循环核酸的鉴定及用途的制作方法
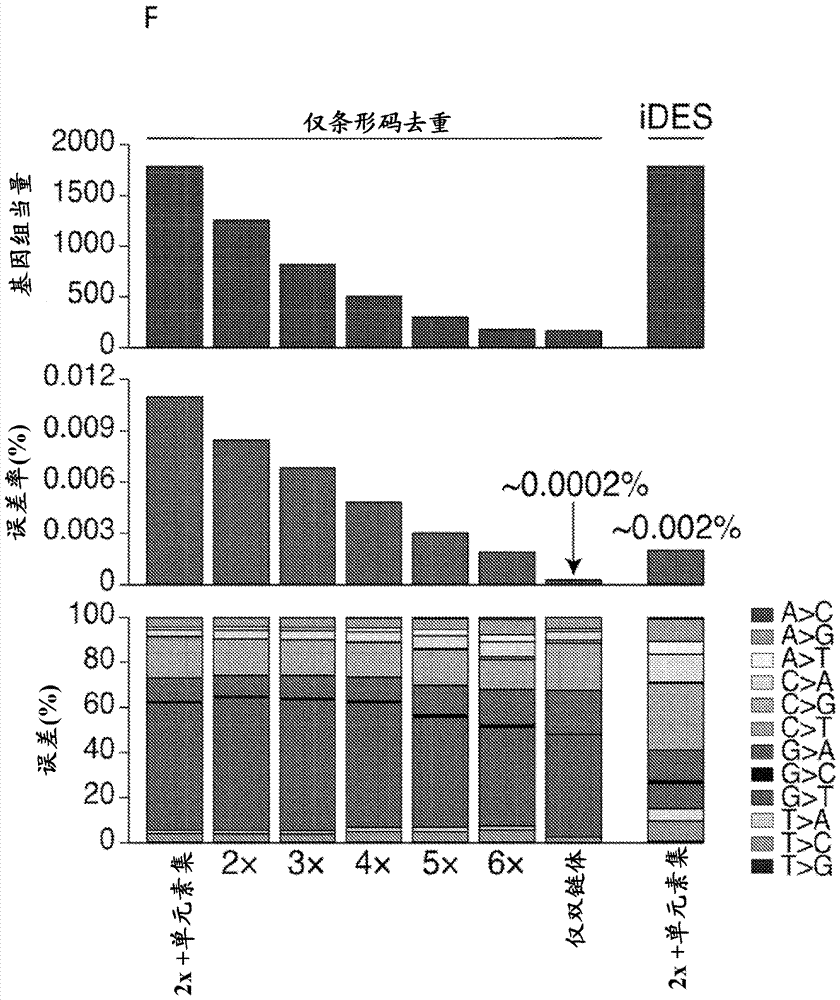
政府支持本发明是根据国防部授予的合同w81xwh-12-1-0285在政府支持下进行。政府在本发明中具有一定权利。通过引用结合本文中所有的出版物、专利及专利申请均通过引用结合,其引用程度如同具体且分别地指出各独立的出版物、专利或专利申请通过引用来结合。本文中的术语和结合的参考文献中的术语有冲突的情况下,以本文中的术语为准。发明背景肿瘤不断地将dna释放到循环中,在所述循环中dna为容易可及的(stroun等,(1987)eurjcancerclinoncol23:707-712)。分析这类癌症来源的无细胞dna(cfdna)具有变革癌症的检测和监测的潜力。非侵入性接近肿瘤来源的dna对于实体瘤而言特别令人关注。然而,循环肿瘤核酸的分析受到低核酸产率和分析技术的人为假象的阻碍。例如,测序误差限制了cfdna的突变谱的分析灵敏性。因此需要用于在癌症患者中检测和监测肿瘤来源核酸的更灵敏且高通量的方法。本发明满足本领域中的这些需求及其它需求。发明概述在一个实施方案中,本发明为用于分析样品中的核酸的独特衔接子库,各衔接子包含:近端的双链部分和远端的两个单链部分,其中所述双链部分包含对所述衔接子特异的至少两个碱基对的双链条形码,且其中所述单链部分包含:对所述样品特异的至少两个核苷酸的预定义单链条形码;和对所述衔接子特异的至少两个核苷酸的随机单链条形码。该衔接子库可具有这样的双链部分,其在所述至少两个碱基对的双链条形码和所述衔接子的近端之间进一步包含一个或多个g/c碱基对。该衔接子库亦可具有在库内的衔接子之间不同的g/c碱基对数量。所述双链条形码可包含2-20个碱基对。所述预定义单链条形码可包含4-20个核苷酸。所述随机单链条形码可包含4-20个核苷酸。在另一个实施方案中,本发明为分析核酸的方法,其包括:经由衔接子的双链部分,将根据权利要求1-6的衔接子库与多个双链核酸的两端连接;使衔接子-核酸的两条链扩增以产生第一扩增子和第二扩增子,其中所述第一扩增子来源于所述双链核酸的第一链且含有所述双链条形码的第一链,和所述第二扩增子来源于所述双链核酸的第二链且含有所述双链条形码的第二链;确定所述第一和第二扩增子的序列;和借助于鉴定所述双链条形码来确定所述第一和第二扩增子是否起源于所述多个双链核酸的单个双链核酸。所述多个双链核酸可包括无细胞dna。所述扩增可包含12-14个pcr循环。在另一个实施方案中,本发明为分析多个双链核酸的方法,所述方法包括:将根据权利要求1-6的衔接子库与多个双链核酸的两端连接;使衔接子-核酸的两条链扩增以产生第一扩增子和第二扩增子,其中所述第一扩增子来源于所述双链核酸的第一链且含有所述双链条形码的第一链,和所述第二扩增子来源于所述双链核酸的第二链且含有所述双链条形码的第二链;确定所述第一和第二扩增子的序列;和鉴定在所述第一和第二扩增子中的突变,其中来自所述第一和第二扩增子的突变为一致的突变;或者消除在第一扩增子中发生但在第二扩增子中未发生的突变;或者消除在来源于双链核酸第一链的至少约90%的第一扩增子上的g至t突变,其中所述g至t突变在来源于所述双链核酸第二链的小于约10%的第二扩增子上未发生;或者消除彼此之间相距少于100个碱基对的突变;或者消除在少于约50%的包含相同预定义单链条形码和随机单链条形码的扩增子上发生的突变;或其任何组合。在此实施方案中,c)的第一扩增子和第二扩增子包含相同的内源条形码和相同的双链条形码,且其中c)的第一扩增子和第二扩增子包含来源于所述衔接子的随机单链条形码的不同随机条形码。另外在此实施方案中,所述方法可包括消除与另一个突变相距少于5个碱基对的突变。另外在此实施方案中,所述方法可包括消除在少于约60%、约70%、约80%、约90%、约95%或约100%的包含相同双链主干(stem)条形码和相同内源条形码的扩增子上发生的突变。在另一个实施方案中,本发明为减小误差的分析核酸的方法,其包括:将来自独特衔接子库中的衔接子与核酸的各端连接,各衔接子包含近端的双链部分和远端的两个单链部分,其中所述双链部分包含对所述衔接子特异的至少两个碱基对的双链条形码,和其中含有5’-端核苷酸的单链部分包含:i)对所述样品特异的至少两个核苷酸的预定义单链条形码;和ii)对所述衔接子的一条链特异的至少两个核苷酸的随机单链条形码;对具有连接衔接子的核酸测序以确定序列,并且如果存在,对核酸变异测序;将共享对所述衔接子的一条链特异的相同随机单链条形码的核酸序列分组,以形成条形码组;消除在少于所述条形码组的所有成员中存在的序列变异;消除在所述条形码组中以低于预定阈值的频率存在的序列变异。所述预定阈值可以是50%。所述阈值可根据包括以下步骤的方法来预定:对多个样品进行单分子测序以确定靶核酸序列;对于各种可能种类的核苷酸置换,确定在所有位置的置换总数(y);以及确定对于具有置换的各位置的支持读出(read)数量(t);定义使y与t相关的函数;通过确定t来解函数,得到y的期望值,其中t为读出的阈值数量,大于该数量时,所述置换可识别为核酸中碱基位置上的序列变异。在另一个实施方案中,本发明为分析样品中的核酸的方法,其包括:将来自独特衔接子库中的衔接子与核酸的各端连接,各衔接子包含近端的双链部分和远端的两个单链部分,其中所述双链部分包含对所述衔接子特异的至少两个碱基对的双链条形码,和其中含有5’-端核苷酸的单链部分包含:i)对所述样品特异的至少两个核苷酸的预定义单链条形码;和ii)对所述衔接子的一条链特异的至少两个核苷酸的随机单链条形码;对具有连接衔接子的核酸测序以确定序列,并且如果存在,对核酸变异测序;将共享相同随机单链条形码的核酸序列分组以形成条形码组;消除在少于条形码组的所有成员中存在的序列变异;对来自对照样品的核酸进行上述步骤以鉴定复发的序列变异;应用统计学分析以确定对于在前述步骤中鉴定的各序列变异频率的置信区间;对于在前述步骤的置信区间内的序列变异频率设定阈值;消除其频率低于在前述步骤中设定的阈值的序列变异。在又另一个实施方案中,本发明为通过前述段落中描述的方法分析患者的无细胞核酸来评价患者的方法,其进一步包括步骤:如果在消除步骤之后仍存在一个或多个序列变异,则将患者评价为患有癌症。在又另一个实施方案中,本发明为设计选择子的方法,所述选择子包含待在患有一种类型的肿瘤的患者样品中分析的多个靶基因组区,所述方法包括:对来自多个患者的该类型肿瘤的基因组进行测序;鉴定包含突变的基因组区;基于在第一步中获得的每千碱基序列中具有一个突变的最高数量的患者,将在步骤b)中鉴定的区域分级;基于每个外显子中具有一个突变的最高数量的患者,将所述区域分级;将来自步骤c)和d)的最高分级区域包含在所述选择子中。所述基因组测序可以是外显子测序。通过所述方法鉴定的区域可以是至少100个碱基对长度。所述突变可包括单核苷酸变异、拷贝数变异、融合、种子区和组织学分类区。包含在所述选择子中的最高分级区域包括前10%的最高分级区域。所述方法可进一步包括从所述选择子消除落入基因组的富含重复序列区域中的区域。在又另一个实施方案中,本发明为在患者中评价癌症的方法,其包括:如上所述设计选择子;从患者中获得包含无细胞核酸的样品;确定患者核酸中的选择子的基因组区的序列;如果至少一个序列含有突变,则将所述患者评定为有可能患有癌症或复发癌症。所述方法可进一步包括将检出突变确认为匹配肿瘤活检中的体细胞突变。在又另一个实施方案中,本发明为设定阈值的方法,所述阈值用于在含有核苷酸置换的靶核酸序列中的碱基位置上识别(calling)序列变体,所述方法包括:对来自多个样品的带条形码的核酸进行单分子测序以确定靶核酸序列;对于各可能种类的核苷酸置换,确定所有位置中的置换总数(y);具有置换的位置的支持读出数量(t);定义使y与t相关的函数;通过确定t来解函数,得到y的期望值,其中t为读出的阈值数量,大于该数量时,所述置换可识别为核酸中碱基位置上的变体。对于多个靶序列中的给定序列g的阈值t,可通过包含以下步骤的方法来针对总体误差率进行调整:确定所述多个靶序列的误差率e,其等于靶序列中具有核苷酸置换的碱基位置的数量除以靶序列中的碱基总数;确定所述多个靶序列的测序深度d;如果序列g的e落入所述多个靶序列的e的前25%之内,则根据下式将序列g的阈值t调整为t’:t’←t×w,其中w=min{q2,5},q=e除以选择子中序列误差率的第75百分位数;如果序列g的d落入所述多个靶序列的测序深度的中位数(dmed)之下,则根据下式将序列g的阈值t调整为t’:t’←t/w*,其中w*=ln(dmed/d)。在又另一个实施方案中,本发明为评价非小细胞肺癌(nsclc)患者的方法,其通过如上所述使用选择子分析所述患者的cfdna以检测突变、和校正误差并评价患者来进行,如果在误差校正之后仍存在一个或多个序列变异,则将所述患者评定为患有nsclc或具有nsclc进展。所述突变可以是表皮生长因子受体(egfr)基因中的突变,其位于所述基因的激酶结构域(外显子19、20和21)中。在又另一个实施方案中,本发明为将核酸测序读出配对以获得双链核酸序列的方法,其包括:确定包含插入序列和含条形码的衔接子序列的多个单链核酸的序列;确定所述插入序列的基因组坐标;如果所述序列具有映射到相反链的互补的条形码和插入片段的基因组坐标,则将所述序列配对成双链核酸。所述方法可进一步包括消除含有序列变体的单成员条形码家族的步骤,如果所述变体未得到至少另一个具有≥2个成员的条形码家族支持。在另一个实施方案中,本发明为用于分析样品中的核酸的独特衔接子库,各衔接子包含:近端的双链部分和远端的至少一个单链部分,其中所述双链部分包含对所述衔接子特异的至少两个碱基对的双链条形码,和其中所述单链部分包含:对所述样品特异的至少两个核苷酸的预定义单链条形码;和对所述衔接子特异的至少两个核苷酸的随机单链条形码。各衔接子可包含远端的两个单链部分;包含5’-端的一个部分和包含3’-端的另一部分,其中所述单链部分彼此为不可杂交的。所述两个单链部分在远端彼此共价连接,例如通过接头连接。所述接头可任选包含切割位点。在一些实施方案中,本发明为独特的衔接子库,其包含两个衔接子亚库的组合:第一亚库,其中各衔接子包含远端的两个单链部分:包含5’-端的一个部分和包含3’-端的另一部分,其中所述单链部分彼此为不可杂交的;和第二亚库,其中各衔接子包含在远端彼此共价连接的两个不可杂交的单链部分。在另一个实施方案中,本发明为减小误差的分析受试者样品中的核酸的方法,其包括:对来自多个对照样品的核酸进行单分子测序以确定靶核酸序列;确定所述对照样品中各位置的各种可能种类的核苷酸置换的频率;对这些频率拟合统计学模型以确定背景误差的频率;对来自受试者样品的核酸进行单分子测序;确定所述受试者样品中各位置的各种可能种类的核苷酸置换的频率;确定所述受试者样品中各靶序列的读出深度;将所述统计学模型应用于所述受试者的序列;消除频率低于背景误差频率的核苷酸置换。附图简述图1:使用分子条形编码减小背景误差。(a)对分离的cfdna进行末端修复和加a尾。(b)将y形衔接子与cfdna连接。(c)所述y形衔接子在其不可杂交部分上包含随机条形码和固定条形码,以及在其可杂交部分上包含引物序列。(d)通过pcr扩增模板核酸并通过下一代测序获得序列信息。*表示真实生物突变。#表示pcr等引起的误差。(e)使用条形码通过生物信息学分析滤除假突变。(f)、(g)突变检测的灵敏性从0.02%增加至0.001%。图2:使用分子条形编码去除g至t突变。(a)对分离的cfdna进行末端修复和加a尾。(b)将y形衔接子与cfdna连接。(c)各y形衔接子在可杂交部分上包含独特的双链条形码。箭头指示主干条形码。图3:内源条形码包括在所述核酸不同基因座上的一个或多个序列。图4:通过深度测序进行癌症个性化谱分析(capp-seq)的开发。示意性描述了capp-seq选择子的设计及其用于评价循环肿瘤dna的应用。图5:来自实体瘤的循环dna的非侵入性超灵敏检测。(a)选择子设计和验证。(b)灵敏性建模。(c)成本优化流程图。图6:含或不含接头(y形和共价闭合末端)的串联条形码衔接子和串联交错条形码衔接子的结构和性能。图7:集成数字误差消除(ides)的设计和性能图8:选择子对多种类型肿瘤的适用性图9:集成数字误差消除的开发。(a)用于误差消除的串联测序衔接子以及单链和双链dna分子的回收;(b)位置-特异性全选择子(selector-wide)误差率的热图;(c)条形码去重(deduping)和背景修正对全选择子误差度量的影响;(d)对于各碱基置换的全选择子llod的密度图;(e)对于所有可能碱基置换的全选择子检测限;(f)ides与不同条形码去重策略的比较。图10:将碱基置换误差定型图11:定型碱基置换误差中的链偏好图12:使用ides对nsclc进行无活检肿瘤基因分型和超灵敏监测。(a)用于无活检基因分型的三种后处理方法的比较;(b)实测等位基因分数和预期等位基因分数;(c)检出snv的热图;(d)使用ides在nsclc患者中检出的白名单变异;(e)d中变体的接受者操作特征(roc)分析;(f)使用ides分析的来自nsclc肿瘤的可作用egfr突变的回收率;(g)用于检测ctdna的后处理方法的比较;(h)监测患者中的肿瘤负荷;(i)双链体测序的检测限评估。图13:验证分析模型的灵敏性图14:各种类型碱基置换的概率分布模型图15:用于ctdna检测和选择子设计的统计框架。(a)作为可得肿瘤报告物(reporter)的函数的ctdna检测限;(b)对于>90%检出可能性,作为所得肿瘤报告基因和测序ge的函数的ctdna检测限;(c)邻接体细胞突变(肺腺癌(luad)来源,癌症基因组图谱(tcga))之间的距离;(d)不同肿瘤报告物的实测数量和预测数量之间的一致性;(e)nsclc选择子覆盖的每个肿瘤中的snv和插入缺失(indel)数量的分析;(f)各选择子的重现性。图16:对检测不同浓度的肿瘤dna的概率建模图17:通过不同方法减小全选择子背景比率图18:使cfdna中的突变与肿瘤相关图19:按照ides分析等位基因特异性检测限发明详述下列描述和实施例详细阐述了本发明的实施方案。要理解的是,本发明不限于本文所述的具体实施方案并因此可改动。本领域技术人员将认识的是,存在本发明的许多变动和修改,所述变动和修改均包含在其范围之内。定义术语“治疗”包括实现治疗性益处和/或预防性益处。治疗性益处可以是根除或缓解正在治疗的潜在病症。并且,治疗性益处可随着与潜在病症相关的一种或多种生理症状的根除或缓解,由此可在患者中观察到改善而实现,尽管所述患者可能仍受所述潜在病症所累。对于预防性益处,可将组合物给予处于发展特定疾病的风险中的患者,或者给予报告疾病的一种或多种生理症状的患者,尽管可能还未诊断该疾病。术语“衔接子”是指可与一个或多个多核苷酸的3’或5’端连接的已知序列或未知序列的多核苷酸。在一些情况下,所述多核苷酸衔接子可与相同或不同的另一个多核苷酸衔接子连接。所述衔接子可包含已知序列、未知序列或二者。多核苷酸衔接子可以是双链的或单链的。双链衔接子可包含两条可杂交的链。或者,衔接子可包含可杂交部分和不可杂交部分。所述不可杂交部分包含至少一个单链区,例如彼此为不可杂交的两个单链区。在所述不可杂交部分内,含有未杂交5’-端的链称为5’-链,而含有未杂交3’-端的链称为3’-链。所述衔接子可在衔接子的一端具有可杂交部分和在衔接子的相对端具有不可杂交部分。所述不可杂交部分可以是开放的(y形衔接子)或经由接头共价闭合(balseq衔接子)。术语“条形码”是指用于鉴定核酸分子的多核苷酸内的核苷酸序列。例如,当将来自数个组的分子组合用于以多重方式加工或测序时,条形码可用于鉴定所述分子。另外,条形码可位于多核苷酸内的特定位置(例如多核苷酸的3’-端、5’-端或中间)并且可包含任意长度的序列(例如1-100个或更多个核苷酸)。此外,条形码可包含一个或多个预定义序列。术语“预定义的”意指条形码的序列为预定的,或者在鉴定包含所述条形码的核酸序列之前或无需鉴定的情况下其便为已知的。在一些情况下,可将预定义的条形码与核酸连接,用于将所述核酸分选到组中。例如,可连接aaaa序列以鉴定分离自患者a的核酸。在另一些情况下,条形码可包含一个或多个随机序列。术语“随机的”意指条形码的序列不是预定的或者在鉴定之前为未知的。术语“随机序列”与术语“简并序列”可交换使用,即,不具有精确定义的序列。例如,可将随机条形码与核酸连接,作为核酸的识别码(identifier)。在一些情况下,条形码可包含人工序列,例如未包含在受试者的未改变基因组中的设计序列。在另一些情况下,条形码可包含内源序列,例如存在于受试者的未改变基因组中的序列。在某些情况下,条形码可以是内源条形码。内源条形码可以是基因组核酸的序列,其中所述序列用作所述基因组核酸的条形码或识别码。不同类型的条形码可组合使用。例如,可将基因组核酸片段与随机条形码连接。基因组dna片段的一个或多个序列可以是内源条形码。所述随机条形码和内源条形码的组合可用作基因组核酸片段的独特识别码。术语“单链条形码”意指包含单链序列的条形码,例如未通过watson-crick氢键与另一个多核苷酸结合的多核苷酸。双链多核苷酸(例如通过watson-crick氢键与另一个多核苷酸结合的多核苷酸),例如双链衔接子,可在一条链上包含单链条形码,其中所述条形码核苷酸在所述多核苷酸的另一条链中不具有互补核苷酸。术语“双链条形码”可意指包含双链序列的条形码,例如通过watson-crick氢键与另一个多核苷酸结合的多核苷酸条形码。在一些情况下,所述双链条形码可包含一个多核苷酸的一部分。双链衔接子可包含一个或多个单链条形码和一个或多个双链条形码。术语“可杂交的”意指核酸的两条多核苷酸链在一个或多个核苷酸位置为互补的,例如所述两条多核苷酸链的含氮碱基可形成两个或更多个watson-crick氢键。例如,如果多核苷酸包含5’atgc3’,则其可与序列5’gcat3’杂交。在一些实验条件下,如果多核苷酸包含5’gggg3’,则序列5’ccac3’和5’ccca3’亦为可杂交的,尽管其并非完全互补。术语“不可杂交的”意指核酸的两条多核苷酸链为非互补的,例如在标准杂交条件下,两个独立的多核苷酸链的含氮碱基未形成两个或更多个watson-crick氢键。例如,如果多核苷酸包含5’atgc3’,则序列5’atgc3’可为不可杂交的。在本文中使用的术语“评价”包括涉及癌症或患有癌症的患者的状态,表示推断肿瘤或肿瘤细胞存在与否,以及对于所述患者,推荐或不推荐疗法或治疗、评估或预测疗法的功效、监测患者状况或评估患者中的癌症预后。术语“诊断”意指测试受试者以确定所述受试者是否具有用于临床决策的具体特性。诊断可包括测试处于发展特定疾病(因传染性生物感染所致)或非传染性疾病的风险中的受试者,例如癌症或代谢病。诊断亦可包括测试已发展特定症状的受试者以确定所述症状的原因。诊断的结果可用于将患者分组,以进行给予特定疗法的临床试验。例如,在一些实施方案中,诊断可包括分析收集自受试者的cfdna以检测癌症相关突变的存在与否。在另一些实施方案中,诊断可包括分析收集自受试者的cfdna以监测癌症进展或癌症阶段。在某些情况下,诊断亦可包括分析收集自接受癌症治疗的受试者的cfdna以检测所述治疗的作用并在治疗期间或治疗之后监测癌症进展或癌症阶段。术语“确定预后”意指测试受试者以预测其是否具有用于临床决策的具体特性。确定预后可包括测试处于发展特定疾病(因传染性生物感染所致)或非传染性疾病的风险中的受试者,例如癌症或代谢病。例如,在一些实施方案中,确定预后可包括分析收集自受试者的cfdna以检测癌症相关突变的存在与否。在另一些实施方案中,确定预后可包括分析收集自受试者的cfdna以监测癌症进展或癌症阶段。在某些情况下,确定预后亦可包括分析收集自接受癌症治疗的受试者的cfdna以检测所述治疗的作用并在治疗期间或治疗之后监测癌症进展或癌症阶段。术语“扩增”是指用于增加核酸序列的拷贝数的任何方法。例如,可使用聚合酶进行扩增,例如以一个或多个聚合酶链反应(pcr)或另外的指数或线性扩增法进行。术语“扩增子”意指扩增反应的核酸产物。例如,术语“扩增子”可不包含未掺入所需核酸产物中的核酸前体(例如未掺入的引物、dntp等)。术语“癌症”、“新生物”、和“肿瘤”在本文中可交换使用以及可以指表现出自发的不受调控生长的细胞或组织,由此所述细胞或组织表现出以增加的细胞增殖(例如显著的细胞增殖)为特征的异常生长。用于本申请的检测、分析或处理的目的细胞可包括但不限于癌前细胞(例如良性细胞)、恶性细胞、转移前细胞、转移性细胞和非转移性细胞。事实上各组织的癌症均为本领域技术人员已知的,包括实体瘤,例如癌、肉瘤、成胶质细胞瘤、黑素瘤、淋巴瘤、骨髓瘤等,以及循环癌症,例如白血病。癌症的实例包括但不限于卵巢癌、乳腺癌、结肠癌、肺癌、前列腺癌、肝细胞癌、胃癌、胰腺癌、子宫颈癌、卵巢癌、肝癌、膀胱癌、泌尿道癌、甲状腺癌、肾癌、癌瘤、黑素瘤、头颈癌和脑癌。短语“癌症负荷”或“肿瘤负荷”是指受试者中癌细胞的数量或肿瘤体积。减小癌症负荷因此可以指减少受试者中癌细胞的数量或肿瘤体积。术语“癌细胞”是指其为癌细胞或来源于癌细胞的任何细胞,例如癌细胞的克隆。术语“癌细胞”亦可以指表现出癌样特性的细胞,例如不可控制的增殖、耐受抗生长信号、能够转移以及丧失经历程序性细胞死亡(例如凋亡)的能力。术语“去重(deduping)”是指一种方法,其包括将核酸序列分组到包含原始存在于样品中的单分子的子代的组中。该原始分子及其子代以相同的独特分子条形码(uid)为特征。去重进一步包括分析子代分子的序列,以降低的误差率间接确定原始分子的序列。术语“突变”是指在生物或细胞的基因组中的遗传改变。例如,目的突变可以是相对于生物生殖系的变化,例如癌细胞特异性变化。突变可包括单核苷酸变异(snv)、拷贝数变异(cnv)、单核苷酸多态性(snp)、插入、缺失和重排(例如融合)。术语“癌症相关突变”意指发生在肿瘤中的突变。例如,癌症相关突变可以是癌症的起因。癌症相关突变亦可促进癌症发展,但并非癌症的唯一起因。癌症相关突变亦可导致或促进癌症转移。术语“无细胞dna(cfdna)”是指在收集时未包含在细胞之内的样品中的dna。cfdna可包括正常细胞和癌细胞来源的dna二者。cfdna通常获自血液或血浆(“循环”)。cfdna可通过分泌或细胞死亡过程释放到循环中,例如细胞坏死或凋亡。某些cfdna为ctdna(见下文)。术语“循环肿瘤dna(ctdna)”或“循环癌症dna”是指起源于肿瘤的无细胞dna(cfdna)级分。术语“基因组当量”是指构成整个基因组所需的dna的量。术语“样品”是指分离自受试者的任何生物样品。例如,样品可包括但不限于体液、全血、血小板、血清、血浆、粪便、红细胞、白血球或白细胞、内皮细胞、组织活检、滑液、淋巴液、腹水以及间质液或细胞外液的等分试样。术语“样品”亦可包括细胞之间的空间中的流体,包括龈沟液、骨髓、脑脊液(csf)、唾液、粘液、痰、精液、汗、尿或任何其它体液。所述样品亦可以是肿瘤样品。样品可通过以下手段获自受试者,包括但不限于静脉穿刺、排泄、射精、按摩、活检、针吸、灌洗、刮除术、手术切口或介入或者本领域已知的其它手段。术语“血样”是指全血或其任何级分,包括血细胞:红细胞、白血球或白细胞、或血小板,或者血清和血浆。术语“基因组区”可以指基因组内的一系列序列。例如,基因组区可包括基因中或基因外的序列,例如内含子、启动子、非编码区等。术语“选择子”是指与一个或多个基因组区杂交的多个寡核苷酸或探针。在一些情况下,所述一个或多个基因组区可能与疾病(例如癌症)有关。术语“连接”是指通过化学键将两个分子相连以生成新分子。例如,将衔接子多核苷酸与另一个多核苷酸连接可以指在所述衔接子和所述多核苷酸之间形成化学键(例如使用连接酶或任何其它方法)以生成包含所述衔接子和所述多核苷酸的单个新分子。术语“基因座”是指基因组中的位置。在一些情况下,基因座可以是一个或多个核苷酸或者一个或多个碱基对。术语“多核苷酸”是指包含在链中共价键合的一个或多个核苷酸单体(天然或非天然)的生物聚合物。在一些情况下,多核苷酸可具有包含基因组核酸序列的序列。在其它情况下,多核苷酸可具有人工序列(例如在基因组核酸中不存在的序列)。多核苷酸可包含基因组核酸序列或/和人工序列二者。人工序列可含有非天然核苷酸。尽管个体与个体之间以及不同类型癌症之间的具体遗传变化不同,但存在可显示复发的变化的基因组区。在所述区域中,任何给定癌症将显示遗传变异的概率增加。癌细胞中的遗传变化可提供一种手段,通过所述手段可将癌细胞与正常(例如非癌的)细胞进行区分。例如,可对cfdna分析肿瘤细胞特有的遗传变异的存在。然而,所述样品中的无细胞肿瘤dna的绝对水平常常很低,且所述遗传变异可能仅代表整个基因组的非常小部分。解决此问题的方法包括在正常细胞核酸的背景中精确检测癌细胞核酸中的癌症相关突变。尽管本文所公开的方法可能具体涉及dna或rna,但明确考虑的是,任何核酸均可代替dna或rna,或者可与一种或多种类型的核酸(例如rna/dna杂交物)组合使用。包含独特条形码的y形衔接子本文提供的是可用于鉴定或分析核酸的组合物。例如,在一些实施方案中,所述组合物可包含y形衔接子库,其中各y形衔接子在y形衔接子的一端包含可杂交部分以及在y形衔接子的相对端包含不可杂交部分,其中所述可杂交部分包含独特且可鉴定的至少两个碱基对的双链主干条形码。本文亦提供这样的组合物,其可包含y形衔接子库,其中各y形衔接子在y形衔接子的一端包含可杂交部分以及在y形衔接子的相对端包含不可杂交部分,其中所述不可杂交部分包含i)至少两个核苷酸的预定义单链条形码,和ii)在与所述预定义单链条形码相同的链上的至少两个核苷酸的随机单链条形码。本文还提供这样的组合物,其可包含y形衔接子库,其中各y形衔接子在y形衔接子的一端包含可杂交部分以及在所述y形衔接子的相对端包含不可杂交部分,其中所述可杂交部分包含至少两个核苷酸的独特双链主干条形码,和其中所述不可杂交部分包含i)至少两个核苷酸的预定义单链条形码,和ii)在与所述预定义单链条形码相同的链上的至少两个核苷酸的随机单链条形码。为了帮助鉴定独特的癌症核酸,在一些实施方案中,衔接子可在衔接子的不可杂交部分的5’链上包含预定义单链条形码和随机单链条形码。另一方面,在其它实施方案中,所述预定义单链条形码和随机单链条形码可位于衔接子的不可杂交部分的3’链上。预定义单链条形码可邻接随机单链条形码。亦明确考虑的是,可通过一个或多个核苷酸将预定义单链条形码与随机单链条形码隔开。预定义单链条形码可包含天然存在的碱基(例如腺苷(a)、胸苷(t)、鸟苷(g)、胞嘧啶(c)和尿嘧啶(u))或非天然存在的碱基,例如氨基烯丙基尿苷、异胞嘧啶、异鸟嘌呤和2-氨基嘌呤,并且介于1-约20个核苷酸长度之间。与预定义单链条形码类似,随机条形码的长度可介于1-约20个核苷酸之间,并且其可包含天然存在的碱基(例如腺苷(a)、胸苷(t)、鸟苷(g)、胞嘧啶(c)和尿嘧啶(u))或非天然存在的碱基,例如氨基烯丙基尿苷、异胞嘧啶、异鸟嘌呤。对于双链主干条形码的长度,其亦可介于1-约20个核苷酸之间。在一些情况下,双链主干条形码可包含预定义序列。在其它实施方案中,双链主干条形码可包含随机序列或包含预定义序列和随机序列二者。双链条形码可包含天然和非天然核苷酸,例如氨基烯丙基尿苷、异胞嘧啶、异鸟嘌呤和2-氨基嘌呤。这可辅助双链条形码的检测。在另外的实施方案中,各y形衔接子可另外包含引物序列。引物序列可以是pcr引物序列或测序引物序列。在一些实施方案中,引物序列可在y形衔接子的不可杂交部分上。在其它实施方案中,引物序列可在y形衔接子的可杂交部分上。在一些实施方案中,引物序列在整个y形衔接子库中可以是相同的。在另一些实施方案中,一个或多个y形衔接子上的引物序列可与其它y形衔接子上的引物序列不同。分析核酸的方法在一些实施方案中,本发明为分析循环肿瘤核酸以用高灵敏性检测癌症相关突变谱的方法。所述方法可应用于含循环肿瘤dna(ctdna)的无细胞dna(cfdna)。所述分析可包括核酸的高通量测序。本文公开的是用于集成数字误差消除(ides)的新型分析建模框架,其使得能够以低误差率对核酸测序。本发明的方法优化了来自低输入样品的ctdna的检测和分析。通过将定式背景误差消除与有效的分子条形编码组合(ides),与先前方法相比在大范围测序方法中得到更低检测限,从而利于无活检定量数百个千碱基中的变体。在一些实施方案中,ctdna分析的灵敏性低至每个典型样品中1-3个突变分子。在一些实施方案中,将所述方法应用于晚期非小细胞肺癌(nsclc)且使得能够以无活检检出egfr基因中的95%激活突变(无假阳性)。在一些实施方案中,所述方法允许在监测环境中回收低至0.004%的ctdna分数。在又另一个实施方案中另外包括的是,通过调整(leveraging)重建的双链体序列,所述方法使得在1,000,000个分子背景中2.5个分子的检测限。本文公开的是分析核酸的方法,包括用于超灵敏检测核酸的方法,所述核酸例如循环无细胞dna,如样品中的循环无细胞肿瘤dna。所述方法精确定量来自早期和晚期肿瘤的核酸,如无细胞肿瘤dna。因为肿瘤来源的dna水平常常与对不同疗法的临床反应相当,所以所述方法可鉴定可作用的突变。所述方法亦可用于非侵入性检测和监测肿瘤,从而利于个性化癌症疗法。用于本发明方法的衔接子在图6中显示。本发明的方法可利用y形衔接子或者亦可使用共价闭合的衔接子。在共价闭合的衔接子中,单链部分可经由接头连接。本发明的方法亦可利用y形衔接子和共价闭合衔接子的组合。所述方法可包括a)使双链衔接子与多个双链核酸的两端连接以产生衔接子连接的核酸,其中双链衔接子各自包含双链主干条形码;b)扩增衔接子连接核酸的两条链以产生第一扩增子和第二扩增子,其中第一扩增子来源于双链核酸的第一链和双链主干条形码的第一链,和第二扩增子来源于双链核酸的第二链和双链主干条形码的第二链;c)确定第一和第二扩增子的序列;和d)使用双链主干条形码分析核酸。分析可包括借助于鉴定双链主干条形码来确定第一和第二扩增子是否起源于多个双链核酸中的单个双链核酸的相同链。本文亦公开的是这样的方法,其包括a)使衔接子经由其双链部分与多个双链核酸的两端连接以产生衔接子连接的核酸,其中各衔接子在一端包含可杂交部分和在相对端包含不可杂交部分,且其中各双链衔接子的可杂交部分包含双链主干条形码;b)扩增衔接子连接核酸的两条链以产生第一扩增子和第二扩增子,其中第一扩增子来源于双链核酸的第一链和双链主干条形码的第一链;和第二扩增子来源于双链核酸的第二链和双链主干条形码的第二链;c)确定第一和第二扩增子的序列;和d)使用双链主干条形码,确定第一和第二扩增子是否起源于多个双链核酸中的单个双链核酸的相同链。本文亦公开的是这样的方法,其包括a)使衔接子经由其双链部分与多个双链核酸的两端连接以产生衔接子连接的核酸,其中各衔接子在一端包含可杂交部分和在相对端包含不可杂交部分,其中可杂交部分包含双链主干条形码,和其中不可杂交部分包含5’链,其包含:i)至少两个核苷酸的预定义单链条形码;和ii)在与预定义单链条形码相同的链上的至少两个核苷酸的随机单链条形码,其中各双链核酸的一个或多个序列提供核酸的内源条形码,且其中随机单链条形码和内源条形码的组合提供用于各双链核酸的独特识别码;b)扩增衔接子连接核酸的两条链以产生第一扩增子和第二扩增子,其中第一扩增子来源于双链核酸的第一链和双链主干条形码的第一链,和第二扩增子来源于双链核酸的第二链和双链主干条形码的第二链;c)确定第一和第二扩增子的序列;和d)通过鉴定双链主干条形码的序列,确定第一和第二扩增子是否起源于多个双链核酸中的单个双链核酸的相同链。本文公开的是用于分析核酸以用于检测、诊断或确定癌症预后的方法。所述方法可包括a)使衔接子经由其双链部分与多个双链核酸的两端连接以产生衔接子连接的核酸,其中各衔接子在一端包含可杂交部分和在相对端包含不可杂交部分,其中可杂交部分包含独特且可鉴定的至少两个核苷酸的双链主干条形码,和其中不可杂交部分包含i)至少两个核苷酸的预定义单链条形码;和ii)在与预定义单链条形码相同的链上的至少两个核苷酸的随机单链条形码;b)扩增模板核酸以产生多个扩增子;c)使扩增子与包含一组寡核苷酸的选择子杂交,所述寡核苷酸与一个或多个样品核酸的全部或其亚组中的基因组区选择性杂交;和d)获得杂交扩增子的序列以检测癌症或癌症相关突变的存在与否。本文公开的是用于分析核酸以用于检测、诊断或确定癌症预后的方法。所述方法可包括a)使衔接子经由其双链部分与多个双链核酸的两端连接以产生衔接子连接的核酸,其中各衔接子在一端包含可杂交部分和在相对端包含不可杂交部分,其中可杂交部分包含独特且可鉴定的至少两个核苷酸的双链主干条形码,和其中不可杂交部分包含i)至少两个核苷酸的预定义单链条形码;和ii)在与预定义单链条形码相同的链上的至少两个核苷酸的随机单链条形码;b)扩增模板核酸以产生多个扩增子;c)使扩增子与包含一组寡核苷酸的选择子杂交,所述寡核苷酸与一个或多个样品核酸的全部或其亚组中的基因组区选择性杂交;和d)获得杂交扩增子的序列以检测癌症或癌症相关突变的存在与否。本发明的方法包括使一个分子与另一个分子连接的步骤,例如使多核苷酸衔接子连接到不同多核苷酸上。所述连接可包括将y形衔接子与一个或多个核酸连接。在一些情况下,用于连接的酶为dna连接酶,例如t4dna连接酶,大肠杆菌(e.coli)dna连接酶、哺乳动物连接酶或其任何组合。哺乳动物连接酶可以是dna连接酶i、dna连接酶iii或dna连接酶iv。所述连接酶可以是耐热连接酶。用于所述方法的衔接子可将本文公开的衔接子及其具体实施方案通过衔接子的可杂交(双链)部分与一个或多个核酸连接。衔接子可具有游离的或连接的单链部分。在一些实施方案中,本发明的方法利用具有游离单链部分的衔接子(y形衔接子)和具有共价连接单链部分的衔接子(bal-seq衔接子)或两种类型衔接子的组合(图6)。在一些实施方案中,共价连接的单链部分通过接头连接。接头可任选含有切割位点,例如限制酶识别序列。本发明的衔接子可具有根据下述的数个不同的实施方案定位的条形码。如在图6中所示,各衔接子可具有数个条形码(在图6中称为“id”)。衔接子可具有各单链部分上的一个或多个条形码和双链部分中的一个或多个条形码。参考图6,在一些实施方案中,单链部分具有分子id条形码,而双链部分可具有样品id条形码。条形码各自可位于(或共同位于)(a)上游(upper)单链区(含5’端),(b)下游(lower)单链区(含3’端),和(c)y形衔接子的双链区或主干,如在表a和图6中所示。表a.条形码布置及在测序中的用途m:多重样品id(mid);u:独特分子id(uid);ss-单链;ds-双链*ds测序,即,仅在条形码并非随机的且具有已知序列因而其可进行匹配的情况下,使用条形码将两条链配对方为可能的。在一些实施方案中,uid位于衔接子的两条链上:上游链和下游链,或者在双链区中。如果uid可匹配为起源于相同的衔接子,双链测序(即配对单链为可能的)。位于双链区中的uid通过watson-crick配对进行匹配。存在于单链部分上的已知序列(非随机)uid可交叉引用为属于相同的衔接分子。在一些实施方案中,随机单链条形码与内源条形码组合,可提供用于各模板核酸的独特识别码。内源条形码可包含任何长度的序列并且可在核酸上包含一组或多组核苷酸序列。所述序列可位于核酸的不同基因座。在一些实施方案中,内源条形码可包含位于核酸一端的序列(图3(a))。在一些实施方案中,内源条形码可在核酸的一端包含第一序列和在核酸的相对端包含第二序列(图3(b))。在其它实施方案中,内源条形码可包含内部序列(图3(c))。在某些实施方案中,内源条形码可包含其为内部序列的第一序列,和位于核酸一端上的第二序列(图3(d))。在又其它的实施方案中,内源条形码可包含均为内部序列的第一和第二序列(图3(e))。在本发明的情况下,来源于相同模板核酸的扩增子含有相同的独特识别码(uid)。这些不同的独特识别码可用于在起始样品中鉴定和计数不同的模板核酸。例如,uid可用于计数含有相同突变的起始模板核酸。在其它情况下,uid可用于对来自相同起始模板核酸的扩增子进行鉴定和分组。主干条形码可位于衔接子主干的任何部分。例如,主干条形码可邻接核酸上的衔接子与之连接的碱基对,或者与核酸上的衔接子与之连接的碱基对距离一个或多个碱基对。独特的双链主干条形码亦可鉴定核酸的链。例如,在衔接子与核酸连接之后,所得核酸的两条链均含有独特的主干条形码,尽管核酸的各链可能含有不同的随机单链条形码或不同的独特识别码。在扩增之后,来源于核酸的一条链的扩增子含有与来源于相同核酸的另一条链的扩增子相同的主干条形码和相同的内源条形码。因此,在一些实施方案中,主干条形码可用于鉴定来源于相同模板核酸的两条链的扩增子。在某些实施方案中,独特的主干条形码可用于鉴定在核酸的一条链上但不在另一条链上的突变。在另一些实施方案中,在模板核酸的一条链上发生但在另一条链上未发生的突变可能是扩增误差,且可忽略为人为假象。在一些实施方案中,本发明包含新型衔接分子。本文公开的是含有两个从根本上不同的条形码的“串联”测序衔接子,其允许追踪单个dna分子以将体内产生的真实体细胞突变与在包括高通量测序在内的离体程序期间引入的误差进行区分。衔接子可包含含有确定序列或随机序列或者随机序列和确定序列的组合的条形码。如在图1和图6中所示,衔接子的单链部分包含由在样品的衔接分子之间共享的多重样品id(mid)部分组成的条形码,和对各衔接分子而言为独特的条形码(独特id或uid)。在一些实施方案中,所述独特条形码为随机条形码。具有这类复合条形码的衔接子称为“索引(index)衔接子”。在一些实施方案中,衔接子为“串联衔接子”。串联衔接子包含具有添加核苷酸的索引衔接子。在一些实施方案中,向各衔接子寡核苷酸的各端添加2个或更多个碱基以得到串联衔接子。在一些实施方案中,添加核苷酸在3’端包含t以使连接成为可能。在其它实施方案中,衔接子为“交错串联衔接子”。交错串联衔接子包括具有添加核苷酸的串联衔接子。在一些实施方案中,向衔接子的内部末端的远端添加2个或更多个碱基。(图6)。在衔接子的一些实施方案中,将典型的样品多重条形码(mid)替换成简并分子条形码,作为独特识别码或uid。在另一个实施方案中,靠近衔接子的连接端的短uid(2个或更多个核苷酸)产生“插入片段”或内部条形码或内部uid。(图6(a))。通过借助(leveraging)各分子的不同基因组坐标,本发明的内部uid允许更短的条形码,使测序通量达到最大。这些内部uid允许有效回收双链体分子,与类似的现有技术方法相比改进约2倍,参见(图7(a))。本发明的方法(即ides、仅条形编码或修正、仅双链体)与来自现有技术的误差消除方法相比为更有利的。(图7(a))参考lou,d.i.等high-throughputdnasequencingerrorsarereducedbyordersofmagnitudeusingcirclesequencing(使用循环测序将高通量dna测序误差降低数个数量级).procnatlacadsciusa110,19872-19877(2013),(“lou”);kennedy,s.r.等detectingultralow-frequencymutationsbyduplexsequencing(通过双链体测序检测超低频突变).natprotoc9,2586-2606(2014),(“kennedy”);和schmitt,m.w.等detectionofultra-raremutationsbynext-generationsequencing(通过下一代测序检测超罕见突变).procnatlacadsciusa109,14508-14513(2012),(“schmitt”。)串联衔接子和交错串联衔接子在一些实施方案中,本发明为以使测序读出内的可用测序深度达到最大而设计的条形编码方法。不是将独特的分子条形码(uid)插入靶序列的读出中,而是将分子条形码置于靠近样品条形码(mid)处,由此以独立的“索引”读出读取分子条形码。与需要给条形码贡献各读出的15个或更多个测序碱基的其它分子条形编码方法(kennedy,s.r.等detectingultralow-frequencymutationsbyduplexsequencing(通过双链体测序检测超低频率突变).natprotoc9,2586-2606(2014))相比,此方法允许将两个末端配对的测序读出整个用于读取来自靶核酸的dna碱基。本发明方法的另一个优点为,本文所用的测序衔接子在其它方面未经修饰。与使用什么的其它方法报道的文库制备的较低效率(kennedy,上文)相比,本发明衔接子的设计允许在文库制备期间的有效连接。在一些实施方案中,将随机条形码的长度设计成允许有足够数量的不同条形码,所述条形码彼此足够不同以同时使用。例如,随机2聚体允许有16种不同的分子条形码,而随机4聚体允许有256种不同的分子条形码。在一些实施方案中,将随机条形码与片段起点和终点的基因组坐标组合,形成内源或内部条形码。总之,内源条形码可提供足够复杂的独特分子条形码(uid)库。在本公开内容的情况下,足够的复杂性意指足以区分预期数量的相同输入分子的许多条形码。例如,本领域技术人员可基于患者样品的典型大小和含量来确定独特核酸分子(或人类基因组当量)的数量。在此情况下,本领域技术人员可确定获自特定体积的无细胞人血浆样品的核酸分子的预期数量并从而确定所需独特条形码的数量。在一些实施方案中,衔接子亦具有多重样品条形码(mid)。在一些实施方案中,mid具有足以设计使用所需数量的样品需要的条形码数量的长度。例如,4个碱基长度的多重条形码允许设计具有至少为2的成对编辑距离的24种多重条形码。在一些实施方案中,本发明为使用索引条形码以减少的误差率测序核酸的方法。本发明的方法包括正确配对来自相同双链体分子的相反链的读出。在此实施方案中,本发明使用位于衔接分子各链的末端的条形码。在一些实施方案中,这些条形码为一个或多个碱基长度,例如2个碱基长度。条形码出现在配对末端读出的各端。如在图6(a)中所示,存在2-碱基标点标记,包括从本文的连接步骤中得到的衔接子中的一个碱基对(g/c碱基对)和靶分子中的一个碱基对(a/t碱基对)。我们称这些衔接子为串联衔接子以反映其含有多重样品条形码(mid)和独特分子条形码(uid)二者的事实。在一些实施方案中,使独特分子条形码尽可能短。在一些实施方案中,独特分子条形码为1或2个碱基长度。例如,具有2-碱基条形码使有信息的测序容量达到最大,同时仍允许区分独特分子。本发明公开了分析单个读出中的核酸的最大长度的方法。在一些实施方案中,条形码和标点仅用完一对读出中的8个碱基,相比之下,现有技术方法使用每对读出中的30个或更多个碱基(kennedy,上文)。由于本发明的串联衔接子含有多重样品条形码(mid)和独特分子条形码(uid)二者,本发明包括使用二者以获得协同作用结果的方法。在一些实施方案中,所述方法包括将两种策略组合(uid去重接着mid去重)以实现最低的误差率。(图6(b))。在一些实施方案中,所述方法使用单链读出,而在其它实施方案中,所述方法使用双链读出,即两条链均经测序和配对的分子的读出。如通过实施例所证实,本发明的方法使用双链读出实现了低误差率。实测2x10-6的误差率(参见实施例18)优于几乎所有先前所示的误差消除机制(kukita,y.,等high-fidelitytargetsequencingofindividualmoleculesidentifiedusingbarcodesequences:denovodetectionandabsolutequantitationofmutationsinplasmacell-freednafromcancerpatients(使用条形码序列鉴定的单个分子的高保真靶向测序:从头检测和绝对定量来自癌症患者的血浆无细胞dna中的突变).dnaresearch(2015);kinde,i.,等detectionandquantificationofraremutationswithmassivelyparallelsequencing(使用大规模平行测序检测和定量罕见突变).procnatlacadsciusa108,9530-9535(2011);lou,d.i.,等high-throughputdnasequencingerrorsarereducedbyordersofmagnitudeusingcirclesequencing(使用循环测序将高通量dna测序误差降低数个数量级).procnatlacadsciusa110,19872-19877(2013),以及类似于使用双链测序的先前所示的误差率(kennedy和schmitt,上文)。结果进一步证实,本发明的方法在回收双链体分子方面优于本领域现有水平。双链体分子以先前报道比率的约两倍回收,测序总读出中有多达3.3%双链分子。在一些实施方案中,本发明使用单链分子或无双链支持的分子。如在实施例中所示,使用ides测序使分子的回收率增加一个数量级,同时将误差率维持在1.5x10-5,其仍优于已公布的非双链体分子条形码误差消除技术(图7(a),实施例18)。在一些实施方案中,将衔接子设计成具有2-碱基条形码和4-碱基条形码的组合。本领域已知的是,在待测序的核酸库中具有大量序列多样性为有益的。当缺乏所述多样性时(因例如核酸的非随机片段化或使用短条形码所致),可向样品掺入高度多样性的核酸库(例如illumina,sandiego,cal.提供的phix文库)。在一些实施方案中,本发明包括使用交错串联衔接子,其比串联衔接子更具多样性。交错串联衔接子库包含具有2-碱基条形码和4-碱基条形码的衔接子的组合。如在实施例中所示,使用交错衔接子得到的误差率至少与使用串联衔接子并掺加phix的误差率一样好(图6(c),实施例12)。样品本文所公开的方法可包括分析一个或多个样品。样品可以是分离自受试者的任何生物样品,例如体液、全血、血小板、血清、血浆、粪便、红细胞、白血球或白细胞、内皮细胞、组织活检、滑液、淋巴液、腹水、间质液或者和/或细胞外液的等分试样。样品亦可包括细胞之间的空间中的流体,包括龈沟液、骨髓、脑脊液(csf)、唾液、粘液、痰、精液、汗、尿或任何其它体液。血样可以是全血或其任何级分,包括血细胞(红细胞、白血球或白细胞、和血小板)、血清和血浆。样品可获自人类或非人类。样品亦可以是肿瘤样品。肿瘤样品可通过以下手段获自受试者,包括但不限于静脉穿刺、排泄、按摩、活检、针吸、灌洗、刮除术、手术切口或介入或者本领域已知的其它手段。其它样品来源可包括汗、呼吸、眼泪和/或羊水。例如,样品可以是脑脊液样品。在一些情况下,样品不为巴氏涂片流体样品。在一些情况下,样品不为囊内液样品。在一些情况下,样品不为胰液样品。可在一段时间内反复从个体收集样品(例如每天一次、每周一次、每月一次、每半年一次或每年一次)。在一段时间内从个体获得多个样品可用于验证来自早期检测的结果或者用于鉴定作为例如药物治疗结果的变化。样品可包含核酸。核酸可以是肿瘤核酸。核酸亦可以是循环核酸,例如无细胞核酸。例如,循环核酸可来自肿瘤,例如ctdna。核酸可以是基因组核酸。对本发明的方法而言有用的样品核酸可包括cfdna,例如未包含在细胞内的样品中的dna。所述dna可以是片段化的,例如可平均长为约170个核苷酸,其可与缠绕单个核小体的dna长度相一致。cfdna可以是来自正常细胞和肿瘤细胞的dna的异种混合物,且cfdna的起始样品可能并非富含癌细胞dna和癌细胞基因组的复发的突变区。术语循环肿瘤dna(ctdna)或无细胞肿瘤dna可用于指来源于肿瘤的样品中的cfdna的级分。本领域技术人员应理解的是,尽管在肿瘤来源和正常细胞来源之间不能区分未突变的生殖系序列,但含有体细胞突变的序列很可能来源于肿瘤dna。在一些情况下,样品可包含对照生殖系dna。样品亦可包含已知的肿瘤dna。另外,样品可包含从疑似在样品中具有ctdna的个体中获得的cfdna。此外,样品可包含从未疑似在样品中具有ctdna的个体中获得的cfdna,例如作为常规测试的部分。本文所公开的方法可包括从受试者中获得一个或多个样品,例如核酸样品。所述一个或多个样品核酸可以是肿瘤核酸。例如,可从肿瘤活检中提取核酸。肿瘤核酸亦可从肿瘤细胞释放到血流中,例如作为对肿瘤免疫应答的结果。释放到血液中的肿瘤核酸可以是ctdna。所述一种或多种样品核酸可以是基因组核酸。应理解的是,从患有特定癌症的受试者中获得肿瘤核酸和基因组核酸的步骤可同时进行。例如,收集血液、血浆或血清的静脉穿刺,可同时收集基因组核酸和肿瘤核酸二者。从患有特定癌症的受试者中获得肿瘤核酸和基因组核酸,亦可在独立的场合进行。例如,有可能从患者获得单个组织样品,例如活检样品,其包含肿瘤核酸和基因组核酸二者。亦有可能从独立的样品、独立的组织中或者在独立的时间从受试者中获得肿瘤核酸和基因组核酸。从患有特定癌症的受试者中获得肿瘤核酸和基因组核酸亦可包括从患有特定癌症的受试者中提取生物流体或组织样品的过程。获得核酸可包括增加核酸的产率或回收率的程序,例如将核酸与可能存在于生物流体或组织样品中的其它细胞组分和污染物分离,例如通过酚氯仿提取、有机溶剂沉淀或结合dna的spin柱进行。如上所述,该程序可改进产率并且可利于测序反应。从患有特定癌症的受试者获得肿瘤核酸和基因组核酸,亦可通过商业实验室进行,其可以不与受试者直接接触。例如,商业实验室可从医院或其它临床机构获得样品,样品在医院或其它临床机构获自受试者。因此商业实验室可在受试者进行治疗或诊断的机构的要求或指示下进行本文公开方法的所有步骤。有时,核酸为混合的或不纯的。因此,如果需要鉴定核酸的来源,则预定义单链条形码可用于将核酸分选到不同的组中。例如,预定义单链条形码可用于鉴定核酸来源的样品。在一些情况下,来自第一样品的核酸可与第一预定义单链条形码相关,反之来自第二样品的核酸可与第二预定义单链条形码相关。在其它情况下,两个或更多个样品的预定义单链条形码可以是不同的。在另一些情况下,两个或更多个样品可来自相同受试者。在某些实施方案中,两个或更多个样品可来自相同受试者的不同组织。例如,一个样品可来自肿瘤,而另一个样品可来自相同受试者的血液,其中肿瘤可以是实体瘤。此外,两个或更多个样品可来自两个或更多个受试者。样品可在相同时间获得或者在两个或更多个时间点获得。扩增核酸扩增可导致将核苷酸掺入核酸分子或引物中,从而形成与模板核酸互补的新核酸分子。新形成的核酸分子及其模板可用作合成另外的核酸分子的模板。被扩增的核酸可以是dna,包括基因组dna,cdna(互补dna)、无细胞dna(cfdna)和循环肿瘤dna(ctdna)。被扩增的核酸亦可以是rna。如在本文中所用,一个扩增反应可由多轮dna合成组成。本文所公开的方法可包括扩增包含与y形衔接子连接的样品核酸的模板核酸。用于核酸(例如dna和rna)扩增的任何已知技术均可与本文的测定法一起使用。一些扩增技术为聚合酶链反应(pcr)方法,其可包括但不限于溶液pcr和原位pcr。或者,扩增可包括非指数扩增,例如线性扩增。模板核酸的扩增可包括使用珠粒扩增接着光纤检测,如描述于美国申请公布号20020012930、20030058629、20030100102、20030148344、20040248161、20050079510、20050124022和20060078909。模板核酸的扩增可包括使用一种或多种聚合酶。例如,聚合酶可以是dna聚合酶或者rna聚合酶。在一些情况下,聚合酶可以是高保真聚合酶(kapahifidna聚合酶)。聚合酶亦可以是phusiondna聚合酶。选择子设计体细胞突变,其为在除生殖系细胞之外的身体的任何细胞中发生的突变,可以是癌细胞的特征。对于单个基因中的体细胞突变而言,大多数人类癌症为相对异种的。选择子可用于从总基因组核酸中富集肿瘤来源的核酸分子。对于具有给定癌症的患者,选择子的设计可指示何种突变可以高概率检出。选择子大小亦可直接影响成本和序列覆盖的深度。例如,选择子的设计和用途部分描述于pct申请号pct/us14/25020(us20140296081)和newman等(2014),natmed.20(5):548-54),通过引用以其整体结合到本文中。本文所公开的方法可包括一个或多个选择子或者一个或多个选择子的用途。选择子可包含与一个或多个基因组区杂交的多个寡核苷酸或探针。基因组区可包含一个或多个突变区。基因组区可包含与一种或多种癌症相关的一个或多个突变。多个基因组区可包含不同的基因组区。在一些实施方案中,多个基因组区可包含从少数基因组区至高达5000个不同的基因组区。基因组区可包含蛋白质编码区或其部分。蛋白质编码区可指编码蛋白质的基因组的区域,例如基因。基因亦可包含非编码序列,例如内含子,或者非翻译区(utr)或其部分。基因组区可包含两个或更多个基因、蛋白质编码区或其部分。在一些情况下,基因组区不包含整个基因。基因组区可包含非蛋白质编码区。在一些情况下,非蛋白质编码区可转录成非编码rna(ncrna)。在一些情况下,非编码rna可具有已知功能。例如,非编码rna可以是转运rna(trna)、核糖体rna(rrna)或调节rna、小核rna(snrna)、小核仁rna(snorna)、微小rna、小干扰rna(sirna)、piwi相互作用rna(pirna)和长ncrna(例如xist、hotair)。基因组区可包含假基因、转座子或反转录转座子。基因组区可包含复发的突变区。“复发的突变区”可以指这样的基因组(通常为人类基因组)的区域,其中总体而言,相对于基因组,在目的癌症中的基因突变概率增加。复发的突变区可以指这样的基因组的区域,其包含在群体中复发的一个或多个突变。复发的突变区可通过“复发指数(ri)”来表征。ri通常是指具有以下突变的单个受试者(例如癌症患者)的数量,所述突变在基因组序列的给定千碱基对之内发生(例如具有突变的患者数量/kb基因组区长度)。基因组区亦可通过每个外显子中具有一个突变的患者数量来表征。可针对各度量(例如ri和患者/外显子或基因组区)选择阈值,以在统计学上富集目的癌症(例如nsclc)的已知驱动基因或疑似驱动基因。亦可通过针对各度量任意选择最高百分位数来选择阈值。选择子中的基因组区数量可根据癌症的性质而变化。将大量基因组区包含在内通常可增加鉴定出独特的体细胞突变的可能性。然而,将太多的基因组区包含在文库中并非没有成本。例如,基因组区的数量可与可能在分析中测序的核酸长度直接相关。最极端的情况下,可对肿瘤样品和基因组样品的整个基因组进行测序,且可将所得序列进行比较以记录与非肿瘤组织的任何差异。本发明的选择子可解决此问题,其通过以下进行:鉴定在特定癌症中复发突变的基因组区,并随后将所述区域分级以使所述区域将包含特定肿瘤中的区别性体细胞突变的可能性达到最大。复发突变基因组区的文库(或“选择子”),可在整个群体中使用用于给定癌症或癌症类别,且无需针对各受试者优化。所述方法可进一步包括杂交反应,例如使扩增子与包含一组寡核苷酸的选择子杂交,所述寡核苷酸与一个或多个样品核酸的基因组区选择性杂交。在一些实施方案中,杂交反应可包括使多个核酸与固体支持物(例如多个珠粒)杂交。所述方法可进一步包括在酶促反应之后进行杂交反应。例如,在一些情况下,酶促反应可包含一个或多个连接反应、片段化反应、末端修复反应、加a尾反应或扩增反应。选择子亦可包含一组寡核苷酸。该组寡核苷酸可与基因组的小于100kb至高达1.5百万碱基(mb)杂交。该组寡核苷酸可能能够与5个至高达500个或更多个不同的基因组区杂交。选择子亦可与一系列不同的基因组区杂交,例如与介于约10至约1000个不同的基因组区杂交。选择子亦可与多个基因组区杂交,例如与50个至最多5000个不同的基因组区杂交。选择子可与包含突变的基因组区杂交,所述突变在群体中并非为复发的。例如,基因组区可包含存在于给定受试者中的一个或多个突变。在一些情况下,受试者中包含一个或多个突变的基因组区,可用于产生用于受试者的个性化选择子。选择子可与包含一个或多个突变的多个基因组区杂交,所述突变选自snv、cnv、插入、缺失和重排。选择子可与已知与癌症相关的基因组区中的突变杂交。已知与癌症相关的基因组区中的突变可称为“已知体细胞突变”。已知体细胞突变可以是位于已知与癌症相关的一个或多个基因中的突变。已知体细胞突变可以是位于一个或多个癌基因中的突变。例如,已知体细胞突变可包括位于p53、egfr、kras或brca1中的一个或多个突变。选择子可与预测与癌症相关的基因组区中的突变杂交。另外,选择子可与未报道与癌症相关的基因组区中的突变杂交。基因组区可包含大小足以捕获一个或多个复发突变的人基因组的序列。当突变在基因组区的序列之内时,可以说该基因组区“鉴定突变”。本发明的方法可涉及cfdna,其通常长度小于约200bp,并因此基因组区通常可能小于约10kb。通常snv的基因组区可相当短,长度从约45bp至约500bp,而融合或其它基因组重排的基因组区可较长,长度从约1kbp至约10kbp。选择子中的基因组区可小于10kbp,例如100bp-10kbp。在一些实施方案中,选择子覆盖的总序列小于约1.5百万碱基对(mb),例如10kb-1.5mb。在一些实施方案中,本发明包括一种改进的选择子设计,其改进在患者样品中检测肿瘤突变(变体)的灵敏性。用于本发明方法的选择子包含从肿瘤的全基因组测序获得的变体。任选的是,选择子可排除位于含重复序列或其它技术障碍的区域中的变体。例如,可从对来自肿瘤样品集合的核酸进行外显子组测序来获得变体列表,所述集合例如肺鳞状细胞癌(scc)肿瘤集合或肺腺癌肿瘤集合或者对于测序分析而言可得的一种或多种类型肿瘤的任何其它集合。可对序列进行过滤以消除位于富含重复序列的基因组区中的变体(例如简单重复序列、微随体、间断重复序列和片段复制)。亦可对序列(或相反地)进行过滤以消除位于间隔区中的具有低映射率或低k-mer唯一性的变体。根据本发明的方法,用于本发明的选择子可设计成以最少量的基因组间隔覆盖尽可能多的患者和每个患者中的突变。在一些实施方案中,本发明包括构建选择子的方法,即选择待在患者中分析的基因组区。根据下述试验步骤将基因组区包含在内。选择子设计成基于本文所定义的“复发指数”(ri)度量来优先考虑将基因组区包含在内。在一些实施方案中,待包含在选择子中的基因组区为含有已知损伤的外显子或外显子的较小部分。待包含在内的基因组区包含已知损伤并且侧翼为一个或多个碱基对至最少100bp的区块(tile)大小。根据本发明的方法,通过渐减的ri将基因组区分级,并将最高级的ri和每个外显子的患者数量二者中的基因组区包含在选择子中。在一些实施方案中,最高级为大于或等于前10%。在此实施方案中,选择子具有最大的额外患者覆盖度且最小的间隔。在一些实施方案中,在降低的严格性下重复选择基因组区的过程,即百分等级低于前10%,例如选择前33%。在此实施方案中,所述方法导致将每个患者的突变的中位数增加最大的区域包含在内。在一些实施方案中,当达到预定大小时,停止将另外的基因组区包含到选择子中。在一些实施方案中,预定的所需大小为约100-200kb(例如对于nsclccfdna选择子为175kb)。在其它实施方案中,当满足上述过滤的所有基因组区均被耗尽时,停止将另外的基因组区包含到选择子中。在一些实施方案中,如上所述包含含有单核苷酸变异(snv)的基因组区的选择子,进一步包含含有其它类型突变的临床上相关的区域,例如融合、种子区、拷贝数变异(cnv)区和组织学分类区。临床适应症本文提供的方法可进一步包含获得杂交扩增子的序列信息以检测癌症的存在与否。例如,序列信息可以是实际的核苷酸序列或者在一些情况下为基因的拷贝数。在一些情况下,癌症可以是实体瘤。例如,实体瘤可以是非小细胞肺癌(nsclc)。癌症亦可以是乳腺癌。例如,乳腺癌可与brca基因中的突变相关,例如brca1。选择子可针对特定癌症进行设计,例如非小细胞癌、子宫内膜子宫癌等。选择子亦可针对癌症的通用类别进行设计,例如上皮癌症(癌)、肉瘤、淋巴癌、黑素瘤、胶质瘤、畸胎瘤等。选择子亦可针对癌症亚属进行设计,例如腺癌、鳞状细胞癌等。选择子亦可包含关于多个基因组区的信息,所述基因组区包含存在于患有癌症的至少一个受试者中的一个或多个突变。例如,选择子可包含关于下列多个基因组区的信息,所述基因组区包含存在于患有癌症的至少一个受试者中的高达20个突变。在一些情况下,选择子可包含关于下列多个基因组区的信息,所述基因组区包含存在于患有癌症的至少一个受试者中的高达200个或更多个突变。选择子亦可包含关于多个基因组区的信息,所述基因组区包含存在于患有癌症的至少一个受试者中的一个或多个突变。在一些情况下,多个基因组区内的一个或多个突变可存在于来自患有癌症的受试者群体中的至少1%至高达20%或更多(例如高达95%或更多)的受试者中。测序ctdna的基因分型、检测、鉴定或定量可利用测序。可使用高通量系统完成测序。可使用诸如基因组dna、来源于rna转录的cdna或rna等本文所述的核酸所为模板来进行测序。例如,无细胞dna样品的序列信息可通过大规模平行测序来获得。在一些情况下,大规模平行测序可使用基因组的亚组进行,例如来自cfdna样品的cfdna亚组。序列信息可使用流动池通过平行测序获得。例如,可将用于扩增的引物与流动池中的载玻片共价连接,并随后将流动池暴露给用于核酸延伸和测序的试剂。高流量测序亦可包括使用获自helicosbiosciencescorp.(cambridge,mass.)的技术,例如通过合成的单分子测序(smss)法。在一些实施方案中,高通量测序包括使用获自454lifesciences,inc.(branford,conn.)的技术,例如picotiterplate装置,其包含光纤板,所述光纤板传送通过测序反应生成的化学发光信号,其通过仪器中的ccd相机记录。光纤的使用允许在4.5小时内检测最少2千万个碱基对。在一些情况下,高通量测序可以是下一代测序技术,例如使用获自illumina(sandiego,cal.)的hiseq或miseq仪器。该测序法是基于使用折返(fold-back)pcr和锚定引物在固体表面上扩增dna。所述测序可包括文库制备步骤。可将基因组dna片段化,并可将剪切末端进行修复和腺苷酸化。可向片段的5’和3’端添加衔接子。可对片段进行大小选择和纯化。测序可包括簇生成步骤。通过与连接在流动池通道表面的一群寡核苷酸杂交,使dna片段与流动池通道的表面连接。可通过桥扩增将片段延伸和克隆扩增,以产生独特的簇。片段变为双链的,且双链分子可以是变性的。固相扩增接着变性的多次循环,可在流动池的各通道中建立相同模板的单链dna分子的约1,000个拷贝的数百万个簇。可使反义链裂解并洗去。可将末端进行封闭,并可使引物与dna模板杂交。可对数亿个簇同时测序。可将引物、dna聚合酶或四种荧光基团标记的可逆终止核苷酸用于进行序贯测序。全部四种碱基均可彼此竞争模板。核苷酸掺入之后,可使用激光来激发荧光基团,捕捉图像并记录第一个碱基的身份。将来自各掺入碱基的3’终止子和荧光基团去除并重复掺入、检测和鉴定步骤。在每个循环中均可读取单个碱基。在一些实施方案中,使用hiseq系统(例如hiseq2500、hiseq1500、hiseq2000或hiseq1000)进行测序。rna或dna的高通量测序亦可使用anydot-芯片(genovoxx,germany)进行,其允许监测生物学过程(例如mirna表达或等位基因变异性(snp检测))。例如,anydot-芯片允许将核苷酸荧光信号检测增强10倍-50倍。其它高通量测序系统包括在venter,j.,等science162001年2月;adams,m.等,science242000年3月;和m.j,levene,等science299:682-686,2003年1月;以及美国专利公布号2003/0044781和2006/0078937中公开的系统。可重复核酸链的生长和添加核苷酸类似物的鉴定,以使核酸链进一步延伸并确定靶核酸的序列。本文所公开的方法可包括基于来自选择子的一个或多个基因组区进行测序反应。在一些情况下,可针对来自选择子的基因组区的亚组获得测序信息。例如,可针对来自选择子的10-500个或更多个基因组区获得测序信息。在一些情况下,可针对小于5%或高达95%的来自选择子的基因组区获得测序信息。末端修复所述方法的实施方案可包括对多个核酸进行末端修复反应以产生多个经末端修复的核酸。例如,可在将衔接子与多个核酸连接之前进行末端修复反应。在一些情况下,可在扩增衔接子修饰的核酸之前进行末端修复反应。在其它情况下,可在扩增衔接子修饰的核酸之后进行末端修复反应。在一些实施方案中,可在将多个核酸片段化之前进行末端修复反应。在其它实施方案中,可在将多个核酸片段化之后进行末端修复反应。末端修复反应亦可通过使用一种或多种末端修复酶进行。在一些情况下,用于修复dna的酶可包括聚合酶和外切核酸酶。例如,聚合酶可从5’至3’方向对dna链填补缺失碱基。所得双链dna可与起始的最长dna链一样长。外切核酸酶可去除3’突出端。所得双链dna可与起始的最短dna链一样长。加a尾所述方法的实施方案可包括对多个核酸进行加a尾反应以产生多个经加a尾的核酸。例如,可在将衔接子与多个核酸连接之前进行加a尾反应。另外,可在扩增衔接子修饰的核酸之前进行加a尾反应。在其它情况下,可在扩增衔接子修饰的核酸之后进行加a尾反应。在一些实施方案中,可在将多个核酸片段化之前进行加a尾反应。在一些情况下,可在将多个核酸片段化之后进行加a尾反应。在其它情况下,可在多个核酸的末端修复之前进行加a尾反应。在一些实施方案中,可在多个核酸的末端修复之后进行加a尾反应。加a尾反应亦可通过使用一种或多种加a尾酶进行。例如,可通过将dna片段与datp和非校正dna聚合酶一起孵育来添加a残基,其将添加单独的3’a残基。减小背景误差本文提供的方法可进一步包括减小背景误差的方法。背景误差可包括例如在扩增或测序期间并非体内发生而是人为产生的突变。背景误差突变,例如单核苷酸变化,如鸟嘌呤至胸腺嘧啶(g至t)突变,可能由pcr或测序的误差导致。这些突变在双链核酸的一条链上发生,但在另一条链上不发生。可对这些人为的g至t突变进行检测并忽略。本文亦公开的是用于减少来源于多个核酸的多个扩增子的序列中的背景误差的方法,其包括a)鉴定来自至少一个第一序列读出和至少一个第二序列读出的突变,其中来自第一序列读出和第二序列读出的突变为一致突变;b)消除在来源于单核酸的小于50%的扩增子上发生的突变;c)消除在来源于双链核酸的第一链的第一扩增子上发生的g至t突变,其中g至t突变在来源于双链核酸的第二链的第二扩增子上未发生;d)消除彼此之间相距少于100个碱基对的突变;e)消除这样的扩增子上的突变,其中扩增子的第一亚组包含第一双链主干条形码,且扩增子的第二亚组包含第二双链主干条形码,其中第一双链主干条形码与第二双链主干条形码不同;或者f)其任何组合。本文所用的术语“消除”可以指忽略来自序列信息的突变数据。减小背景误差可包括鉴定来自至少一个第一序列读出和至少一个第二序列读出的突变,其中来自第一序列读出和第二序列读出的突变为一致突变。在此情况下,突变可为真实突变,即并非背景误差。在一些实施方案中,从双链核酸的一条链的第一序列读出鉴定的核苷酸突变,与从双链核酸的相同链的第二读出鉴定的核苷酸突变一致。例如,如果突变为真实的,例如并非背景误差,则从双链核酸的一条链的序列读出鉴定的a突变(例如突变成a的核苷酸),应与从双链核酸的相同链的序列读出鉴定的a突变(例如突变成a的核苷酸)一致。在其它实施方案中,从双链核酸的一条链的第一序列读出鉴定的核苷酸突变,与从双链核酸的另一条链的第二序列读出鉴定的互补核苷酸的突变一致。例如,如果突变为真实的,例如并非背景误差,则从双链核酸的一条链的序列读出鉴定的a突变(例如突变成a的核苷酸),应与从双链核酸的另一条链的序列读出鉴定的t突变(例如突变成t的核苷酸)一致。减小背景误差可包括从2个或更多个(例如高达20个或更多个)序列读出中鉴定一致突变。被认为是序列信息中的背景误差的突变,可随机发生在各个基因座上,并因此可能不会存在于含有所述突变的基因座的所有扩增子中。含有突变的相同基因座的扩增子可通过扩增子上的预定义单链条形码、扩增子上的随机单链条形码或其任何组合来鉴定。在一些实施方案中,含有突变的相同基因座的扩增子可以是含有相同独特识别码的扩增子。在另一个实施方案中,可进行生物信息学分析以去除并非在含有相同基因座的所有扩增子中均发生的突变。背景误差可包括并非在来源于单个核酸的所有扩增子中均发生的突变。例如,减小背景误差可包括消除在小于约50%至小于约75%或小于约100%的来源于单个核酸的扩增子中发生或者以低于经试验确定的截止水平发生的突变。突变的空间接近性其为背景误差的一些突变可彼此靠近,例如以靠近的空间接近性存在。例如,减小背景可包括去除(例如忽略)彼此邻接的突变。在其它实施方案中,减小背景误差可包括消除彼此相距一个或更少碱基对或者高达100个碱基对的突变。来源于单个核酸的扩增子可包含一个或多个条形码,例如一个或多个相同的条形码(相同的随机单链条形码,或者相同的预定义单链条形码,或者相同的内源条形码)或者两个或更多个相同的条形码的组合。g至t突变有时,背景误差可包括人为鸟嘌呤(g)至胸腺嘧啶(t)突变。在一些实施方案中,减小背景可包括消除人为g至t突变。作为人为假象的g至t突变可发生在双链核酸的双链之一上。然而,并非人为假象的g至t突变,例如真实突变,将有可能发生在双链核酸分子的两条链上。因此,去除因人为因素所致的g至t突变可包括鉴定扩增子来源的亲代核酸链。条形码可用于鉴定扩增子来源的亲代链。可将一个或多个双链主干条形码与双链核酸连接。可将不同的单链条形码与双链核酸的两条链连接。在一些情况下,来源于核酸的一条链的扩增子和来源于核酸的另一条链的扩增子可包含相同的双链主干条形码,以及核酸上的一个或多个序列(例如内源条形码)。在另一些情况下,来源于核酸的一条链的扩增子和来源于核酸的另一条链的扩增子可包含不同的随机单链条形码。因此,在一些实施方案中,减小背景可包括消除发生在来源于双链核酸的第一链的第一扩增子上的g至t突变,其中g至t突变在来源于双链核酸的第二链的第二扩增子上未发生。在某些实施方案中,第一扩增子和第二扩增子可包含相同的内源条形码和相同的双链条形码,但包含来源于双链核酸的随机单链条形码的不同随机条形码。在一些实施方案中,减小背景可包括消除这样的g至t突变,其发生在来源于双链核酸的第一链的第一扩增子的部分上,但在来源于双链核酸的第二链的第二扩增子的较大部分上未发生。来源于双链核酸的第一扩增子的数量和第二扩增子的数量可使用条形码来确定。在一些实施方案中,第一扩增子可包含来源于亲代双链核酸的第一链上的第一单链条形码的第一条形码。第二扩增子可包含来源于亲代双链核酸的第二链的第二单链条形码的第二条形码。在一些实施方案中,第一条形码和第二条形码可包含不同的序列。在其它实施方案中,第一条形码和第二条形码可包含相同的序列。第一和第二扩增子的数量可通过计数第一和第二条形码来确定。本文所公开的方法可进一步包括扩增子数量的计数。在一些实施方案中,例如第一扩增子的数量可通过计数以下扩增子来确定,所述扩增子包含与亲代双链核酸相同的双链主干条形码和相同的内源条形码,以及来源于亲代核酸的第一链上的随机单链条形码的第一随机条形码。类似地,第二扩增子的数量可通过计数以下扩增子来确定,所述扩增子包含与亲代双链核酸相同的双链主干条形码和相同的内源条形码,以及来源于亲代核酸的第二链上的随机单链条形码的第二随机条形码。因此,基因座上携带g至t突变的第一扩增子的数量以及相同基因座上携带g至t突变的第二扩增子的数量,可使用条形码来确定。减小背景亦可包括消除未在双链核酸的两条链上均发生的突变。在一些实施方案中,减小背景误差可包括消除在小于约50%(或更高阈值)的包含相同双链主干条形码和相同内源条形码的扩增子上发生的突变。估算llod在一些实施方案中,本发明包括估算ctdna检测的灵敏性的步骤,如在图7(b)中所述。在一些实施方案中,所述灵敏性取决于报告基因的数量。通过利用可得的肿瘤基因组当量和肿瘤特异性报告基因(即体细胞变体)的数量的知识,以不依赖于测定法的方式估算灵敏性。所述方法利用数个肿瘤特异性报道的组合(如描述于pct/us14/25020(us20140296081)),与单个报告基因(例如dpcr)相比,其使按比例调节检测限成为可能。例如,与单个报告基因检测相比,使用8个报告基因使最低检测限(llod)减小8倍。在一些实施方案中,报告基因独立起作用。在一些实施方案中,检测方法的llod低于可得肿瘤基因组当量的数量。图15阐述了用于ctdna检测和选择子设计的统计框架。图15(a):在从10ml血液得到的典型cfdna产率内(假设约50%捕获效率),简易分析模型允许将ctdna检测限估算为可得肿瘤报告基因的函数。在图15(b)中,对于>90%检测可能性,ctdna的检测限显示为可得肿瘤报告基因和测序ge的函数。在图15(c)中,存在于肺腺癌(luad)肿瘤的编码区中的邻接体细胞突变之间的距离,通过癌症基因组图谱(tcga)进行谱分析(n=381)。垂直虚线指示cfdna分子的长度中位数。图15(d)显示实测的不同肿瘤报告基因数量与利用预处理nsclc血浆和来自先前研究的确定掺入物(spike-ins)通过分析建模预测的数量之间的一致性。图15(e)在tcga训练组(luad)和独立的肺腺癌组二者中分析nsclc选择子覆盖的每个肿瘤中的snv和插入缺失(indel)的数量。图15(f)通过capp-seq谱分析的在tcga训练数据集和验证组之间,各选择子区块在捕获nsclc患者上的重现性。复发指数等于每千碱基覆盖的独特患者的百分数。在一些实施方案中,本发明为使用改进的最低检测限(llod)将循环肿瘤核酸(例如ctdna)基因分型的方法。在一些实施方案中,本发明为在患者中评价癌症的方法,其通过使用靶向0.01%或更小(低至0.0001%)llod的选择子设计(即,具有数量足以实现llod0.01%或更小(低至0.0001%)的报告基因的选择子设计)将来自患者的ctdna基因分型来进行。在一些实施方案中,本发明进一步包括将通过本领域已知的任何方法获得的来自患者的匹配肿瘤样品进行基因分型,所述样品例如外科手术样品或细针活检;冷冻样品和福尔马林固定标本。在一些实施方案中,本发明进一步包括将来自患者的非肿瘤样品(例如外周血白细胞)基因分型,以确认临床上确定的变体的肿瘤关联。本发明人发现的是,许多癌症被至少一个共有突变覆盖。在一些实施方案中,根据本发明的方法使用一种类型的肿瘤设计选择子并将其用于不同类型的肿瘤,如在图8中所述。去除重复“去重”在一些实施方案中,本发明为对ctdna测序的方法,其包括通过将共享相同独特分子id(uid)的分子分组到多个家族来减小背景误差的步骤。该步骤确定了起始测序分子的数量(作为共享uid的家族的数量)并消除了未被该家族所有成员共享的误差。这些误差可通过氧化核苷损伤、pcr和在离体拷贝或加工靶分子期间的其它外源性来源而引入。通过uid将分子分组并评价误差的方法在本文中称为“去重(deduping)”。使用不同条形码的误差消除在一些实施方案中,本发明为通过使用分子条形编码消除误差以分析ctdna来评价癌症的方法。在此方法的改动中,本发明为使用分子条形编码在将来自患者的ctdna进行基因分型中消除误差的方法。在此实施方案中,根据本文所述的方法,从受试者例如患者中获得并制备ctdna用于测序。例如,可从受试者提取低至30ng的ctdna并测序至1000-10,000倍(例如6,000倍)的深度。然后可如本文所述使用uid将序列“去重”。为了将来自分子条形编码的误差消除基准化,本发明人对来自12个健康成年人的cfdna样品进行谱分析。对于各受试者,对一致的dna输入(中位数32ng)测序至约6,000倍的深度中位数(在去除重复之前)。考虑到在临床血浆样品中通常较低的cfdna产率,可使用全部回收分子来评价误差率和条形编码性能(即不论uid拷贝数或成链情况(strandedness))。在一些实施方案中,基于评价误差校正方法,本发明的方法包括将未去重数据进行比较以评价内部(插入)或外部(衔接子)分子条形码是否使全选择子背景大量减小。在一些实施方案中,所述方法包括使用内部(插入)uid用于误差消除或条形码去重策略。误差定式化在一些实施方案中,本发明为在对包含ctdna的核酸测序中减小背景的方法,其通过鉴定误差并去除来自以下基因组位置的核苷酸变化来进行,所述基因组位置在诸如种群研究等先前测序研究中确定为易错的。在一些实施方案中,易错位置为g(鸟嘌呤),且去除的核苷酸变化为t(胸腺嘧啶)。本发明人观察到,在健康供血者中,易错位置在其基因组定位和碱基置换图谱中为显著定式化的(图9(b)和图10)。即便在使用条形码去重消除误差之后,大多数剩余误差仍由这些定式化低频等位基因(<0.1%)组成,大部分因g>t颠换所致,以及较低程度为c>t或g>a转换(图9(b))。不受特定理论束缚,本发明人假定的是,在文库制备期间核酸的氧化损伤可能发生,导致8-氧鸟嘌呤形成和胞嘧啶脱氨。本发明人进一步观察到,当映射到参照人基因组的相反(正)链时,与交互的c>a事件相比,高度倾向于g>t变化(图9(b)),且此失衡并非由测序链偏移所致(图11(b))。因此本发明人检验了富集步骤,并且鉴定出在g>t误差与c>a误差的比率上的梯度增加,其在介于0.1天时长和3天时长的靶向序列捕获持续时间内重现地增加2.5倍(图11(c))。对于仅在两条dna链中见到的误差亦观察到类似趋势(仅双链体数据,图11(d))。数据显示,与仅靶向正链的捕获试剂耦联的活性氧物质,在很大程度上驱动g>t颠换(图11(e))。在一些实施方案中,本发明包括在核酸测序中减小背景误差的步骤,其通过使样品与一种或多种核酸修复酶接触以从样品的核酸中去除受损核酸来进行。所述修复酶的实例包括(i)尿嘧啶dna糖基化酶(udg),其留下替代尿嘧啶的无碱基位点(胞嘧啶氧化产物),阻止pcr继续通过氧化位点,消除因胞嘧啶氧化所致的c>t误差;(ii)8-氧鸟嘌呤dna糖基化(fpg),其去除受损嘌呤并在受损碱基位点切割,消除因鸟嘌呤氧化所致的g>t误差。通过应用这些酶减小误差率,阐述于图11(a)。用于误差消除中变体识别(calling)的阈值单分子测序误差率为不均匀的,其在靶基因组间隔之间和在碱基置换类型之间在量级上不同。此外,在样品之内和在样品之间的测序深度通常不同。总之,这些问题使选择用于变体识别的稳健阈值复杂化,导致在灵敏性和特异性之间的次优权衡。为了改进低频等位基因的检出率,我们开发了适当考虑背景误差率中的局部变差和总体变差的通用基因分型方法,其使自动确定各样品中的位置特异性变体识别阈值成为可能。所述方法包括针对具有变体(snv)的测序读出的最小数量确定阈值t,以将变体鉴定为真实变体而非人为假象。在一个实施方案中,本发明包括使用对照样品确定对于各类碱基置换的总体误差率的步骤。在一些实施方案中,针对24个可能的碱基置换确定总体误差率。在一些实施方案中,阈值设定步骤将具有>5个支持读出的候选序列变异排除在外,以使真实变体的混杂影响最小。考虑到碱基置换类别具有不同的背景分布(图2b),我们试图针对各种类别分别控制假阳性率。为此,我们针对各碱基置换类别对背景误差的累积分布建模。本发明人发现,幂级数和指数函数较好地拟合实测数据(图19(a)),且针对各种类别,我们使用对数线性空间中的线性回归选择最佳捕获数据的函数。为了增加灵敏性,我们对含和不含第二核酸链支持的候选序列变体分别建模,每个样品有总计24个碱基置换模型(2条链×12个置换类别)。所述模型容易地阐述背景修正对置换特异性误差率的影响(图19(a))。所述方法进一步包括定义将误差数与实测支持读出数相关联的函数的步骤。在一些实施方案中,针对试验参数调整误差数,例如,每个碱基数量测序的“累积误差”。所述方法进一步包括对24个函数的每一个求解的步骤,以鉴定得到y累积误差所需的最小支持读出数t。在一些实施方案中,y=0。(图19(a))。在一些实施方案中,本发明进一步包括如上所述调整阈值t。在一个实施方案中,本发明包括基于局部误差率和测序深度调整总体误差率的步骤。所述步骤包括针对各基因确定误差率e(定义为含有非参照碱基的位置数量除以测序碱基的数量)并确定测序深度d。(图19(b))。如果基因g落入全选择子基因水平误差率的前25%内,则根据下式将阈值t调整为t’:·t’←t×w,其中w=min{q2,5}且q=e除以所有可评价基因的误差率的第75百分位数。如果基因g具有小于全选择子测序深度中位数dmed的测序深度,则根据下式将阈值t调整为t’:·t’←t/w*,其中w*=ln(dmed/d)在一些实施方案中,本发明为以减小的误差率对包括cfdna的核酸测序的方法,其包括鉴定为真实snv序列变体,所述变体超出经试验建立的阈值(即超出阈值的变体读出数),其中阈值如上所述确定。去除低频等位基因在一些实施方案中,本发明包括应用启发式过滤来检测和去除具有较低等位基因频率的snv的步骤。(图19b)所述步骤包括建立候选snv列表,根据等位基因频率将snv分级并去除最低频snv。在一些实施方案中,所述方法包括确定用于将误差与真实snv区分开的阈值点的统计学分析。所述方法可包括将列表分成至少两个部分,统计学评价两个部分之间的方差差异,得到p值和最小p值。通过渐增的af将l分级之后,将迭代器i用于遍历列表。对于各i,将l分成两部分,af小于li的snv和af≥li的snv。使用双向f检验来统计学评价两个列表之间在方差上的差异,得到p值。然后以渐增af的顺序遍历小于最小列表l的snv,以鉴定对应于局部最小值(如果存在)的第一p值的指数i*。所述最小值(如果检出)指示噪声(左尾)和信号(较高af)之间的可能拐点。如果对应于i*的p值小于0.05且如果li比li-1大至少10%,则我们随后使用单边z检验(针对snvaf调整实测的给定正态性)评价li和可能背景事件分布之间的差异(l1至li-1)。如果对应的p值<0.01,则将候选snv列表拆分并去除左尾(l1至li–1)。在经验分析中,发现该程序改进特异性(未显示数据),表明其可有效检测残留的背景变体。集成数字误差消除(ides)在一些实施方案中,本发明为消除位置特异性测序误差的计算方法。在此实施方案中,所述方法包括获得序列变异的基线分布以针对单核苷酸变异(snv)设置经试验确定的阈值,接着对经条形码去重的数据进行“计算机(insilico)修正”(消除等位基因分数落入阈值之下的变异)。在此实施方案中,本发明包括在包含ctdna的核酸测序中减小误差的方法,其包括将共享相同独特分子id(uid)的分子分组到各家族并将频率低于预定阈值的核苷酸变化作为误差消除。可在诸如种群研究等测序研究中建立阈值。所述方法阐述于图9。图9(a)为描述与假定双链(双链体)dna分子连接的串联测序衔接子的示意图,所述dna分子在两条链中均含有真实生物突变,以及仅在一条链(上)中含有未复制的不对称碱基变化。内部/插入条形码的应用允许(i)误差消除和(ii)回收单链(中)和双链(下)dna分子。图9(b)(上图)显示热图,描述了分配到所有可能碱基置换(行)中并通过来自健康对照的12个cfdna样品之间减小平均等位基因分数(针对各置换类型)来组织(列)的位置特异性全选择子误差率。对于未去重数据(左)、条形码去重数据(中)和背景修正之后的条形码去重数据(右),显示背景图谱。误差定义为将生殖系snp除外的非参照等位基因。深色表示无背景,浅色表示0.03%丰度分数上限。图9(b)下图:全选择子误差度量。图9(c)显示对于分为训练组(n=12)和测试组(n=18)的30个正常对照cfdna样品,条形码去重和背景修正对全选择子误差度量的影响。显示了中位值和四分位距。(d)对于各碱基置换的全选择子llod的密度图。图9(e)(上图)显示对于所有可能碱基置换的全选择子检测限的直方图。图9(e)(下图)显示对于上述直方图中各单元(bin)的碱基置换类型的分布。饼形图显示对于来自与我们的nsclc选择子交叉的癌症体细胞突变目录(cosmic)数据库的频率为前1%变体的单碱基变化的分布。图9(f)显示30个cfdna对照样品中将ides与不同的条形编码去重策略进行比较。图9b-f中的所有分析均使用nsclc临床选择子进行。本发明包括通过以误差消除获得的低误差率对ctdna基因分型来评价癌症,所述误差消除包括“计算机修正”经条形码去重的数据,消除等位基因分数小于位置特异性阈值的变体(图9(b))。本发明人将计算机修正和分子条形编码的组合应用称为“集成数字误差消除”(ides),以分别减小或消除定式化误差和随机误差的影响。在一些实施方案中,全选择子误差率降至1.5×10-5,且无误差位置增加至约98%。在此实施方案的改动中,所述方法仅包括对去重或未去重的cfdna样品进行背景修正(即在使用或未使用条形码的情况下进行修正),或者仅进行条形码去重而不修正。然而,注意的是,当一起进行时,两种方法协同作用。(图10)。在一些实施方案中,本发明为用以横跨人类基因组的大量区域表征等位基因特异性检测限的误差概况的分析,所述区域在癌症基因组中为复发突变的。本发明包括分析12种可能的核苷酸置换类别,以确定其误差率、背景和llod。本发明进一步包括确定具体的置换类别是否描述于癌症体细胞突变目录(cosmic)中(图9(d))。如在本文中所述,所有可能snv的近80%被无误差检出(图9(e))。在剩余误差中,g>t检测最值得注意,其仍保持横跨选择子约0.3%的合理的低llod中位数(图9(d))。此外,g>t变化,其包含大部分检测限>0的等位基因,仅包含覆盖我们的nsclc选择子的经cosmic注释的5%的突变热点,表明对基因分型性能的最小影响(图9(e))。在一些实施方案中,本发明为通过以减小的误差率和最大灵敏性对患者cfdna测序来评价患者病况的方法,所述方法包括使用双链体分子(如果可得)的步骤,但另外使用单链分子获得经背景修正的测序数据。在一些实施方案中,本发明为使用集成数据误差消除(ides),经由对肿瘤的无活检基因分型来评价患者状态的方法。所述方法可包括首先评价ides对体细胞改变(snv和插入缺失)的作用的步骤,所述体细胞改变在癌症中为高度复发且临床上相关的。可通过将基因分型应用于来自健康受试者的cfdna来进行评价。具有双链体支持的识别表明体内发生的真实的变体等位基因,而条形编码的未去重数据指示假阳性识别。本发明可进一步包括模拟ctdna的基准评价,其使用掺加到对照cfdna中的突变dna的确定输入进行。如果经掺加dna中的突变为已知的,则本发明可包括评价用于检测罕见变体的灵敏性、特异性、阳性预测值和阴性预测值以及假阳性率。在一些实施方案中,本发明为用于从血浆对患者进行无活检基因分型的集成数据误差消除(ides)的方法。所述方法可包括确认突变在匹配的肿瘤活检中为体细胞突变。例如,本发明为在例如nsclc患者(例如(ib-iv)期)中评价癌症的方法,其包括以低误差率非侵入性评价肿瘤基因型。在一些实施方案中,基因分型包括检测egfr突变,包括在激酶结构域(外显子19、外显子20和21)中的突变,其包括激活突变和抗性突变。在一些实施方案中,本发明为用于从患者血浆样品对nsclc患者进行无活检基因分型的误差消除(ides)的方法,其中患者肿瘤的基因型为已知的。在一些实施方案中,本发明包括通过对cfdna基因分型来研究患者肿瘤内的克隆动态。在另一个实施方案中,本发明为对于检测响应或耐受埃罗替尼的可能性或者耐受埃罗替尼的机制进行无活检基因分型的方法。在另一个实施方案中,本发明为根据本文所述的低误差方法,通过对来自血浆样品的cfdna进行基因分型,在受癌症所累的患者中进行预后或检测疾病进展的方法。在一些实施方案中,本发明为根据本文所述的低误差方法,通过分析患者血浆样品中的cfdna来检测对疗法(例如埃罗替尼)的耐受性的机制。双链测序在一些实施方案中,本发明为分析核酸的方法,其包括将测序读出配对以获得双链(双链体)序列的步骤。此实施方案中的第一步为读取核酸序列以确定条形码。在一些实施方案中,两条链上的条形码彼此互补(例如,如果uid位于衔接子的主干区)。在其它实施方案中,两条链上的非随机条形码交叉引用为位于相同衔接分子的单链上。在又另一个实施方案中,条形码为外部和内部条形码的组合。将所述序列配对的第一步为读取和匹配短的(2个或更多个碱基对)外部条形码。其次,亦对一部分插入序列测序以确定插入序列的基因组坐标。如果短条形码彼此互补且插入序列的基因组坐标映射到相反链,则读出代表双链体分子的交互链。在一些实施方案中,本发明为分析核酸的方法,其包括使用条形码消除误差的步骤。所述方法包括将序列映射到参照基因组并鉴定所有单核苷酸变体(snv)(即与参照序列不同的碱基)的步骤。所述方法进一步包括使snv经历质量过滤的步骤。在一些实施方案中,质量过滤为使用阈值q为30的phred质量过滤,其消除由测序人为因素引起的99.9%的误差。所述方法进一步包括减小误差的步骤,其通过计数各基因组位置的snv数量(经历并通过了前述步骤中的质量过滤)并选择最大丰度变体来进行。所述方法进一步包括从定义为共享相同uid的条形码家族的序列组减去snv未通过质量过滤的序列的步骤。所述方法进一步包括将条形码家族的所有成员合并到单个序列中的步骤,仅保留通过步骤3的具有≥2个成员的变体。作为最终误差消除步骤,消除单元素集(singleton)条形码家族(即具有一个序列的家族)中的所有非参照变体,除非得到来自至少另一个具有≥2个家族成员的dna分子支持所述变体的证据支持。我们称此去重策略为“2x+单元素集”(图9(f))。评估肿瘤负荷在一些实施方案中,本发明为通过对患者cfdna中的双链体分子测序来确定患者中的肿瘤负荷的方法。考虑到双链体测序的优秀误差率,我们试图确定其对于定量循环肿瘤负荷的llod。所述方法包括设计覆盖足够数量的(例如>1,500个)序列变异例如非同义突变的选择子的步骤。选择子可通过任何测量法设计,例如肿瘤的外显子组测序。在一些实施方案中,可通过对患者肿瘤进行外显子组测序来设计个性化选择子。所述方法进一步包括对患者cfdna进行双链体测序的步骤。在一些实施方案中,在此步骤中可回收少至1,000基因组当量。如通过实施例所证实,所述方法能够以高线性度精确检出从0.025%至0.00025%的靶序列(图12(i)),其进一步验证了我们的分析模型(图13)并证实在每1,000,000个分子背景中检出2.5个分子的检测限,是单个等位基因dpcr的llod的近1/100。这表明个性化选择子为用于深度监视显微镜下残留疾病、特别是用于具有高突变负荷的肿瘤的有前景的方式。总之,本文所公开的方法提供用于超灵敏ctdna谱分析的稳健且灵活的框架,其使用检测限的精确分析模型、集成数字误差消除和自适应cfdna基因分型来实现。通过将用于误差减小和有效双链体链回收的实际分子条形编码执行与用于去除残留背景误差的普遍适用方式组合,ides使误差率降低10-100倍而不会损失回收的基因组产率(图7(a))。与用于具有有限dna含量(例如临床上实际的血液采集量)的样品的先前方法相比,这些特性改进了检测限(图7(b))。此外,通过借助双链体链回收,当可得到足够的肿瘤报告基因和ge时,可将我们的方法进行调整以得到优越的灵敏性。考虑到其对于ctdna谱分析的优点,我们预期的是,对于需要对低频等位基因精确数字定量的多种深度测序应用而言,ides将具有广泛实用性。使灵敏性最高同时使测序成本最小循环dna具有变革例如癌症等疾病的鉴定和监测的潜力,但其在大部分患者的血液、血清和/或血浆中的检测仍较昂贵且具挑战性。本文的公开内容包括可将超深度测序与新型生物信息学方法组合以实现高灵敏性和特异性以及非侵入性评价绝大多数患者中的循环dna的新型经济方法。在一些实施方案中,所述方法可用于任何癌症类型。癌症可以是实体瘤,例如非小细胞肺癌(nsclc)。所述方法亦可包括设计和验证用于这些恶性肿瘤的每一个的选择子(约200kb),其通过将公开可得的体细胞突变数据的生物信息学分析与临床上相关的基因组区和断点热点的现有知识组合来进行。本文亦公开的是用于合理设计选择子以实现所需ctdna检测限,并通过实证掺加试验来验证建模的方法。例如,在一些实施方案中,本文所公开的方法对于所有癌症可实现每50,000个分子中至少检出1个的检测限,所述癌症例如nsclc、食管腺癌(eac)/食管鳞状细胞癌(escc)和胰腺癌(paad),给定约30ngcfdna输入,50%捕获效率(约3ml血浆)。在实施方案中,所述方法包括设计使灵敏性最高同时测序成本最低的算法,其基于(i)在各肿瘤中鉴定的报告基因数量,(ii)输入血浆dna质量,(iii)预计的dna复制率和捕获效率,以及实证推导的ctdna水平和肿瘤体积之间的关系(仅nsclc)。在一些情况下,所述方法可包括通过使用从跨越多种阶段、肿瘤体积和疗法的多个nsclc、eac/escc和paad患者收集的匹配肿瘤活检分析纵向血浆样品,来验证选择子(elector)。本文亦公开的是确定所需双链核酸的量和流动池数量的方法。所述方法可包括:a)确定选自以下的参数:i)选择子中寡核苷酸的大小;ii)癌症报告基因的数量;iii)总基因组当量;和iv)其任何组合;b)将算法应用于a)中所述参数,以确定所需双链核酸的量和流动池数量;和c)调整双链核酸的量和流动池数量以优化成本。在一些实施方案中,癌症报告基因可包含癌症突变。例如,癌症报告基因可以是相对于生殖系序列的变化,例如癌细胞特异性变化。在其它实施方案中,癌症报告基因可包括单核苷酸变体(snv)、拷贝数变体(cnv)、插入、缺失和重排(例如融合)。实施例实施例1:使用分子条形编码减小背景误差将条形码用于消除因背景误差所致的突变,所述误差例如由pcr和测序导致的误差。从患者血样中分离无细胞dna。使cfdna经历末端修复和加a尾(图1(a))。cfdna的起点坐标和终点坐标上的序列形成用于各cfdna分子的独特内源条形码。然后通过t4dna连接酶将y形衔接子与各cfdna分子的两端连接,所述衔接子包含可杂交和不可杂交部分(图1(b))。y形衔接子的不可杂交部分包含四碱基随机条形码和四碱基固定条形码,其中两个条形码彼此邻接(图1(c))。y形衔接子亦包含可与测序引物结合的通用序列(图1(c))。随机条形码和内源条形码的组合用作各所得模板核酸的独特识别码。固定条形码为预定义的,以鉴定cfdna的来源,例如cfdna从其中分离的患者或组织。所得模板核酸通过12-14个pcr循环扩增。然后通过下一代测序获得扩增子的序列信息,例如使用illuminahiseq2000。序列信息包含在体内发生的癌症相关的突变,例如真实的生物学变体。然而,序列信息亦包含因pcr、测序或其它人为因素引入的误差所致的突变(图1(d))。在此实施例中,由于各模板核酸分子通过独特识别码鉴定,所以来源于相同模板核酸的扩增子包含相同的独特识别码并因此进行分选。作为真实生物学变体的突变发生在相同的基因座上,而因背景误差所致的假突变随机发生在不同的基因座上。此外,因背景误差所致的突变发生在来源于相同模板核酸的扩增子的亚组中。基于独特识别码将来源于相同模板核酸的扩增子进行比对,并进行生物信息学分析以滤除因背景误差所致的突变,例如假突变,其随机发生在不同的基因座上,或者发生在来源于相同模板核酸的扩增子的亚组中。将假突变滤除之后,序列信息中的突变检测的灵敏性从0.02%增加至0.001%(图1(e-g)),例如每99,999个正常细胞当量背景中检出1个肿瘤细胞当量。实施例2(预示性的):使用分子条形编码去除g至t突变从患者血样中分离无细胞dna。使cfdna经历末端修复和加a尾(图2(a))。cfdna的起点坐标和终点坐标上的序列形成用于各cfdna分子的独特内源条形码。然后通过t4dna连接酶将y形衔接子与各cfdna分子的两端连接,以产生多个模板核酸,所述衔接子包含可杂交和不可杂交部分(图2(b))。y形衔接子的不可杂交部分包含四碱基的随机条形码和四碱基的固定条形码,其中两个条形码彼此邻接(图2(c))。随机条形码和内源条形码的组合为用于各模板核酸的独特识别码。固定条形码为预定义的,以鉴定cfdna的来源,例如cfdna从其中分离的患者或组织。此外,y形衔接子亦包含2碱基对的独特主干条形码(图2(c))。连接之后,主干条形码与连接位点相距2个碱基对。所得模板通过12-14个pcr循环扩增。然后通过使用illuminahiseq2000的下一代测序获得扩增子的序列信息。序列信息包含在体内发生的癌症相关的突变,例如真实的生物学变体。然而,序列信息亦包含因pcr、测序或其它人为因素引入的误差所致的突变,例如g至t突变。因背景误差所致的g至t突变发生在模板核酸的一条链上,但在另一条链上未发生。因此,为了减小背景,忽略在模板核酸的一条链上发生而在另一条链上未发生的g至t突变。使用双链主干条形码鉴定扩增子来源的链。来源于模板核酸的扩增子包含相同的主干条形码和相同的内源条形码。此外,来源于模板核酸的不同链的扩增子包含不同的随机条形码(图2(c))。基于主干条形码、内源条形码和随机条形码,将来源于相同模板核酸的扩增子进行比对。比对的扩增子包含具有主干条形码、内源条形码和随机条形码的第一组扩增子,和具有相同主干条形码、相同内源条形码但不同随机条形码的第二组扩增子。然后忽略g至t突变,如果其发生在99.9%的第一组扩增子中,但发生在小于1%的第二组扩增子中。实施例3(预示性的):使用双链主干条形码和内源条形码减小背景误差从患者血样中分离cfdna,接着进行末端修复并使其经历加a尾。cfdna的起点坐标和终点坐标上的序列形成用于各cfdna分子的独特内源条形码。然后通过t4dna连接酶将y形衔接子与各cfdna分子的两端连接,所述衔接子包含可杂交和不可杂交部分。各y形衔接子包含可与测序引物结合的通用序列。固定条形码的序列为预定义的,以鉴定cfdna的来源,例如cfdna从其中分离的患者或组织。各y形衔接子的可杂交部分包含与连接位点相距2个碱基对的双链主干条形码。所得连接的模板核酸通过12-14个pcr循环扩增。然后通过使用illuminahiseq2000的下一代测序对扩增子测序。序列信息包含在体内发生的癌症相关的突变,例如真实的生物学变体。然而,序列信息亦包含因背景误差所致的突变,例如由pcr、测序或其它人为因素引入的假突变。连接之后,将各双链cfdna片段与双链主干条形码连接。来源于双链cfdna的一条链的扩增子和来源于另一条链的扩增子包含相同的双链主干条形码和相同的内源条形码。真实的生物突变发生在含有基因座的所有扩增子的相同基因座上。假突变包括发生在仅来源于双链cfdna的两条链之一的扩增子上的突变。因此,假突变可发生在约50%的具有相同主干条形码和相同内源条形码的扩增子上。在此实施例中,基于主干条形码和内源条形码将扩增子进行比对。滤除仅发生在约50%的扩增子上的假突变。序列信息中的突变检测的灵敏性通过滤除假突变来增加。实施例4:非侵入性超灵敏检测来自实体瘤的循环dna我们开发了一种经济的新方法,其将超深度测序和新型生物信息学方法组合以实现高灵敏且高特异性的非侵入性评价绝大部分患者中的ctdna。我们将所述称为capp-seq的方法(图4)应用于三种主要的实体癌症类型,非小细胞肺癌(nsclc)、食管腺癌(eac)/食管鳞状细胞癌(escc)和胰腺癌(paad)。通过将公开可得的体细胞突变数据的生物信息学分析(表1)与临床上相关的基因组区和断点热点的现有知识组合,我们设计和验证了用于这些恶性肿瘤的每一种的capp-seq选择子(约200kb)(图5(a))。我们开发了合理设计选择子以实现所需的ctdna检测限的分析模型,并通过经验掺加试验验证了我们的建模(图5(b))。在此基础上,对于全部三种癌症,我们预期实现每50,000个分子至少检出1个的ctdna检测限,给定约30ngcfdna输入,50%捕获效率(约3ml血浆)。基于(i)在各肿瘤中鉴定的报告基因数量,(ii)输入血浆dna质量,(iii)预计的dna复制率和捕获效率,以及经验推导的ctdna水平和肿瘤体积之间的关系(仅nsclc),我们设计了使灵敏性最高同时测序成本最低的算法(“通道平衡算法”)(图5(c))。通过使用从跨越多种阶段、肿瘤体积和疗法的多个nsclc、eac/escc和paad患者收集的匹配肿瘤活检分析纵向血浆样品,来验证capp-seq选择子(表2)。表1.体细胞突变源数据pmid作者,年方法患者数量疾病-tcgawes381luad22960745tcga,2012wes176lscc22980975imielinski等,2012wgs24luad22980975imielinski等,2012wes148luad总数553/176luad/lscc23525077dulak等,2013wes149eac22877736agrawal等,2012wes12/11escc/eac总数161/11eac/escc未公布的tcgawes59paad总数59paad表2.患者详情患者id原发位点性别确诊年龄组织学阶段肿瘤体积(cc)血浆样品数量snv和插入缺失数量吸烟史治疗和末次随访(如有)lup2肺m61大细胞iii429重度手术,化放疗;完全应答lup6肺m55腺iv28无cis/pem;疾病进展lup18肺m56腺iia150.5225轻度放射治疗;完全应答lup19肺f81小细胞ib43.228重度放射治疗;完全应答lup20肺f80腺ib33.549无放射治疗;疾病进展lup21肺f78nscib24.946吸烟者放射治疗;完全应答lup22肺f46腺iv10.4252无tkico-1686;疾病轻微进展lup23肺m89腺ia9.9752重度tbdlup24肺f68腺iib8.344重度放射治疗;完全应答lup25肺m45nsciiib57.674无埃罗替尼,羟氯喹和co-1686;疾病进展lup26肺f57腺iiib86.4433重度化放疗,辅助化疗,完全应答ep1食管m67腺20.458调节强度的放射治疗ep2食管f68腺45.9416调节强度的放射治疗ep3食管m78鳞状42.754质子疗法ep5食管m64腺96.755诱导化疗;质子疗法ep7食管m55腺19.356质子疗法ep8食管m69鳞状13.956质子疗法ep9食管m60鳞状15.253调节强度的放射治疗ep10食管m57腺23.1415质子疗法ep11食管m59腺70.155诱导化疗;调节强度的放射治疗ep12食管m54腺208.3512调节强度的放射治疗pp1胰m66腺iv64.7345化放疗;疾病进展pp2胰m65腺iib(vsiii)112.8232化放疗pp3胰m65腺iib250.820手术,化放疗;稳定1.25年,化疗后进展pp4胰m58腺iib(vsiv)84.712化放疗;疾病进展pp5胰f76腺iii335.822化放疗;疾病进展pp6胰m56腺iib236.930手术和化疗;疾病进展pp7胰m30腺iii62.617化放疗;疾病进展pp8胰m68腺iib14.110手术,化放疗;疾病进展pp9胰m59腺iii152.352化疗;疾病进展pp10胰f64腺iv25.7413化疗;疾病进展pp11胰f74腺iia55.313手术,化放疗;无疾病迹象->失访pp12胰m66腺iii150.2412化放疗;疾病进展通过将驱动基因、抗性突变和断点热点的现有知识与复发突变(非)编码区的生物信息学选择(例如通过计算复发指数)整合,设计了三种实体瘤capp-seq选择子:i)nsclc选择子(203kb),其覆盖近100%的nsclc肿瘤,具有每个患者中8个突变的中位数;ii)eac/escc选择子(180kb),其覆盖近100%的eac/escc肿瘤,具有每个患者中7-8个突变的中位数;iii)paad选择子(185kb),其覆盖约85%的胰腺癌肿瘤,具有每个患者中8-11个突变的中位数。设计和验证了用以在给定肿瘤报告基因数量和测序基因组当量的血浆中预测ctdna检测限的分析模型。使用优化的泳道上样,过度测序(例如成本)可达到最小,同时对于临床上不同组的nsclc、eac/escc和paad患者的血浆中的ctdna检测限可以达到最大。实施例5.患者和样品患者选择。本研究中的所有患者样品均以知情同意用于研究用途来收集,并依照赫尔辛基宣言经stanford机构审查委员会批准。血液收集和加工。将血液吸入bdvacutainer紫盖10mlk2edta管(bectondickinson,franklinlakes,nj目录号366643)中。以1800xg将管离心10分钟,然后将血浆以1-2ml等份移入1.5或2ml管中,然后于-80℃冷冻直至cfdna分离。将少量血浆与血沉棕黄层和红细胞沉淀混合,然后转移到1.5ml管中,将管于-80℃冷冻直至生殖系dna分离。dna分离。根据制造商说明书,使用qiaamp循环核酸试剂盒从(qiagen,valencia,cal.)血浆样品中分离cfdna。根据制造商说明书,使用qiaampdna微量试剂盒(qiagen)从血液的细胞级分中分离生殖系dna。对于从ffpe肿瘤样品分离dna,首先,使用具有leica窄面一次性刀片的leicarm2155轮转切片机收集10um(手术标本)或20um(细针抽吸物)的4个切片。在此之后,根据制造商说明书,使用qiagenallprepdna/rnaffpe试剂盒从切片中分离dna。分离之后,使用qubitdsdna高灵敏性试剂盒(lifetechnologies,grandisland,ny)定量dna。基因组dna的剪切。在文库制备之前将生殖系dna和来自ffpe的dna进行剪切以达到约170-250个碱基对的中值大小。如果可能,使用100-2000ng的dna作为输入用于剪切,但是capp-seq以更少量的dna成功进行。使用水或trisedta缓冲液ph8.0将输入dna稀释成120ul。使用下列条件,用covariss2超声波仪和covaris微量管(covaris,woburn,mass.目录号520045)进行剪切:10%工作循环,强度水平5,每次脉冲(burst)200个循环,120秒持续时间。剪切之后,使用qiaquickpcr纯化试剂盒(qiagen)纯化dna,并用50ul缓冲液eb洗脱。超声处理之后,使用qubitdsdna高灵敏性试剂盒对dna定量,并使用agilentbioanalyzer高灵敏性dna试剂盒(agilent,santaclara,cal.,目录号5067-4626)验证一些样品的大小分布。捕获前测序文库的制备。用作文库测序而输入的dna的量,根据dna输入的类型(cfdna、生殖系dna或ffpe肿瘤dna)、在一个测序通道中多路复用的样品数量和分离dna的量而不同。通常,对于在illuminahiseq2000高输出通道中12路复用的cfdna样品,输入最多32ngcfdna,而对于24路复用的illuminahiseq2000高输出通道,将高达100ng生殖系dna或肿瘤dna用于输入。对制造商的方案进行一些修改,使用kapaltplibraryprep试剂盒(kapabiosciences,wilmington,mass.)进行测序文库制备。用水或eb将dna稀释成50ul,然后根据制造商说明书进行末端修复和第一次agencourtampurexp(beckmancoulter,pasadena,cal.)珠粒纯化。基本上根据制造商说明书进行加a尾和第二次ampure珠粒纯化,不同之处在于在30℃孵育之后增加70℃下孵育5分钟以灭活加a尾酶。使用相对于输入dna样品100倍摩尔过量的衔接子,以16℃孵育过夜进行连接。连接之后,使用严格ampure珠粒纯化分离dna,并洗脱到24ul的水或eb中。使用kapahifi和illumina通用引物(各引物的终浓度为2um),以60℃30秒的退火步骤和72℃30秒的延伸步骤进行pcr。将pcr循环数优化以进行最小数量的循环,预期其对于整个通道产生3000ngdna(即,如果样品将用于1/12的通道,则进行最小预期数量的pcr循环以获得250ng)。通过一轮或两轮ampure珠粒纯化来纯化pcr并洗脱到60ul水中。通过qubit定量文库并使用agilentbioanalyzer高灵敏性dna试剂盒确定文库的大小分布。如果衔接子二聚体以大于5%的摩尔比率存在,则进行另一次严格ampure纯化来将其去除。如果存在少于所需的dna,则进行更多的pcr循环,接着一次或两次严格ampure纯化。基于杂交从测序文库中富集特异性序列。对于基于杂交富集特异性序列,使用自定义设计的生物素化dna寡聚物合并物(pool)。合并物作为定制seqcap试剂由nimblegen提供,将后用水稀释10倍,等分成4.6ul等份,将其储存于-20℃直至使用。将测序文库合并成1500ng的总质量,然后移出500ng用于后续qc。向另外的1000ng加入5ul1mg/ml人cot1dna(invitrogen,sandiego,cal.)和1mmxgen通用封闭oligo-ts-p5和xgen通用封闭oligo-ts-p7(8个核苷酸)(idt)各自1ul。对于各1000ng样品,将4.6ul选择子等份融化并预热至47℃。根据制造商说明书,使用nimblegenseqcapez杂交和洗涤试剂盒(nimblegen,madison,wisc.)进行杂交和纯化。洗涤之后,用96ul水使珠粒悬浮,并分到4个50ulpcr反应中,所述pcr反应使用kapahifi聚合酶和illumina通用引物(2um终浓度)进行。进行15个pcr循环,将4个反应合并,使用qiaquickpcr纯化试剂盒分离dna。杂交之后评价文库质量和富集。使用qubitdsdna高灵敏性试剂盒对测序文库进行定量。然后使用agilentbioanalyzer高灵敏性dna试剂盒确定文库的大小分布和摩尔浓度。为了评价富集,使用7个扩增子对20倍稀释的终样品和20倍稀释的捕获前合并文库一式三份进行qpcr:nimblegen内参序列nsc1-4、alk1内含子19、kras外显子2、eif2c1阴性对照。为了对各引物对评价富集,计算δct=(捕获前ct)-(捕获后ct),然后使用下列效率值计算未经调整的倍数富集:nsc1,1.84;nsc2,1.8;nsc3,1.78;nsc4,1.93;alk,1.7;kras,1.7。将来自捕获前后的qubit读出用于调整富集值,以考虑用于qpcr的dna的质量。通常,nsc1-4的富集值大于70,而kras和alk的富集值大于800。如果得到较差富集(对于nsc1-4小于约50),则再次对捕获前样品进行捕获并再次进行pcr。实施例6.制备衔接子为了制备索引衔接子,使用标准8-碱基条形码illumina衔接子,替代具有4个随机碱基接着4碱基多重条形码的8-碱基索引。设计24种不同的衔接子序列,由此所有的多重条形码对均具有至少为2的编辑距离。使用索引衔接子作为起点来设计串联衔接子。使用具有至少为3的成对编辑距离的12种索引衔接子。对于各衔接子,向各衔接子寡核苷酸的内部末端添加2个碱基,接着向一个寡核苷酸的3’端添加gt,和在另一个寡核苷酸的5’端添加c。对于gt,需要t以允许连接,且选择g以允许在衔接子的末端存在一致的gc夹碱基对。对于12种多重条形码的每一种,需要16对寡核苷酸,每种二核苷酸需要一对。在使用衔接子之前,如上所述使其退火。使用串联衔接子作为起点来设计交错串联衔接子——串联衔接子中的6种具有靠近远端添加到衔接子内部末端的gt上的2个碱基。这2个碱基的序列通过其它的内部条形码碱基来确定,仅保留16种可能的4碱基条形码。对于这些条形码中的8种条形码,衔接子末端的gt替换成ct。为了使衔接子退火,各2种100um衔接子寡聚物各自20ul与终浓度10mmtris/10mmnaclph7合并到50ul反应体积中。根据制造商说明书,使用eppendorfvapoprotect热循环仪(eppendorf,hamburg,germany)使衔接子退火。退火之后,使用10mmtris/10mmnaclph7.5将衔接子稀释成15um。对于索引衔接子,使illumina通用衔接子寡聚物与24种索引衔接子寡聚物的每一种连接。对于12种串联衔接子的每一种,进行16次退火反应:对于衔接子末端的各种二核苷酸条形码进行一次。在退火之后将这16个退火反应物以等浓度合并,然后稀释成15um。实施例7:分子条形码的加工对读出序列进行加工以提取4-bp索引和/或插入条形码序列。后者最初从给定读出对的各端断裂(图6(a)),并在分析之前连接。为了回收具有插入条形码的双链体序列,我们使用以下标准,通过实例阐述:假设分别在读出1和2中实测at和cg插入条形码,则其对应的dna片段f1匹配参照基因组的正链。如果随后分别在来自匹配负链的另一个片段f2的读出2和读出1中实测at和cg条形码,且如果两个片段共享基因组坐标,则f1和f2可能代表双链体分子的交互链。因此分析所有插入条形码。否则,以相同模式处理两种条形码类型。在条形码去重之前,将所有读出映射到参照基因组并使用30的阈值q对所有单碱基变体(例如与参照不同的碱基)进行phred质量过滤,其消除由测序人为因素产生的99.9%的误差。在碱基质量过滤之后,如下对具有≥2个成员的各条形码家族分别进行分析,以鉴定和消除另外的误差:1)对于给定条形码家族中的各基因组位置i,计数不同的非参照变体的数量vi,仅考虑通过碱基质量过滤的变体。如果在给定位置i以q≥30存在>1个不同的非参照变体,则将vi设置为等于最具丰度的高质量变体,或者在相持的情况下,任选变体之一。2)对于含有来自步骤1的候选变体(即vi>0)的各位置,通过减去未通过phred质量过滤的非参照变体的数量qi来调整条形码家族成员的数量。因此,nj*=ni-qi。3)消除来自步骤2的所有非参照变体,其中vi<(f×ni*),其中默认f=1。4)将条形码家族的所有成员合并到单个序列中,仅保留通过步骤3的具有≥2个成员的变体。作为最终误差消除步骤,消除单元素集条形码家族(即具有一个序列的家族)中的所有非参照变体,除非得到来自至少另一个具有≥2个家族成员的dna分子支持变体的证据支持。我们称此去重策略为“2x+单元素集”(图9(f))。实施例8.统计学分析我们如下对检测ctdna的概率建模。假设n=测序基因组当量的数量,d=检测限(ctdna分子的分数),和k=肿瘤报告基因的数量。在cfdna中实测单个肿瘤报告基因的概率为poisson,平均值λ=n×d,其中λ表示突变等位基因拷贝的期望值。因此,给定1个报告基因,则检出≥1个ctdna分子的概率x等于1-poisson(λ),其简化为:(1)x=1–e–nd推广到k个独立的肿瘤报告基因(图16),可使用几何分布的累积分布函数对观测到成功(即检出≥1个ctdna分子)的概率建模。因此,给定k个报告基因,检出≥1个ctdna分子的概率p为1–(1–x)k。将x插入(1),得到(2)p=1–e–ndk如果规定了另三个参数,则此方程可用于求出任何参数。例如,给定1个报告基因(k)、2,000ge(n)和90%置信度(p),则检测限d等于0.12%(即d=ln(1–p)/(–nk))。最后,在cfdna中观测一个报告基因所需的肿瘤报告基因的数量等于1/x(几何分布的平均值)且血浆中期望报告基因的数量等于k×x。实施例9.背景修正为了对cfdna中的位置特异性背景分布明确建模,我们使用了一种新方法,其根据可得的信息含量,交替采用两种统计学模型。首先,我们编译了具有高背景的12个正常对照cfdna样品的训练组(图9(c))。我们随后迭代通过nsclc临床选择子中的各种可能的snv(约1.2m;将生殖系snp除外),以及对于各snv,我们使用在12个正常cfdna对照组中实测的所有等位基因分数(af)填入(populate)位置特异性和碱基置换特异性一维矢量v。为了减小离群值的影响,我们从v中去除最大af,留下11个剩余元素。如果v中非零af的总数小于4,则我们使用高斯分布对整个矢量建模,并使用全部11个af计算平均值μ和标准差σ。否则,我们使用来自r的fitdistrplus程序包中的fitdist,将weibull分布拟合到v中的非零af集合,并将所得的形状和尺度参数保存到磁盘。鉴于v常常为零过多的,我们亦保存v中的非零af的分数以便将零值观察结果的频率并入最终模型中。因其与其它概率分布相比在拟合位置特异性非零背景误差上观察到的优异性能,我们选择weibull分布(图14)。为了确定在未去重或经条形码去重的数据中是否学习背景模式,我们比较了位置特异性误差的复发率。我们在其之间观察到高度一致性,表明定式背景未被条形码去重可靠消除(例如图9(f)中的2x+单元素集)。因此我们使用未去重数据来对基线分布建模,得到背景数据库φ。为了消除(即“修正”)独立cfdna样品中的定式误差s,我们使用其在φ中的相应背景模型,评价s中各候选snv的丰度分数f。如果模型为高斯的,则我们使用单边z检验评价f,得到p值。否则,使用来自weibull分布的形状和尺度参数来计算累计概率p*,通过模型产生的给定af小于f(使用r中的pweibull函数)。考虑到零过多的训练数据,我们随后使用来自训练组的非零af的分数δ调整p*。具体而言,我们使用下式,p值=1–((1–δ)+(δ×p*)),其在结构上类似于两分量零过多poisson模型。通过z检验或零过多weibull分布计算的候选snvp值,随后使用严格bonferroni校正针对多个假设检验进行调整(其中n=背景数据库中的所有碱基置换)。在训练组中的至少2个正常对照和至少20%的正常对照中发生的候选snv之间,我们消除这样的给定候选,当且仅当(i)其与背景在统计学上为不可区分的(调整的p≥0.05),(ii)其并非与双链体支持一起存在,和(iii)f小于5%或者支持分子数量≤10。实施例10.使用改进的选择子设计估算最低检测限(llod)在此实施例中,估算了本发明方法的最低检测限(llod)。首先,设计改进的选择子。我们获得了来自178个肺鳞状细胞癌(scc)肿瘤(v2.3)和606个肺腺癌(luad)肿瘤(v2.4)的tcga全外显子组测序研究的突变注释格式(maf)文件。使用uscs基因组浏览器特征轨迹(ucscgenomebrowserfeaturetracks)将maf文件预过滤,以消除在(i)富含重复序列的基因组区和(ii)具有低映射率的间隔区中的变体。为了对基因组区的包含排序,我们使用启发式方法,其借助本文所定义的“复发指数(ri)”。先前使用了类似策略,将外显子作为基本的基因组单元,而不考虑插入缺失。鉴于仅一个亚组的外显子可能含有已知的体细胞突变,我们将靶向区限制为侧翼为用户定义的缓冲区(默认1bp)、含有已知缺损的子序列,其具有100bp的最小区块大小。随后通过递减的ri将区域分级,并将在ri和每个外显子中患者数量二者的前10%的区域包含在内,其使额外的患者覆盖度最大且具有最小空间。然后重复此过程,但放松百分位数过滤(例如允许前1/3区域),并加入使每个患者中的突变数量中位数最大增加的区域。当达到所需大小(例如对于nsclccfdna选择子为175kb)或者当满足过滤的所有基因组区均被耗尽时,停止选择子增长。亦将融合和种子区包含在cfdna选择子中。我们亦设计了较大的临床选择子,其包含添加了拷贝数变异和组织学分类区的整个cfdna选择子。使用基因组构建hg19ncbibuild37.1/grch37以及设置为1的优选相近匹配(preferredclosematches)和设置成2的最大相近匹配(maximumclosematches),通过nimbledesign入口(rochenimblegen,madison,wisc.)自动选择用于两个文库的探针。用于预测ctdna检测阈值的稳健模型将在临床分析中设置性能期望并利于改进的设计靶标。因此我们开发了用于在给定可得肿瘤基因组当量(ge)和肿瘤特异性报告基因(即体细胞变异)的知识的情况下评估ctdna灵敏性的不依赖于测定的模型。我们发现的是,ctdna检测限与两个量均成线性比例,因此当固定dna输入时,从1个报告基因(例如dpcr)到8个报告基因(例如capp-seq)时,得到llod的8倍减小(图15(b))。当报告基因独立作用时仍保持该结果,其对于nsclc(图15(c))和其它癌症而言通常为真实的,且允许llod降至低于可得肿瘤ge(图15(b))。通过分析来自我们先前研究的样品,我们发现在血浆中的不同肿瘤来源报告基因的实测数量和预测数量之间极佳的一致性(r2=0.98;图15(d);图16),从而验证了我们的模型。此外,肿瘤变体之间的不均匀性分数未显著改变预测性能,这与复发突变趋于在nsclc肿瘤发生早期出现的报道相一致。实施例11.以较低llod对多个肿瘤类型的ctdna基因分型使用实施例1中所述的方法,我们使用靶向在llod上改进两倍且更好针对非吸烟者的选择子设计,评价了基因分型性能(图15(e))。在对来自87个nsclc患者的95个肿瘤(其包括细针活检和福尔马林固定标本,具有匹配的外周血白细胞)进行谱分析时,我们确认了所有临床上确定的变体,并观察到在我们的组和训练数据中的复发突变区之间的强一致性(图15(f)),包括每个患者的突变数量中位数(图15e)。然后将实施例1的方法应用于其它癌样品,证实了用于多种人类恶性肿瘤的单个选择子(例如图8b-e)。实施例12.比较使用具有内部uid和外部uid的衔接子的误差消除为了将来自分子条形编码的误差消除基准化,我们对来自12个健康成年人的cfdna样品进行谱分析。对于各受试者,我们使用一致的dna输入(中位数32ng),测序至约6,000倍的深度中位数(在去除重复之前)。考虑到在临床血浆样品中通常较低的cfdna产率,我们使用全部回收分子来评价条形编码性能(即不论uid拷贝数或成链情况)。当与“未去重”数据比较时,内部/插入分子条形码使全选择子背景减小60%(从约0.025%减小至约0.01%),以及使无误差基因组位置的分数改进50%(从约60%改进至约90%;图9(b);图6(b),图17)。鉴于该方法通常优于外部索引uid,我们使用内部插入uid作为主要的cfdna误差消除或条形码去重策略(图6(b、c))。此实施例中的数据证实,在健康供血者中,易错位置在其基因组定位和碱基置换图谱中为显著定式化的(图9(b)和图10)。即便在使用条形码去重消除误差之后,大多数剩余误差仍由这些定式化低频等位基因(<0.1%)组成,大部分因g>t颠换所致,以及其次为c>t或g>a转换(图9(b))。不受特定理论束缚,我们假定的是,在文库制备期间氧化损伤可能发生,导致8-氧鸟嘌呤形成和胞嘧啶脱氨。有趣的是,当映射到参照人类基因组的相反(正)链时,与交互的c>a事件相比,高度倾向于g>t变化(图9(b)),且此失衡并非由测序链偏移所致(图11(b))。因此我们检验了富集步骤,并且鉴定出在g>t误差与c>a误差的比率上的梯度增加,其在介于0.1天时长和3天时长的靶向序列捕获持续时间内重现地增加2.5倍(图11(c))。对于仅在两条dna链中见到的误差亦观察到类似趋势(仅双链体数据,图11(d))。因此我们怀疑,g>t颠换的比例过高在很大程度上通过与仅靶向正链的捕获试剂耦联的活性氧物质来驱动(图11(e))。亦使用下列产品来检测损伤dna碱基的酶法去除:(i)尿嘧啶dna糖基化酶(udg;neb目录号m0372s),其留下替代尿嘧啶的无碱基位点(胞嘧啶氧化产物),阻止pcr继续通过氧化位点,消除因胞嘧啶氧化所致的c>t误差;(ii)8-氧鸟嘌呤dna糖基化(fpg;neb目录号m0240s),其去除受损嘌呤并在受损碱基位点切割,消除因鸟嘌呤氧化所致的g>t误差,和(iii)precr修复混合物(neb目录号m0309s),其设计成去除多种受损碱基,包括氧化的鸟嘌呤和胞嘧啶。在文库制备之前,使用udg(1个单位)、fpg(8个单位)(udg和fpg一起)、precr修复混合物(1ul)或补充有1mg/mlbsa的precr修复混合物处理来自健康对照的cfdna样品。将样品于37℃处理30分钟,然后通过于60℃加热10分钟来灭活udg和fpg。使用ampure珠粒将样品纯化,并洗脱到50ul水中用于文库制备(图11(a))。实施例13.消除位置特异性测序误差(ides)我们实施了分子条形编码和计算机消除的一系列应用,“集成数字误差消除”(ides)。使用一组健康供者cfdna样品研究基线分布(图9c),我们对条形码去重的数据进行“计算机修正”,消除等位基因分数低于位置特异性阈值的变体(图9b),如在实施例中所述。因此,全选择子误差率降至1.5×10-5,且无误差位置增加至约98%。出人意料的是,本发明人观察到,将背景修正应用于未去重cfdna样品,得到与单独条形码去重类似的误差率(图9c)。此外,所述两种方式在组合时协同作用(图9c)。此实施例进一步研究了定式背景误差的概况并比较了误差消除技术的性能。图10(上图)显示描述173个cfdna样品中的全选择子背景误差模式的热图,所述样品包含30个正常对照,其中12个用作训练组以研究定式背景误差,和143个cfdna样品收集自nsclc患者。显示了条形编码、修正及其组合的影响。图10(下图)显示对应于上述热图中的样品的碱基置换分布和全选择子误差率。使用ides,我们表征了跨越人类基因组的大量区域的等位基因特异性检测限,所述区域在癌症基因组中为复发突变的。在12个核苷酸置换种类中,大部分在很大程度上不受背景影响(图9d)且所有可能snv的近80%为无误差的(图9e)。在剩余误差中,g>t检测最值得注意,其仍保持横跨选择子约0.3%的合理的低llod中位数(图9d)。此外,g>t变化,其包含大部分检测限>0的等位基因,仅包含覆盖我们的nsclc选择子的经癌症体细胞突变目录(cosmic)注释的5%的突变热点,表明对基因分型性能的最小影响(图9e)。实施例14.将ides与现有技术误差消除条形编码策略进行比较我们接着将ides与数种误差消除条形编码策略进行比较。参见图9f、图10。与使可用基因组当量(ge)最大且平衡碱基置换分布的ides不同,单独的条形码去重需要每个uid>5个家族成员以实现相当的误差谱。即便在我们相对高的测序深度下,这亦导致大量ge损失,且仅得到在g>t比例过高上的适度改进。相比之下,双链体分子在健康对照cfdna中实现格外低的误差率2×10-6。将用于本研究报道的方法(即ides,单独的条形编码或修正,单独的双链测序)的误差率(x轴)和分子回收率(y轴;每个测序读出中的一致读出数量),与来自数个其它研究的误差消除方法进行比较(lou,d.i.等high-throughputdnasequencingerrorsarereducedbyordersofmagnitudeusingcirclesequencing(使用循环测序将高通量dna测序误差降低数个数量级).procnatlacadsciusa110,19872-19877(2013),(“lou”);kennedy,s.r.等detectingultralow-frequencymutationsbyduplexsequencing(通过双链测序检测超低频突变).natprotoc9,2586-2606(2014),(“kennedy”);和schmitt,m.w.等detectionofultra-raremutationsbynext-generationsequencing(通过下一代测序检测超罕见突变).procnatlacadsciusa109,14508-14513(2012),(“schmitt”。)。注意的是,双链体测序的理论误差率近似等于单链条形码测序的误差率乘以其本身再除以3(考虑了所有可能的碱基置换)。然而,单链分子的相应损失将可能阻碍对于从限定输入的核酸中检测罕见变体的灵敏性(图7)。因此我们设计了借助于双链分子(如果可得)的基因分型方法,但另外依靠背景修正的单链分子以增加可用基因组当量的数量并使灵敏性最高。实施例15.使用ides在健康受试者中评价基因分型为了使用ides评价无活检基因分型,我们首先对约300个体细胞改变(snv和插入缺失)评价其性能,所述体细胞改变在预定义的“白名单”内的癌症中为高度复发且临床上相关的。(参见实施例20)。图12阐述了使用ides对nsclc进行无活检肿瘤基因分型和超灵敏监测。对5%hd500的4组重复查询29种已知hd500变体连同近300个额外热点和/或临床上相关突变的存在,以评价特异性。图12(a)(左)显示条形编码、修正和ides对单个代表性重复的基因分型结果的差别影响。仅显示具有至少2个支持读出的变体识别。图12(a)(中)显示跨越所有四个重复的性能度量。图12(a)(右)显示在30个正常cfdna对照和25个预处理nsclccfdna样品中,用于每个样品检出变体的平均数的误差消除方法的比较。除对hd500特异的变体之外,评价了在左图中分析的所有变体。使用双边wilcoxon秩和检验进行组间比较。(ns-非显著的)以95%置信区间将数据表示为平均数。图12(b)显示在capp-seq(实测)和真实分数(groundtruthfraction)(预期)之间比较对于跨越四个重复的13个变体的hd500等位基因分数(af),如使用微滴式数字pcr(ddpcr)所校正。将数据表示为平均数±s.e.m。图12(c)显示使用全选择子基因分型在5%hd500样品检测snv,并跨越重复和低十倍的hd500掺加物追踪snv。结果呈现为热图,下面显示相应的等位基因分数。水平线表示平均等位基因分数(maf)。图12(d)显示使用ides进行无活检基因分型,在来自i-iv期nsclc患者的连续的cfdna样品中识别的白名单变体。通过递减的maf从左到右将样品分级,仅显示具有≥1个变体识别且具有匹配肿瘤活检的样品。误差条表示范围。图12(e)显示d中变体的受试者工作特征(roc)分析,连同具有已知临床状况的egfr识别。auc,曲线下面积。图12(f)显示使用ides的无活检基因分型的来自晚期nsclc肿瘤的预处理血浆的可作用egfr突变的回收率。图12(g)显示用于在来自33个nsclc患者的预处理血浆中检测ctdna的后处理方法的比较。在包含30个正常对照的各血浆样品(行;n=63)中评价患者来源的肿瘤报告基因(列;n=33组),以评价特异性。对相同样品分析各后处理方法(例如ides),并在热图中同样排序。红色方块,真阳性;蓝色方块,假阳性,白色方块,未检出。图12(h)显示在经历egfr靶向疗法的iia期nsclc患者中监测肿瘤负荷。pre,预处理;carbo,卡铂;pem,培美曲塞;cetux,西妥昔单抗;dod,因病死亡。‘监测’表示使用所有肿瘤报告基因来计算ctdna检测指标。星号‘*’表示其中egfrt790m和del19未能检出的时间点。然而,使用监测框架可显著检出ctdna。图12(i)显示评价双链体测序的检测限的探索性掺加分析。maf,突变等位基因分数。sn,灵敏性;sp,特异性;ppv,阳性预测值;npv,阴性预测值。a和d的x轴的刻度标记表示独立的cfdna样品。将选择子应用于来自30个健康受试者的cfdna时,ides仅得到8个识别,其中5个具有双链体支持,表明体内产生的真实变体等位基因。相比之下,条形码去重和未去重数据在相同健康受试者中分别得到10倍和50倍的识别,表明高假阳性率(图12a,左)。为了将性能基准化,我们使用确定输入的掺加到对照cfdna中的商业化参照标准突变dna(hd500horizondiscovery,cambridge,uk)来模拟ctdna。除我们的白名单之外,还评价了所有验证的hd500变体。与单独的条形码去重相比,对于检测低至1-3个突变分子的变体,ides表现出相当的灵敏性(96%)但实现更高的特异性(99.9%),阳性预测值(ppv=99.1%)和阴性预测值(npv=99.5%)(在此情况下,对于单个变体的llod为约0.04%给定测序ge;图12a,右)。此外,hd500等位基因的丰度分数在各重复之间为一致的,且与输入浓度相一致(图12b)。这些数据表明,ides为用于cfdna的无活检基因分型的稳健框架。与dpcr不同,ides可同时访问多个变体而不损失灵敏性。为了扩展超出预定义白名单的变体检测的范围,我们测试了使用ides的全面的全选择子基因分型的性能。使用新型snv检测方法,我们在含5%突变dnahd500的cfdna样品中鉴定了324个snv,回收了21个(27个中的)真实的等位基因。从我们的分析中,我们计算得到0.6%假阳性率(图12),表明对于无活检监测应用的适用性。实施例16.使用ides在nsclc患者中进行肿瘤基因分型我们接着检验了ides用于从血浆对nsclc患者进行无活检基因分型的临床潜能。发现71个连续血浆样品中近72%含有白名单变体(图12d),所述样品包括50%的早期样品和92%的晚期预处理样品。在匹配的肿瘤活检中,近2/3的检出变体被确认为体细胞变异。大部分剩余变体与其为亚克隆的肿瘤相关突变相一致,因为2/3独立存在于连续的时间点或者具有双链体支持。实际上,所述变体在具有更晚期nsclc阶段的患者中更普遍,且与在健康成年人中相比,其在从未吸烟的nsclc患者中显著更普遍(图18)。实施例17.使用ides在nsclc患者的血浆中检测egfr突变考虑到其对于现有和新兴靶向疗法的重要性,我们接着关注egfr突变。在对患有ib-iv期肿瘤的nsclc患者进行谱分析时,我们证实在血浆样品中检出145个变体的100%(图12e)。在来自晚期患者的预处理cfdna中,对于可作用egfr变体的检出率较高,对于激活突变具有平均100%特异性和95%灵敏性,以及对于亚克隆t790m抗性突变具有83%灵敏性(图12f)。鉴于ides优于其它方法,这些数据证实其对于无需肿瘤基因型的现有知识来鉴定cfdna中的临床上相关突变的前景。实施例18.使用ides检测nsclc患者的血浆中的已知肿瘤基因型我们接着研究了使用肿瘤基因型的现有知识,ides是否可得到对于ctdna监测的类似改进。通过经验调整前述ctdna检测指标的性能,在包含100%ib期肿瘤的预处理血浆样品中可检出94%的病例,在考虑健康成年人对照时允许100%特异性(图12g)。与ides相比,单独的双链体测序允许相当的特异性,但受较低的灵敏性和显著较低的单个变体回收所累。相反地,其它方法实现相当的灵敏性,但较低的特异性(图12(g))。对于处理后样品亦观察到类似性能。单独看来,在首先针对del19及随后针对t790m经历靶向egfr治疗的患有iia期nsclc的患者中(图12h),直接血浆基因分型显示与埃罗替尼之后的优势抗性机制相一致的亚克隆动态。在对化疗的临床反应之后,两种突变均未能独立检出。然而,通过将多个报告基因整合到一个监测背景中,ides,但未进行双链体测序,在此时间点检出新出现的分子残留疾病,且预示在该患者中的临床进展,所述患者最终死于nsclc(图12h)。在患有iiib期nsclc的另一个患者中,在临床进展前ides检出0.004%ctdna,相对于我们先前实施中实测的llod有五倍改进。这些数据突出了ides用于发现罕见ctdna分子的潜在适用性,其具有用于监测最小残留疾病和非侵入性检测抗性基因的应用。实施例19.使用双链体测序检测血浆中的肿瘤突变负荷考虑到双链体测序的优秀误差率,我们试图确定其对于定量循环肿瘤负荷的llod。为了克服单链分子的损失(图9f),我们设计了“个性化”选择子以覆盖通过复发的人成胶质细胞瘤的外显子组测序而鉴定的>1,500个非同义突变。然后基于对报告基因数量和输入质量的考虑,我们将确定量的肿瘤基因组dna掺加到对照cfdna中。我们获得含有具有已知频率的变体的dna参照标准(hd500,horizondiscovery)并以5%和0.5%浓度将其掺加到健康供者cfdna中。以各掺加浓度制备四个capp-seq文库并进行测序。通过ddpcr分析编码egfrl858r、krasg13d和brafv600e的变体,以校正预期的掺加浓度。对于图12(a),我们访问了所有hd500变体,其(i)被我们的nsclc临床选择子靶向和(ii)存在于horizondiagnostics提供的真实突变列表(即‘多重完整突变列表’)中。对于图12(b),我们分析了经horizondiagnostics内部验证且被我们的nsclc临床选择子靶向的hd500变体的亚组。尽管回收<1,000个具有双链体支持的ge,但所述方法以高线性度精确检测了从0.025%到0.00025%的确定输入(图12i),进一步验证了我们的分析模型并证实每1,000,000个分子背景检出2.5个分子的检测限,是单个等位基因dpcr的llod的近1/100。实施例20.“白名单”基因分型在此实施例中,通过使用用户定义变体的“白名单”,脚本(script)从cfdna和肿瘤样品中执行突变回收(snv和插入缺失)而无需匹配的生殖系样品,以减少假设空间并增加灵敏性。“白名单”为采用下列优先次序的支持证据:双链体支持(1x)>>链支持(2x)>>无链支持(3x)。使用下式设置识别snv所需的最小aff:f=ln(1–p)/–n,其中p=检出概率(默认0.95),n=给定基因组位置上的总ge。实施例21.以不同的cfdna浓度实施所述方法对于此研究中的分析,我们需要最小的位置特异性深度,对于肿瘤为20ge,对于cfdna为1,000ge。为了并入匹配的生殖系样品,我们消除了这样的候选变体识别,如果其以≥1%af、≥4个支持读出存在于匹配生殖系中,以及存在于≥10的总ge的位置中。我们接着评价了本方法的技术性能。首先,我们建立了计算机(insilico)稀释系列,其中对深度中位数为3,861ge的对照cfdna样品进行操作,以引入100个均匀分布的纯合snv。然后以5%和0.5%比例向起始cfdna样品中添加各合成分子(numerator)。为了模拟cfdna的中值长度,从而维持其在测序数据中的分布,将基因组区随机掺入170bp连续片段中。观察到稳健的性能(图9(c))。单独看来,与我们先前用于肿瘤基因分型的方法相比,我们发现自适应方法对于肿瘤的体细胞基因分型表现出更高灵敏性和特异性,在ctdna监测框架中对其变体识别进行评价(与图9(g)中的分析相同)。尽管已参考具体实施例详细阐述本发明,但对于本领域技术人员而言明显的是,在本发明的范围内可作出各种修改。因此本发明的范围不应限于本文所述的实施例,而应受限于所附权利要求书。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1