用于从甘油和酰氯生产氯醇的方法与流程
本发明涉及用于从甘油和酰氯直接开始生产氯醇(chlorohydrin)的新方法。酰氯同时发挥反应物和催化剂的作用。一旦以化学计算量(stoichiometricamout)引入酰氯时,其与甘油反应以形成足以用于生产α,γ-二氯丙醇(α,γ-dichlorohydrin)之量的酯和气态盐酸。
背景技术:
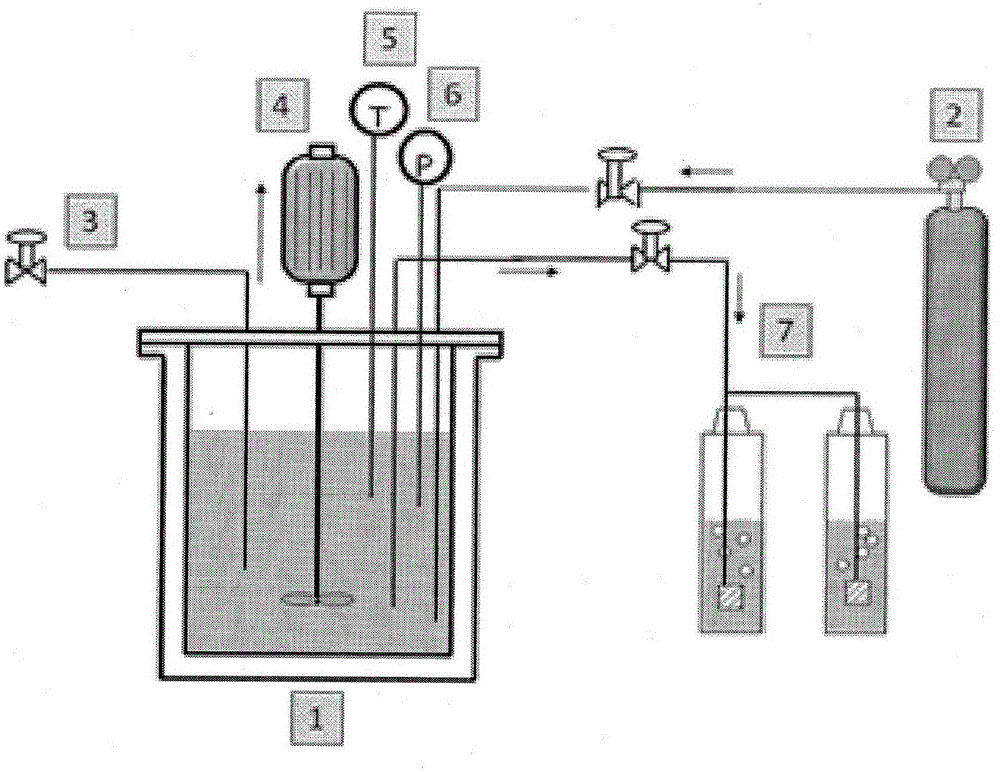
:甘油用于精细化学领域,并且其用途对于生产α,γ-二氯丙醇是特别重要的,所述α,γ-二氯丙醇构成生产表氯醇(epichlorohydrin)的方法中的中间产物,所述表氯醇进而用于合成环氧树脂[1-7]。在最近开发的方法中[8-11],为了从甘油开始获得α,γ-二氯丙醇,在作为催化剂的羧酸的存在下,根据以下反应使甘油与气态盐酸反应:正如可注意到的,这些方法产生作为副产物的水;如已经观察到的,这使反应速率降低。事实上,所述反应经历甘油的第一次氯化,这导致首先形成α-单氯甘油(α-monochlorohydrin)和水,含有少量的β-单氯甘油(β-monochlorohydrin);接着进行第二次氯化,从其中主要获得的有期望产物α,γ-二氯丙醇以及非常适中量的α,β-二氯丙醇。因此,更详细的反应方案如下:追溯到很久之前的使用甘油作为原料的传统方法考虑在作为催化剂的乙酸的存在下,在约80-100℃的温度下使甘油与盐酸在水溶液中反应[12-15]。一些专利描述了使用不互溶的惰性有机溶剂的方法,其中α,γ-二氯丙醇是可溶的,其中反应在反应混合物的沸点下进行,因此,所述沸点可根据所使用的特定溶剂而变化,但是在任何情况下都不超过110℃[16]。另一些专利描述了反应之后进行复杂的中和、萃取和蒸馏,用于回收α,γ-二氯丙醇[17,18]。这些方法都存在相当大的缺点,例如:-由于使用盐酸水溶液在反应混合物中引入水和未能除去在所述反应后形成的水二者所引起的反应减慢;以及-难以从反应混合物中分离α,γ-二氯丙醇。这些缺点以及作为原料的甘油的高成本在过去阻碍了该方法的发展。目前,最广泛采用的方法使用丙烯作为原料,其在混合物中得到70%的α,β-二氯丙醇和30%的α,γ-二氯丙醇。该方法同样存在缺点。事实上,大量的α,β-二氯丙醇带来了困难,原因是在后续的脱氯化氢生成表氯醇中,该物质比α,γ-二氯丙醇慢得多。这对设备(plant)的大小有影响并导致产率降低,形成副产物[5,6]。在假设甘油可在低成本获得的条件下,显然从甘油开始的反应可潜在地更有优势。在这一点上,应当提到产生约10wt%的作为副产物的甘油之生物柴油的增产通常引起可用性提高,因此甘油的成本逐渐降低。此外,因为甘油生产过程中主要的成本之一是其纯化,应指出,可直接对粗制(即,未纯化)的甘油进行氯化反应,进一步地降低成本。最近公布了多个考虑从甘油和气态(无水)盐酸开始生产二氯丙醇的专利。这些专利都使用羧酸作为催化剂,而且通过所使用的羧酸的类型和/或通过所采用的操作条件彼此区分开。在作为催化剂的羧酸的存在下,从甘油开始生产氯醇比从丙烯开始生产氯醇的优势之一是:对生产α,γ-二氯丙醇实现的高选择性,正如已经提到过的,在形成表氯醇的反应中,α,γ-二氯丙醇比α,β-二氯丙醇具有强得多的反应性。因此,可使得脱氯化氢反应器的体积显著减小,或者假定同样的体积,生产率提高。在由Solvay提出的专利[8]中,例如建议使用己二酸。由EurochemEngineering提出的专利[9]描述了使用以下羧酸:丙酸、丙二酸、乙酰丙酸、柠檬酸(cytricacid)和琥珀酸,此外,其声称通过提馏(stripping)来分离α,γ-二氯丙醇的可能性。在DOWGlobalTechnologies提出的专利[10]中,建议了可能的基于羧酸之催化剂的极长列表。但是,考虑到反应速率和产率受到HCl分压的显著影响,首要是强调了在压力下操作的优势[19,20]。在由CONSERS.p.A.等提出的专利[11]中,建议了这样的可能性,即依次使用两种不同的催化剂(首先是苹果酸,然后是琥珀酸),在低压(1-2巴)下开始反应,并第二阶段中升高压力(8-10巴)。对羧酸性能进行了深入地研究[19,21,22],并且发现了酸的pKa(其范围应为4-5)和催化活性之间一定的相关性。例如乙酸、单氯乙酸和三氯乙酸的系列显示出活性降低,原因是酸性力(acidicforce)随着含氯量而提高[22]。已经再次证明一些包含羧酸官能团的催化剂在生产单氯甘油中具有选择性,原因使只有在甘油已被完全转化成单氯甘油后才可发生后续的氯化[21-23]。已经阐述了能够描述实验观察到的动力学行为的反应机制[23]。然而,这些专利同样存在缺点,即形成相当大量的副产物水,这造成反应逐渐减慢。最近,一个公开专利中声称使用了一类新的催化剂,所述催化剂由酰氯构成,先前从未实验过[24]。在该专利中,重点在于如何使用酰氯,假定相同的摩尔浓度,与基于羧酸的传统催化剂相比,其具有更高的活性和选择性。通过研究这些催化剂的性能,出人意料地发现所述催化剂不必供应盐酸就可产生期望的反应产物α,γ-二氯丙醇,所述盐酸在甘油与酰氯反应形成酯的反应后在原位形成。然后盐酸与酯反应以得到反应产物和作为副产物的羧酸,而非传统方法中的水。鉴于羧酸是反应的良好催化剂,其进行快速且具有良好的选择性,而已知水对反应速度有反作用。然后使盐酸与酯反应以得到以下反应。即,总的来说然后使用1∶2比例的甘油/酰氯,普遍地以高反应速度获得α,γ-二氯丙醇,其表征在使用酰氯作为催化剂中已经观察到了催化作用[24]。然后形成高浓度的作为反应的良好催化剂的羧酸而不是水,水对酯的形成有负面作用并因此影响反应速率。技术实现要素:因此,形成本发明的对象是用于从甘油和酰氯生产氯醇的方法。以化学计算量使用的酰氯既用作反应物又用作催化剂。事实上,将通式RCOCl的酰氯与甘油设置为以化学计算量比1∶2接触,则立即发生以下反应:即,对于形成氯醇的反应所必需的盐酸与甘油酯在原位同时形成。然后使所形成的盐酸与酯反应以得到以下反应即,总的来说然后使用1∶2比例的甘油/酰氯,普遍地以高反应速率获得α,γ-二氯丙醇,这表征在使用酰氯作为催化剂中已经观察到了催化作用[24]。然而,在这种情况下,形成高浓度的羧酸(而不是水),因为在传统的方法中反应是由羧酸催化的。羧酸对所述反应具有良好的催化作用;而已知水对酯的形成有负面作用并因此影响反应速率。因此所述反应更快速发生,并且具有良好的选择性。可使用的酰氯具有以下通式其中R为具有2至10个碳原子的直链、支链或环状烷基链,其可被一个或更多个R’基团取代,所述R’基团彼此相同或者不同,选自:直链、支链或环状脂肪族C1-C10基团;芳香族C1-C10基团;以及一个或多于一个另外的酰氯基团。作为副产物形成的羧酸可经分离、纯化并返回到待进行氯化的酰氯的供应体(supplier)中并再次使用,具有明显的经济优势。事实上,以下事实代表了该方法的一个优势:期望的反应在生产氯醇的位置处无需提供气态盐酸即可发生反应。更容易连续操作代表了另一个优势。本发明的另一个目的是可同样使用作为生物柴油生产之副产品的粗制甘油(rawglycerol),其将初步处理的操作限于:使用无机酸(优选盐酸)中和作为催化剂的碱,以及除去残留的甲醇和任何可能的湿气。本发明的另一个目的是提供将能使甘油几乎完全转化的方法。本发明的另一个目的是提供将在热力学有利的条件下发生的方法,其具有相应提高的产率和反应速率。从本发明随后的描述中将更清楚地体现的上述和其他目的是通过用于生产氯醇并且特别是α,γ-二氯丙醇的方法实现,所述方法包括在甘油和酰氯之间化学计算量反应的步骤。本发明的另一些目的将在随后本发明的一些实施方案的描述中清楚地体现出。附图简述为了能够更好地理解,将参考附图对本发明进行描述,所述附图仅通过非限制性实例举例说明其一个优选的实施方案。在附图中:图1是通过举例的方式提供的在测试中使用的设备的示意图,其中:由1表示的是哈氏合金反应器(Hastelloyreactor),容积为300mL;由2表示的是HCl缸(cylinder);由3表示的是用于取出待分析的样品的阀门;由4表示的是磁搅拌器;由5表示的是温度读取器;由6表示的是压力读取器;并且由7表示的是用于中和未反应HCl的系统(含有氢氧化钠溶液的两个串联的Drechsel鼓泡器)。图2是示出了根据实施例1的形式在供应乙酰氯期间和之后反应器中压力变化的图;和图3是示出了根据实施例2的形式在供应乙酰氯期间和之后反应器中压力变化的图。发明详述根据本发明的方法包含引起甘油与酰氯(例如乙酰氯)以化学计算量比1∶2反应的基本操作。甘油与乙酰氯的反应在室温条件下已经非常快速,但在100-120℃下几乎是瞬间的。这意味着,如果两种试剂从一开始就完全接触,由于迅速产生了HCl,反应器中的压力显著提高。因此,将一种试剂逐步添加到另外一种中(优选地,将酰氯添加至甘油)是有利的。以这种方式,保持了4-6巴的适度压力值,并且可根据调节酰氯流量的方法之期望的持续时间进行调整。因此,根据本发明的方法可在半连续的反应器中进行,随着酰氯的消耗,向所述反应器中连续地供应酰氯。然而,所述方法也可连续进行,连续供应酰氯和甘油二者。反应温度可保持在60-250℃,优选100至175℃。可使用具有如上所述通式的酰氯。优选将在先专利中已经使用的酰氯作为催化剂,其可以是单官能团的且包含2至10个碳原子,或者是双官能团/多官能团的,具有2至10个碳原子。可用于本发明中的酰氯的非限制性实例包括:乙酰氯、丙酰氯、己二酰二氯(hexandioyldichloride)、丙二酰二氯(propandioyldichloride)、丁二酰氯(butandioyldichloride)、苯乙酰氯。在实验室水平和本发明中提供的实施例中,在先前引入了甘油的反应器中,在搅拌下用泵供应酰氯。在不同的时间从该反应器中采样以通过化学分析确定所采的不同样品中产物的分布以追踪在反应时间中的变化。在下文中提供的实例中,仅通过非限制性举例说明本发明,详细地描述了以不同的方式进行的用于生产氯醇(特别是α,γ-二氯丙醇)的测试。图1示出了用于进行实施例中提供的实验的装置的示意图。反应器1为300-cm3Parr高压釜,利用经温度调节的烘箱外部加热以达到预设的温度。将甘油以预先确定的量(例如100g)引入到反应器中。通常保持在约1000r.p.m.的磁搅拌器4使反应混合物保持均匀。然后将反应器加热直到达到反应温度。然后以预设的恒定流量(1-2cm3/分钟)引入酰氯,持续供应酰氯直至达到化学计算量值(酰氯摩尔数∶甘油摩尔数=2)。此时,中断供应酰氯,并让混合物再反应几个小时直到发现反应器中HCl的压力不再降低。在不同的反应时间,通过阀门3取出少量的反应混合物,并进行分析以确定其组成。用读取器5和6来读取温度和压力。使未反应的HCl通过Drechsel鼓泡器从高压釜中释放出来,所述Drechsel鼓泡器中填充有氢氧化钠饱和溶液并且包含当达到中性时发出信号的指示器。在以下实施例中描述了所得到的结果。实施例1中描述的是通过向图1中包含100g甘油的反应器以1cm3/分钟的流量供应乙酰氯持续154分钟而获得的系统的行为。乙酰氯的供应量符合乙酰氯/甘油的化学计算量比例2。反应温度保持在100℃。测试持续共4小时。实施例2中描述的是通过向图1中包含100g甘油的反应器以2cm3/分钟的流量供应乙酰氯持续80分钟而获得的系统的行为。乙酰氯的供应量符合乙酰氯/甘油的化学计算量比例2。反应温度保持在100℃。测试持续其4小时。在实施例3中,将在前面两个实例中结束时得到的反应混合物进行蒸馏,并按顺序分离出:残留的盐酸、形成的乙酸以及可能的痕量水。将从蒸馏得到的残留物进行化学分析,其表明主要形成期望产物α,γ-二氯丙醇。实施例实施例1-通过以1cm3/分钟的恒定流量供应乙酰氯的甘油与乙酰氯之间的反应测试在图1的反应器中进行,维持以下条件:起始甘油量100g;乙酰氯的流量1cm3/分钟(在室温下供应),供应的持续时间:154分钟;测试的持续时间:4h;操作温度100℃。在这些条件中,由于生成了盐酸而在反应器中设定的压力是形成酯和随后形成HCl的反应与消耗HCl形成氯醇的反应之间的动力学平衡的结果。当停止供应乙酰氯,反应再持续86分钟。反应器中保留的残余压力对应于少量未反应的HCl。该HCl通过捕集器(trap)被释放和减弱,对小样本分析后得到的液体保留下来用于进行产物的纯化(参见实施例3)。图2中示出了反应器中在供应乙酰氯期间和随后测试持续期间的压力图。正如可从图2的图中注意到,形成氯醇的反应极其快速,就约80-100分钟而言,压力升高几乎可以忽略不计,即,所有已经形成的盐酸都已反应。另一个观察结果是,在供应乙酰氯结束时,设定为刚好高于7巴值的HCl压力没有降低,这是反应结束的标志。表1示出了分别在180分钟和240分钟采样的分析:表1-在不同反应时间时采样的分析时间αβαγαβ甘油000001001801.88.281.48.602400.55.982.311.30这些分析证实,在达到化学计算量的酰氯量不久之后,可认为反应完成。实施例2-通过以2cm3/分钟的恒定流量供应乙酰氯的甘油与乙酰氯之间的反应测试再次在图1所示的反应器中进行,维持以下条件:起始甘油量100g;乙酰氯流量2cm3/分钟(在室温下供应),供应的持续时间:80分钟;测试的总持续时间:4h;操作温度:100℃。由于生成了盐酸而在反应器中设定的压力同样是形成酯和随后形成HCl的反应与消耗HCl形成氯醇的反应之间的动力学平衡的结果。在停止供应乙酰氯后,反应再进行160分钟。同样地,在这种情况下,反应器中保留的残余压力对应于少量未反应的HCl。所述HCl通过捕集器释放和减弱,并且在对小样本分析后得到的液体保留下来用于产物的纯化(参见实施例3)。图3示出了在供应乙酰氯期间和整个测试的持续期间反应器中压力的图。在这种情形下,乙酰氯的添加非常快,因此,系统中压力更迅速升高,尽管最终的平台(plateau)较低,这是消耗了更多盐酸的标志。另一个观察是,在供应乙酰氯结束时,设定为刚好高于5巴值的HCl压力没有进一步降低,这是反应结束的标志。表2中示出的是分别在180分钟和240分钟采样的分析。表2-在不同反应时间采样的分析时间αβαγαβ甘油0000010018004.091.94.1024003.792.34.00图3表明,在130分钟后,即在达到化学计算量的乙酰氯后,反应器中的HCl压力保持稳定(settle),因此认为反应终止。此外,该测试还表明可通过适当地调节操作条件(例如甘油的起始量、乙酰氯的添加速率、以及反应温度)减少反应时间,而反应器中的压力将根据预先选择的条件自动调节。此外,以所述的方式操作,即,更快地添加反应性酰氯,提高了对α,γ-二氯丙醇的选择性。实施例3-实施例1和2中反应产物的纯化假设反应进行直到完成,将实施例1和2中得到的产品进行蒸馏。低沸点产物为HCl、乙酸以及可能的痕量水,因此将这些从混合物中除去。将以这种方式得到的残余物进行验证分析,在下表3中提供了所考虑的两种情况的组成。表3-在通过蒸馏将低沸点组分纯化后反应产物的分析样品αβαγαβ甘油表105.882.112.10表203.992.23.90参考文献[1]SolvayInc.Processfortheproductionofalpha-epichlorhydrin.GB799,567(1958).[2]P.Bruin;Epoxyethers,theirpolymersandderivatives.GB814,695(1959).[3]Z.Brojer,P.A.Penczec,;J.H.Sas;Processofmanufacturingepoxideresins.GB1,001,364,(1965)[4]T.Kawabe,M.Kadoma,K.Murakami,H.Itoh;Processforcontinuousproductionofepichlorohydrin;US4113746(1978)[5]S.Carrà,E.Santacesaria,andM.Morbidelli,Ind.Eng.Chem.ProcessDes.Dev.,18(3),424-427,(1979).[6]S.Carrà,E.Santacesaria,andM.Morbidelli,Ind.Eng.Chem.ProcessDes.Dev.,18(3),428-433,(1979)[7]Clarke,H.T.;Hartman,W.W.Epichlorohydrin.InOrganicSyntheses,Coll.;(1941);Vol.1,p233.[8]P.Krafft,P.Gilbeau,B.Gosselin,S.Claessens,(Solvay).Processforproducingdichloropropanolfromglycerol,theglycerolcomingeventuallyfromtheconversionofanimalfatsinthemanufactureofbiodiesel.PCTPatent,WO054167A1,2005.[9]Siano,D.;Santacesaria,E.;Fiandra,V.;Tesser,R.;DiNuzzi,G.;DiSerio,M.;Nastasi,M..(EurochemEngineeringsrl);Processfortheproductionofα,γ-dichlorohydrinfromglycerolandhydrochloricacid.WO111810A2,2006,US2009/0062574,EP1879842B1[10]D.Schreck,W.Kruper,R.D.Varjian,M.E.Jones,R.M.Campbell,K.Kearns,B.D.Hook,,J.R.Briggs,J.G.Hippler,(DowGlobalTech.Inc.).Conversionofmultihydroxylated-aliphatichydrocarbonoresterthereoftoachlorohydrin.PCTPatent,WO2006/020234andEP1771403B1(2007)[11]S.Cassarino,F.Simola;(CONSERSpA)Conversionofglyceroltodichlorohydrinsandepichlorohydrin.WO2009/066327[12]J.B.Conant,O.R.Quayle;Glycerolαγ-dichlorohydrin.InOrganicSyntheses,Coll.;(1941);Vol.1,p292.[13]J.B.Conant,O.R.Quayle;Glycerolα-monochlorohydrin.InOrganicSyntheses,Coll.;(1941);Vol.1,p294.[14]Fauconnier,Sanson,Bull.Soc.Chim.,48,(1887),pp.236-240[15]A.J.Hill,E.J.Fischer,Asynthesisofbeta-chloro-allylchloride;J.Am.Chem.Soc.,(1922)Vol.44,pp.2582-2595[16]E.G.Britton,R.X.Heindel,Preparationofglyceroldichlorohydrin;US2,144,612(1939)[17]E.C.Britton,H.R.Slagh;Glyceroldichlorohydrin;US2,198,600(1940)[18]W.P.Thompson;Recoveryofaliphaticchlorohydrinshavingfrom3to5carbonatomsfromaqueoussolution.GB931,211(1963).[19]R.Tesser,M.DiSerio,R.Vitiello,V.Russo,E.Ranieri,E.Speranza,E.Santacesaria;GlycerolChlorinationinGas_LiquidSemibatchReactor:AnAlternativeRouteforChlorohydrinsProduction;Ind.Eng.Chem.Res.51(2012)8768-8776.[20]B.M.Bell,J.R.Briggs,R.M.Campbell,S.M.Chambers,P.D.Gaarenstroom,J.G.Hippler,B.D.Hook,K.Kearns,J.M.Kenney,W.J.Kruper,D.J.Schreck,C.N.Theriault,C.P.Wolfe,GlycerolasaRenewableFeedstockforEpichlorohydrinProduction.TheGTEProcess,Clean36(2008)657-661[21]E.Santacesaria,R.Tesser,M.DiSerio,L.Casale,D.Verde,NewProcessforProducingEpichlorohydrinviaGlycerolChlorination;Ind.Eng.Chem.Res.49(2010)964-970.[22]R.Tesser,E.Santacesaria,M.DiSerio,G.DiNuzzi,andV.Fiandra;KineticsofGlycerolChlorinationwithHydrochloricAcid:ANewRoutetoαγ-Dichlorohydrin;Ind.Eng.Chem.Res.46(2007)6456-6465.[23]E.Santacesaria,M.DiSerio,R.Tesser,(EurochemEngineeringsrl).Processformonochlorohydrinsproductionfromglycerolandhydrochloricacid.PCTPatentWO132770A1,2008.[24]E.Santacesaria,R.Tesser,R.Vitielio,M.DiSerio;(EurochemEngineeringsrl)Processoperlaproduzionedicloridrine;ItalianPatentRM2013A000488.当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1