水合物(0.005g),并将所述混合物在同样的温度下搅拌过夜。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.018g)。实施例19{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-羟基甲基吡啶-2-基]环己基}乙酸在室温下向{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-羟基甲基吡啶-2-基]环己基}乙酸乙酯和{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-4-(4-氟-2-甲基苯基)-6-羟基甲基吡啶-2-基]环己基}乙酸甲酯(0.025g)、四氢呋喃(0.50mL)、甲醇(0.25mL)和水(0.25mL)的混合物添加氢氧化锂一水合物(0.007g),并将所述混合物在同样的温度下搅拌过夜。所述反应混合物通过添加乙酸中和。所生成的混合物用乙酸乙酯提取。有机层用水和饱和盐水洗涤,并经过无水硫酸镁干燥。在减压下浓缩溶剂,获得标题化合物(0.023g)。实施例20和21反式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸(实施例20)和顺式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸(实施例21)反式-和顺式-{4-[5-{[2-(3,5-双三氟甲基苯基)-2-甲基丙酰]甲氨基}-6-氰基-4-(4-氟-2-甲基苯基)吡啶-2-基]环己基}乙酸(实施例8)(0.18g)的混合物通过液相色谱在以下条件下分离,分别获得实施例20(0.035g)和实施例21(0.037g)。[液相色谱的分离条件](A)分离装置装置名:制备型HPLC系统(Gilson,Inc.)(B)分离条件柱:InertSustain(注册商标)C18;型号:20mmI.D.x50mmL.;粒度:5μm流动相:乙腈/10mM乙酸铵水溶液=45/55<v/v>;流速:30mL/min;温度:室温;检测波长:220nm(C)保持时间实施例20:约10.4分钟,实施例21:约9分钟实施例20和21在下列分析条件下分析。[液相色谱的分析条件](A)分析系统泵:LC-10AT(岛津制作所);检测器:SPD-10A(岛津制作所);自动进样器:SIL-10A(岛津制作所)(B)分析条件柱:Inertsil(注册商标)ODS-3;型号:4.6mmI.D.x250mmL.;流动相:乙腈/10mM乙酸铵水溶液=40/60-80/20<v/v>流速:1.0mL/min;温度:40℃;检测波长:225nm;注入量:5μL(C)保持时间实施例20:16.786分钟,实施例21:17.286分钟表1至11显示了上述参考例1至63的化合物的化学结构,和上述实施例1至21的化合物的化学结构和物理性质。这些表中的缩写:“RefNo.”、“ExNo.”、“Str.”、“Physicaldata”、“1H-NMR”、“DMSO-d6”和“CDCl3”分别表示参考例编号、实施例编号、化学结构、物理性质、氢核磁共振谱、二甲亚砜-d6和氯仿-d1。并且,“MS”和“ESI_APCI”分别表示质谱和电喷雾电离-大气压化学电离测定。[表1][表2][表3][表4][表5][表6][表7][表8][表9][表10][表11]试验例1对人NK1受体的亲合性(1)人NK1受体表达载体的制备PCR利用人类成人正常组织来源的脑cDNA(BioChain)作为模板,以SEQIDNO:1为正向引物和SEQIDNO:2为反向引物,利用PCR酶PrimeSTARMaxDNA聚合酶或PrimeSTARGXLDNA聚合酶(注册商标,TakaraBio)进行。扩增的产物利用ZeroBluntPCR克隆试剂盒(注册商标,LifeTechnologies)插入质粒(pCR-BluntII-TOPO(注册商标),LifeTechnologies)中。通过常规方法,大肠杆菌(Escherichiacoli)(OneShotTOP10感受态细胞,LifeTechnologies)被所述质粒转化为其中已插入所述扩增产物的质粒。所述大肠杆菌细胞在含有50μg/mL卡那霉素的LB琼脂培养基上培养一天。培养后,选择菌落并在含有50μg/mL卡那霉素的LB培养基中培养。培养后,利用QuantumPrep质粒微量制备试剂盒(Bio-Rad)提纯所述质粒。所述质粒利用限制性内切酶XhoI和HindIII(NewEnglandBiolabs)双重消化约两个小时。然后,进行利用1%琼脂糖凝胶的电泳,收集被切开的片段并利用TaKaRaRICOCHIP(TakaraBio)提纯。另外,也从已经被载体(pcDNA3.1(-)(注册商标),LifeTechnologies)转化的大肠杆菌提纯质粒,并将所述质粒利用限制性内切酶XhoI和HindIII(NewEnglandBiolabs)双重消化约两个小时。然后,进行利用1%琼脂糖凝胶的电泳,收集被切开的载体并利用TaKaRaRICOCHIP(TakaraBio)提纯。从用所述限制性内切酶处理的pCR-Blunt-II和pcDNA3.1(-)载体中切下的片段利用DNA连接试剂盒<MightyMix>(TakaraBio)连接。通过常规方法,由通过所述连接得到的质粒转化大肠杆菌(OneShotTOP10感受态细胞,LifeTechnologies)。所述大肠杆菌细胞在含有50μg/mL氨苄青霉素的LB琼脂培养基上培养一天。培养后,选择菌落并在含有50μg/mL氨苄青霉素的LB培养基中培养,然后利用QuantumPrep质粒微量制备试剂盒(Bio-Rad)提纯所述质粒。得到的质粒的蛋白质编码核苷酸序列(SEQIDNO:3)与已知的数据库(NCBI)上登记的人速激肽受体1(TACR1,NK1R)的核苷酸序列(NM_001058.3)完全一致。因此,证实了所述克隆的基因序列是人NK1受体的核苷酸序列并且所述将被翻译的氨基酸序列是人NK1受体。其中插入了SEQIDNO:3的核苷酸序列的pcDNA3.1(-)(注册商标)用作人NK1受体表达质粒。(2)人NK1受体表达细胞的制备(2-1)293T细胞的培养使用补充有抗生素青霉素-链霉素溶液(LifeTechnologies,最终青霉素浓度100U/mL和最终链霉素浓度100μg/mL)和胎牛血清(最终浓度10%)的液体D-MEM(Dulbecco改良的Eagle培养基)培养基(低葡萄糖,含有L-谷氨酰胺,和光纯药工业),在恒温箱中在5%CO2气体的条件下37℃培养293T细胞(RIKEN)。(2-2)293T细胞的传代培养几乎汇合的细胞用PBS(磷酸盐缓冲盐水,和光纯药工业)洗涤,利用0.05%胰蛋白酶-EDTA(LifeTechnologies)脱离并悬浮在所述液体培养基中。所述细胞悬液用上述液体培养基以分散比变成1:10这样的方式稀释,然后培养所述细胞。(2-3)人NK1受体表达细胞的制备汇合的细胞用PBS洗涤,利用0.05%胰蛋白酶-EDTA(LifeTechnologies)脱离并悬浮在补充有胎牛血清(最终浓度10%)的液体D-MEM培养基(低葡萄糖,含有L-谷氨酰胺,和光纯药工业)中。所述细胞悬液用所述液体培养基稀释,并且所述细胞以5x104细胞/孔的密度和100μL/孔的液体培养基量接种在聚-D-赖氨酸涂层的96-孔微孔板(BDBiocoat(注册商标),NipponBectonDickinson)的孔中。接种后,所述细胞在恒温箱中在5%CO2气体的条件下在37℃培养约四至五小时,从而制备准备用所述人NK1受体表达质粒转染的细胞。(2-4)人NK1受体表达质粒的293T细胞转染使用Lipofectamine2000(注册商标,LifeTechnologies)转染所述人NK1受体表达质粒。所述人NK1受体表达质粒用Opti-MEM(注册商标)I血清减少的培养基(LifeTechnologies)稀释到导致浓度为0.2μg/25μL/孔。同时,Lipofectamine2000(注册商标,LifeTechnologies)用Opti-MEM(注册商标)I血清减少的培养基(LifeTechnologies)稀释到导致浓度为0.4μg/25μL/孔并在室温下温育五分钟。五分钟后,为了形成人NK1受体表达质粒/Lipofectamine2000的复合体,将所述稀释的人NK1受体表达质粒和所述稀释的Lipofectamine2000混合并在室温下温育20至25分钟。温育后,50μL/孔的所述复合体溶液添加到准备用所述人NK1受体表达质粒转染的细胞,并将所述细胞在恒温箱中在5%CO2气体的条件下在37℃培养约48小时。培养48小时的所述细胞作为人NK1受体表达细胞用于分析。(3)与人NK1受体结合亲合性的测定(3-1)从人NK1受体表达细胞制备膜级分在175cm2培养瓶(NipponBectonDickinson)中制备人NK1受体表达细胞。人NK1受体表达质粒和Lipofectamine2000的复合体的形成是通过计算培养面积比率并将上文2-4中描述的方法的规模按所述比率增加来实施的。在用于膜级分制备的缓冲液(50mMTris(WakoPureChemical),120mM氯化钠(和光纯药工业),5mM氯化钾(和光纯药工业),1mM乙二胺四乙酸(Sigma),0.002mg/mL糜蛋白酶抑制素(PeptideInstitute),0.04mg/杆菌肽(和光纯药工业),0.005mg/mL磷氨米酮(PeptideInstitute)和0.5mM苯甲基磺酰氟(和光纯药工业),pH7.4)中收集人NK1受体表达细胞并以1,880g离心10分钟,并将细胞沉淀悬浮在用于膜级分制备的缓冲液中。在所述细胞冻融一次后,所述细胞利用Dounce型匀浆器(在冰上冷却,1000rpm,20次)匀化。匀化的细胞的悬液以20,000rpm离心10分钟,并除去上清液而得到细胞沉淀。所述细胞沉淀再次悬浮在所述用于膜级分制备的缓冲液中并利用Dounce型匀浆器(在冰上冷却,1000rpm,30次)匀化。所述细胞悬液以20,000rpm离心10分钟,并除去上清液而得到细胞沉淀。再次重复同样的匀化和离心,并得到最终的细胞沉淀。所述最终的细胞沉淀悬浮在用于受体结合试验的缓冲液(50mMTris(和光纯药工业),3mM氯化镁(和光纯药工业),0.002mg/mL糜蛋白酶抑制素(PeptideInstitute),0.04mg/杆菌肽(和光纯药工业)和0.02%牛血清白蛋白(Sigma),pH7.4)中,并利用BCA蛋白分析试剂盒(Pierce)测定蛋白质浓度。(3-2)受体结合试验所述用于受体结合试验的缓冲液以22.5μL/孔分配到96-孔分析板(Greiner)的孔中。试验化合物的DMSO溶液,以高80倍的浓度利用100%二甲基亚砜(DMSO)制备,以2.5μL/孔添加到所述孔中(最终浓度1nM至100nM),并将所述溶液混合。使用125I-P物质(P物质,[125I]Tyr8-,PerkinElmer)作为放射标记配体。125I-P物质用所述用于受体结合试验的缓冲液稀释到导致浓度为125pmol/25μL/孔并添加到所述96-孔分析板,并将所述溶液混合。从所述人NK1受体表达细胞制备的膜级分用所述用于受体结合试验的缓冲液稀释到产生8至10μg/孔的浓度,悬浮直到所述悬液变成该悬液可以顺利流过27G注射针这样的均匀状态,然后以150μL/孔添加到所述96-孔分析板。然后,在振荡所述板的同时,将所述板在室温下温育60分钟。所述反应溶液通过已经用0.3%聚乙烯亚胺预处理的多筛96-孔滤板(Millipore)吸滤,并通过用洗涤液(50mMTris和0.02%牛血清清蛋白,pH7.4)洗涤四次来终止反应。所述微孔板的底部在60℃干燥,然后向所述孔分配100μL/孔的MicroScint20(PerkinElmer)。所述板的上部用TopSealA(PerkinElmer)密封,并将所述板振荡5至10分钟。然后,用TopCountNXT(注册商标)(PerkinElmer)测定放射性。各孔的放射性通过减去添加10μM阿瑞吡坦的孔的放射性(非特异性结合)来计算。计算125I-P物质的结合率(%)=(添加试验化合物的组的放射性)/(添加介质的组的放射性)x100。利用分析软件,GraphPadPrism(GraphPadSoftware),将所述结合率(%)针对试验化合物的浓度绘图并线性近似,计算50%抑制浓度,IC50。这些结果显示在表12和13中。在表中,Ex.No.是指实施例编号,IC50(nM)是50%抑制浓度。(4)结果[表12]Ex.No.IC50(nM)11.4521.7832.1042.3550.9861.0071.6384.0192.35102.79116.75124.46131.62142.21[表13]Ex.No.IC50(nM)162.78175.51186.45194.96203.27212.36如表12和13所示,证明了本发明的化合物表现出对人NK1受体的高结合亲合性。试验例2对人NK1受体的抑制作用(1)人NK1受体表达细胞的制备人NK1受体表达细胞按与试验例1的2-3同样的方法制备。(2)对细胞内钙浓度上升抑制作用的研究人NK1受体表达细胞用300μL/孔的洗涤液(20mMHEPES/Hank’s平衡盐溶液(HBSS)pH7.3)洗涤。荧光钙指示剂(Fluo-4直接钙分析试剂盒,LifeTechnologies,含有0.42mM丙磺舒和0.1%牛血清清蛋白,按照该产品的方案调制)以150μL/孔添加到孔中,并将所述板在恒温箱中37℃温育30分钟。然后,试验化合物的DMSO溶液,以高80倍的浓度利用100%二甲基亚砜(DMSO)制备,以2.5μL/孔添加到所述孔中(最终浓度0.1、1和10μM),并将所述溶液混合。然后,所述板在恒温箱中37℃进一步温育30分钟。30分钟后,立即测定细胞内钙浓度。细胞内钙浓度各自使用FDSS(注册商标)7000(HamamatsuPhotonics)作为荧光信号测定。在开始读数后10秒,P物质(PeptideInstitute,Inc.)溶液,利用分析缓冲液(20mMHEPES/Hank’s平衡盐溶液(HBSS)pH7.3,含有0.1%牛血清白蛋白)以0.4μM或4μM制备,以50μL/孔(最终浓度0.1或1μM)自动添加到每个孔,并测定荧光信号直到120秒。添加了试验化合物的细胞的细胞内钙浓度(%)通过下面的方程式计算,其中添加介质(DMSO)的组的荧光信号被视为100%,而添加P物质前的荧光信号被视为0%。细胞内钙浓度(%)=(试验化合物添加组的荧光信号)/(介质添加组的荧光信号)x100所计算的细胞内钙浓度(%)被视为P物质残存的激动剂活性(残存P物质响应:SPRR)。这些结果在表14和15中显示。Ex.No.是指实施例编号。SPRR(%)是当P物质浓度是1μM和化合物浓度是0.1μM时得到的值。(3)结果[表14]Ex.No.SPRR(%)12027.533645752662179.4831942108.6115.41253135.11422153.7[表15]Ex.No.SPRR(%)16371926202.82136如表14和15所示,证明了本发明的化合物表现出强效的人NK1受体拮抗剂活性。试验例3对CYP3A4的抑制作用制备评价浓度的1000倍浓度的试验化合物二甲亚砜(DMSO)溶液,并稀释所述溶液制备反应溶液。通过在含有1nM至20μM试验化合物、3.2mM氯化镁、0.2pmol人CYP3A4(BDBiosciences)、0.5mM还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)和3μM荧光素-IPA(Promega)的磷酸钾缓冲液(pH7.4)中37℃温育10分钟,进行酶反应。反应液的量是50μL/孔。30-分钟预温育组在添加底物荧光素-IPA溶液(12.5μL/孔)之前37℃温育30分钟。在所述酶反应结束时,向所述孔添加50μL/孔的荧光素检测试剂(Promega),并将所述板在室温下留置20分钟。然后,用InfiniteM1000(TECAN)测定发光强度。计算相对于没有添加所述试验化合物的组的值的酶活性(%)。利用分析软件GraphPadPrism(GraphPadSoftware)作出剂量-反应曲线,并计算表现出50%抑制时的各化合物浓度,IC50。作为比较例,以同样方式试验NK1受体拮抗剂,阿瑞吡坦。使用所述试验化合物的30-分钟预温育组的IC50值通过上述测定方法测定,结果在表16和17中显示。表中,Ex.No.是指实施例编号,IC50(μM)是50%抑制浓度。[表16]Ex.No.IC50(μM)11326.839.447.151366.171186.196.91011112.7122.2138.2147.5阿瑞吡坦0.02[表17]Ex.No.IC50(μM)163.1173.3184.9205.5214.2如表16和17所示,证明了本发明的化合物的CYP3A4-抑制活性与阿瑞吡坦相比降低。因此,预期本发明的化合物具有比阿瑞吡坦少的基于CYP3A4抑制作用的药物间相互作用。试验例4对叩足的作用(1)对叩足的作用试验化合物溶液通过所述试验化合物溶解在介质(50%N,N-二甲基乙酰胺(和光纯药工业)、30%丙二醇(和光纯药工业)、4%2-羟丙基-β-环糊精(和光纯药工业)和16%蒸馏水的混合物)中制备。雄性沙鼠(JapanSLC)用异氟烷麻醉,并从颈静脉施用0.1mg/kg的试验化合物。四小时后,在用异氟烷麻醉下,在头部前囟侧方1mm部位和4.5mm深度将NK1受体激动剂GR73632(5pmol/5μl生理盐水)施用到脑室中。施用后,将沙鼠移动到观察笼,并测定在翻正反射恢复后30分钟期间的叩足时间。各试验化合物的叩足抑制率(%)通过以下方程式计算。叩足抑制率(%)={1-(施用试验化合物时的叩足时间)/(施用溶剂时的叩足时间)}x100(2)药物浓度测定叩足结束后,立即在用乙醚麻醉下进行开腹术,并从腹部大静脉取血样。同时,摘取脑。通过利用液相色谱-质谱法(LC/MS)定量分析,测定试验化合物在血浆和脑中的浓度。(3)结果对叩足的作用通过上述试验方法测定,结果在表18和19中显示。表中,Ex.No.是指实施例编号。抑制(%)是叩足抑制率,Conc.(nM)是脑中药物浓度。[表18]Ex.No.抑制(%)Conc.(nM)1982421001931004847735790248100599754710974211957213100511499401510039[表19]Ex.No.抑制(%)Conc.(nM)169364179657181004719952420100922110092如表18和19所示,本发明的化合物穿透中枢神经系统并还在体内表现出优异的NK1受体拮抗剂活性。试验例5雪貂药代动力学试验(1)方法用于静脉施用的试验化合物溶液通过所述试验化合物溶解在介质(50%N,N-二甲基乙酰胺(和光纯药工业)、30%丙二醇(和光纯药工业)、4%2-羟丙基-β-环糊精(和光纯药工业)和16%蒸馏水的混合物)中制备。作为口服施用溶液,使用悬液(0.5%甲基纤维素)。在异氟烷麻醉下,从股静脉向雄性雪貂(MarshallBioResourcesJapan)静脉内施用0.1mg/kg的试验化合物。在口服施用的情况下,向清醒动物经口施用1mg/kg的试验化合物。施用试验化合物后,在施用后接连7天从头臂静脉取血样。通过利用液相色谱-质谱法(LC/MS)定量分析,测定血浆中试验化合物的浓度。(2)结果通过上述试验方法在雪貂中实施药代动力学试验,结果在表20和表21中显示。表中,Ex.No.是指实施例编号。t1/2、CLtot和Vss分别是在静脉内施用的情况下基于血浆浓度的半衰期、全身清除率和稳态分布容积。Cmax、AUC和BA分别是在口服施用的情况下,最高血浆试验化合物浓度、施用后7天内血浆试验化合物浓度-时间曲线下面积、和生物利用度。[表20]Ex.No.t1/2(min)CLtot(mL/min/kg)Vss(mL/kg)132,4890.13401[表21]Ex.No.Cmax(ng/mL)AUC(ng·min/mL)BA(%)131,2585,294,06666如表20和表21所示,本发明的化合物表现出优异的口服吸收性伴低清除率。试验例6对顺铂诱发的急性和延迟性呕吐反应的作用(1)方法试验化合物溶液通过所述试验化合物溶解在介质(50%N,N-二甲基乙酰胺(和光纯药工业)、30%丙二醇(和光纯药工业)、4%2-羟丙基-β-环糊精(和光纯药工业)和16%蒸馏水的混合物)中制备。对照组只施用所述介质。在异氟烷麻醉下,从颈静脉向雄性雪貂(MarshallBioResourcesJapan)静脉内施用0.01mg/kg或0.1mg/kg的试验化合物。在所述药物施用后一小时,腹腔内施用5mg/kg加热到40-50℃的顺铂生理盐水溶液。所述雪貂从施用顺铂后立即观察72小时,并计数干呕(没有胃内容物吐出的腹部周期性收缩)和呕吐次数。(2)结果结果在图1中显示。在对照组中,观察到在急性期(直至顺铂施用后24小时)和延迟期(顺铂施用后24小时到72小时)中干呕和呕吐次数增加。在静脉内施用实施例13的化合物的组中,观察到在急性期和延迟期中干呕和呕吐次数抑制。这证明了本发明的化合物具有优异的药效持续性以及对顺铂诱发的急性和延迟性呕吐反应的抑制作用。试验例7hERG电流的评价(1)方法制备评价浓度(10μM)的1000倍浓度的试验化合物二甲亚砜(DMSO)溶液,并稀释所述溶液制备最终应用浓度的溶液。hERG电流通过全细胞方法利用膜片钳系统测定,其中接种了表达hERG通道的人胚肾(HEK)293细胞的盖玻璃放在灌流浴上并使灌注液流动。测定由数据获取/分析软件pCLAMP9(AxonInstruments,Inc.)的脉冲方案(保持电位-80mV,+20mV的去极化脉冲1.9秒,-50mV的复极化脉冲2秒,以15秒间隔刺激)引起的hERG通道来源电流的变化。测定条件是流速约1.5ml/min和温度约33℃。分析施加试验化合物之前紧接的两个波形和施加10分钟后紧接的两个波形,并进行统计分析。施加试验化合物之前的值被视为100%,确定基于所述值的变化率。(2)结果通过上述方法评价试验化合物对hERG电流的作用(结果在表22中显示)。表中,Ex.No.是指实施例编号,变化率是平均值±标准误差。[表22]Ex.No.%(n=3)1381.1±5.61480.3±3.9本发明的化合物与介质对照(0.1%DMSO)相比,在hERG电流上没有引起任何统计显著性的变化。产业实用性本发明的化合物或其可药用盐具有优异的NK1受体拮抗活性,因此也可用作预防或治疗癌症化疗所致恶心和呕吐的药剂。序列表自由文本<序列表1>SEQIDNO:1是用于DNA扩增SEQIDNO:3的正向引物的序列。<序列表2>SEQIDNO:2用于DNA扩增SEQIDNO:3的反向引物的序列。序列表<110>橘生药品工业株式会社(KisseiPharmaceuticalCO.,Ltd.)<120>环己基吡啶衍生物(cyclohexylpyridinederivative)<130>SCT165884-80<150>JP2014-095776<151>2014/05/07<160>3<170>PatentIn版本3.5<210>1<211>28<212>DNA<213>人工序列<220><223>正向引物<400>1tacctcgagagatagtagggctttaccg28<210>2<211>28<212>DNA<213>人工序列<220><223>反向引物<400>2gccaagcttctaggagagcacattggag28<210>3<211>1224<212>DNA<213>智人<400>3atggataacgtcctcccggtggactcagacctctccccaaacatctccactaacacctcg60gaacccaatcagttcgtgcaaccagcctggcaaattgtcctttgggcagctgcctacacg120gtcattgtggtgacctctgtggtgggcaacgtggtagtgatgtggatcatcttagcccac180aaaagaatgaggacagtgacgaactattttctggtgaacctggccttcgcggaggcctcc240atggctgcattcaatacagtggtgaacttcacctatgctgtccacaacgaatggtactac300ggcctgttctactgcaagttccacaacttctttcccatcgccgctgtcttcgccagtatc360tactccatgacggctgtggcctttgataggtacatggccatcatacatcccctccagccc420cggctgtcagccacagccaccaaagtggtcatctgtgtcatctgggtcctggctctcctg480ctggccttcccccagggctactactcaaccacagagaccatgcccagcagagtcgtgtgc540atgatcgaatggccagagcatccgaacaagatttatgagaaagtgtaccacatctgtgtg600actgtgctgatctacttcctccccctgctggtgattggctatgcatacaccgtagtggga660atcacactatgggccagtgagatccccggggactcctctgaccgctaccacgagcaagtc720tctgccaagcgcaaggtggtcaaaatgatgattgtcgtggtgtgcaccttcgccatctgc780tggctgcccttccacatcttcttcctcctgccctacatcaacccagatctctacctgaag840aagtttatccagcaggtctacctggccatcatgtggctggccatgagctccaccatgtac900aaccccatcatctactgctgcctcaatgacaggttccgtctgggcttcaagcatgccttc960cggtgctgccccttcatcagcgccggcgactatgaggggctggaaatgaaatccacccgg1020tatctccagacccagggcagtgtgtacaaagtcagccgcctggagaccaccatctccaca1080gtggtgggggcccacgaggaggagccagaggacggccccaaggccacaccctcgtccctg1140gacctgacctccaactgctcttcacgaagtgactccaagaccatgacagagagcttcagc1200ttctcctccaatgtgctctcctag1224当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1