有效的坎普他汀类似物的制作方法
本申请要求2005年11月28日提交的美国临时申请号60/740,205的优先权,其全部内容在此引入作为参考。
政府支持
依据35U.S.C.ξ202(c),承认美国政府可以享有在此描述的本发明的某些权利,本发明一部分是使用来自国家卫生研究所授权号GM 62134的基金做出的。
发明领域
本发明涉及体内补体级联的激活。尤其是,本发明提供了能够结合C3蛋白并抑制补体激活的肽和模拟肽。
发明背景
说明书自始至终引用各种出版物,包括专利、公开的申请、技术文献和学术文章。这些引用的每篇出版物在此全部引入作为参考。说明书中未完全引用的出版物的完全引证列在说明书的结尾。
补体系统是对抗外来病原体的第一道免疫防线。其通过经典的、旁路的或凝集素途径的激活引起过敏毒性(anaphylatoxic)肽C3a和C5a产生和C5b-9膜攻击复合体形成。补体成分C3在所有三个途径的激活中起着重要作用。补体途径C3转变酶引起的C3激活及其随后附着于靶表面引起膜攻击复合体装配,并最终引起靶细胞损害或裂解。C3是独特的,因为它具有提供多重不同配体结合位点的丰富的构造,所述位点在免疫监视和免疫反应途径中很重要。
不适当的补体激活可以引起宿主细胞损害。补体牵连几种病态,包括各种自身免疫疾病,并且已经发现其促进另外的临床病症,例如成人呼吸综合征、心脏病发作、异种移植后排斥和烧伤损害。还已发现补体介导的组织损伤是由生物不相容情况,例如经受透析或心肺分流术的患者遭遇的情况引起的。
补体介导的组织损伤通过膜攻击复合体直接介导,和通过产生C3a和C5a间接介导。这些肽通过它们对各种细胞,包括嗜中性白细胞和肥大细胞的影响而引起损害。在体内,对C3和C5激活步骤的补体调节通过血浆和膜蛋白来提供。血浆蛋白抑制剂是因子H和C4结合蛋白,而位于细胞表面的调节膜蛋白是补体受体1(CR1),衰变加速因子(DAF)和膜辅因子蛋白(MCP)。这些蛋白通过促进多亚基复合物解离和/或通过蛋白酶解(由因子I催化)使复合物失活来抑制C3和C5转变酶(多亚基蛋白酶)。通过体外测定已经鉴定出调节或调整补体活性的几种药剂,但是大多数在体内显示出活性低或有毒性。
迄今为止,没有补体激活抑制剂被批准用于临床,尽管存在某些供临床使用的候选物,具体地说,被称为可溶补体受体1(sCRl)的重组形式的补体受体1和人源化抗C5单克隆抗体(5Gl.l-scFv)。这两种物质在体内动物模型中已经显示出抑制补体激活(Kalli KR等,1994;和Wang等,1996)。然而,每种物质都具有作为大分子量蛋白(分别是240kDa和26kDa)的缺点,难以制备和必须通过输液给药。因此,最近的研究着重于较小活性剂的开发,其易于递送、更稳定和制造成本更低。
Lambris等的美国专利号6,319,897描述了噬菌体展示组合随机肽文库用于鉴定与C3结合并抑制补体激活的27个残基的肽的用途。这个肽被截短为保留了全部活性的13个残基的环状片段,其在本领域中被称为坎普他汀(compstatin)。坎普他汀抑制C3被C3转变酶切割为C3a和C3b。已在一系列体外、体内、离体和体内/离体交界实验中测试了坎普他汀,并已证明:(1)在人血清中抑制补体激活(SahuA等,1996);(2)在灵长类动物中抑制肝素/鱼精蛋白引起的补体激活,而无显著的副作用(Soulika AM等,2000);(3)延长输注人血的猪至人异种移植物的寿命(Fiane AE等,1999a;Fiane AE等,1999b;和Fiane AE等,2000);(4)在心肺分流术、血浆去除术和透析体外循环模型中抑制补体激活(Nilsson B等,1998);和(5)具有低毒性(Furlong ST等,2000)。
坎普他汀是包含序列ICVVQDWGHHRCT-NH2(SEQ ID NO:1)的肽,其中Cys2和Cysl2形成二硫键。使用同核2D NMR核磁共振与两个分开的实验上受约束的计算方法相结合来确定它的三维结构。第一种方法涉及距离几何学、分子动力学和模拟退火(Morikis D等,1998;WO99/13899)而第二种方法涉及总体优化(Klepeis等,J.Computational Chem.,20:1344-1370,1999)。坎普他汀的结构揭示了一个由极性斑和非极性斑组成的分子表面。极性部分包括I型β转角且非极性斑包括二硫键。此外,合成了具有丙氨酸置换(丙氨酸分区)的一系列类似物并测试了活性,揭示β转角的四个残基和带环绕的疏水簇的二硫键在坎普他汀的抑制活性中起着重要作用(Morikis等,1998;WO99/13899)。
使用包括测定旁路途径介导的红细胞裂解的补体活性试验,已经测定了坎普他汀的IC50为12μM。已经证明某些以前测试的类似物具有相当于或大于坎普他汀的活性。公开的国际申请号WO2004/026328公开了具有N-和C-末端和在第4和9位变异的坎普他汀类似物和模拟物,其在上述试验中显示活性提高。已报道某些类似物活性的提高超过坎普他汀达99倍{也参见,Mallik等,2005)。具有甚至更高活性的坎普他汀类似物或模拟物的开发将构成本领域的显著进步。
发明概述
本发明提供了补体抑制肽坎普他汀(HOOC-ICVVQDWGHHRCT-NH2;SEQ ID NO:1)的类似物和模拟物,其与坎普他汀相比具有提高的补体抑制活性。
在一个方面,本发明的特征是抑制补体激活的化合物,其包含具有下述序列的肽:
Xaa1-Cys-Val-Xaa2-Gln-Asp-Xaa3-Gly-Xaa4-His-Arg-Cys-Xaa5(SEQ ID NO:26);其中:
Xaa1是Ile、Val、Leu、Ac-Ile、Ac-Val、Ac-Leu或包含Gly-Ile的二肽;
Xaa2是Trp或Trp的类似物,其中Trp的类似物与Trp相比疏水特性增加,条件是,如果Xaa3是Trp,那么Xaa2是Trp的类似物;
Xaa3是Trp或在其吲哚环上包含化学修饰的Trp的类似物,其中化学修饰增加吲哚环的氢键能;
Xaa4是His、Ala、Phe或Trp;
Xaa5是L-Thr、D-Thr、Ile、Val、Gly、包含Thr-Asn的二肽、或包含Thr-Ala的二肽、或包含Thr-Ala-Asn的三肽,其中L-Thr、D-Thr、Ile、Val、Gly或Asn中任何一个的羧基末端-OH任选被-NH2替代;并且
两个Cys残基通过二硫键相连。
在某些实施方案中,Xaa2参与与C3的非极性相互作用。在其他实施方案中,Xaa3参与与C3氢键键合。在其他实施方案中,Xaa2参与与C3的非极性相互作用,和Xaa3参与与C3氢键键合。
在各种实施方案中,Xaa2的Trp的类似物是卤代色氨酸,例如5-氟代-L-色氨酸或6-氟代-L-色氨酸。在其他实施方案中,Xaa2的Trp类似物在第5位包含低级烷氧基或低级烷基取代基,例如5-甲氧基色氨酸或5-甲基色氨酸。在其他实施方案中,Xaa2的Trp类似物在第1位包含低级烷基或低级烷酰基取代基,示例实施方案包含1-甲基色氨酸或1-甲酰基色氨酸。在其他实施方案中,Xaa3的Trp的类似物是卤代色氨酸,例如5-氟代-L-色氨酸或6-氟代-L-色氨酸。
在某些实施方案中,Xaa2在色氨酸第1位包含低级烷酰基或低级烷基取代基,Xaa3任选包含卤代色氨酸和Xaa4包含丙氨酸。在特定实施方案中,Xaa2是1-甲基色氨酸或1-甲酰基色氨酸和Xaa3任选包含5-氟代-L-色氨酸。本发明的一些示例化合物包含SEQ ID NOS:15-25中的任何一个。
在一些实施方案中,该化合物包含由编码肽的多核苷酸表达而生产的肽。在其他实施方案中,化合物至少部分通过肽合成来生产。还可以使用合成法的组合。
在某些实施方案中,坎普他汀类似物是其中化合物是聚乙二醇化的,如包含SEQ ID NO:36的化合物示例。
在其他实施方案中,坎普他汀类似物进一步包含延长化合物体内保留的另外的肽成分。例如,另外的肽成分可以是白蛋白结合肽。一个示例的坎普他汀-白蛋白结合肽缀合物包含SEQ ID NO:39。
本发明的另一个方面的特征是抑制补体激活的化合物,包含SEQ ID NO:26或在上文描述的类似物和缀合物的任何其他序列的非肽或部分肽模拟物。这些非肽或部分肽模拟物被设计用来结合C3并抑制补体激活,在同等试验条件下,其活性比包含SEQ ID NO:1的肽大至少100倍。
本发明的坎普他汀类似物、缀合物和模拟物具有实用性,用于应用坎普他汀本身的任何目的,如本领域已知和本文更详细描述的。某些这些用途包括化合物配制入给予患者的药物组合物。这种制剂可以包含该化合物的药物可接受盐,以及一种或多种药物可接受稀释剂、载体、赋形剂等等,如在本领域技术人员权限内的。
通过参考下述详细描述、附图和实施例会理解本发明的各种特征和优点。
附图简述
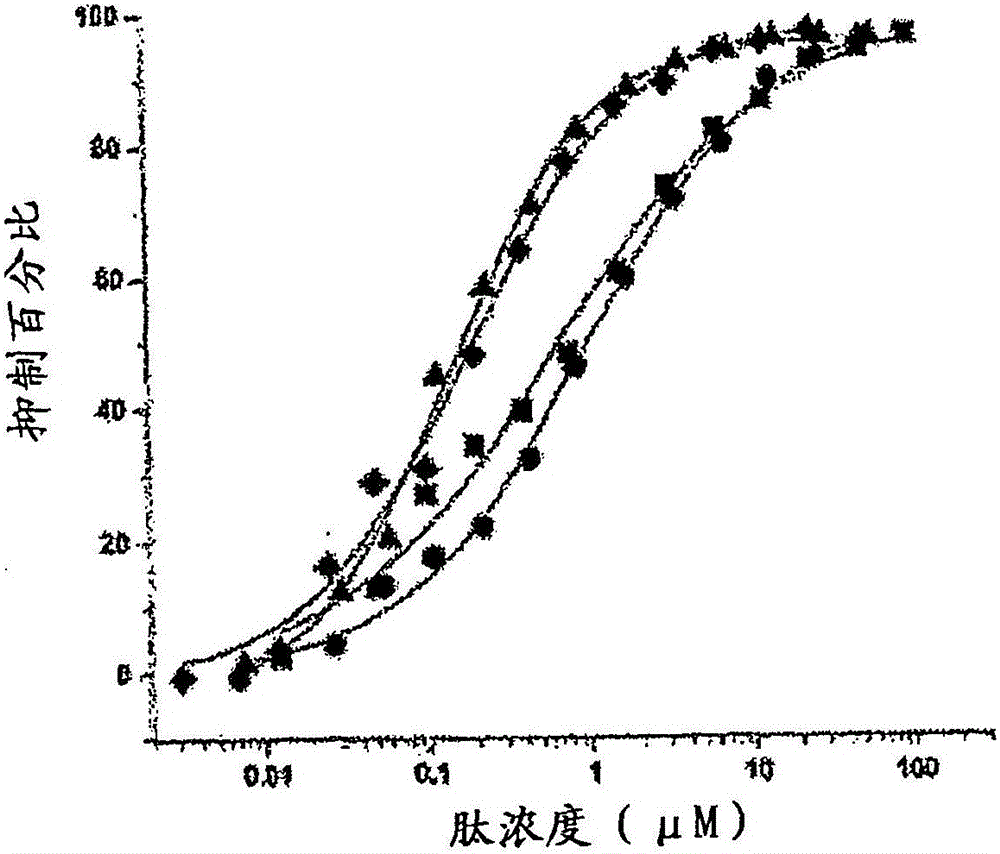
图1.表达的坎普他汀及其类似物的活性。补体抑制百分比对Ac-V4W/H9A(SEQ ID NO:5)(正方形)和表达的具有色氨酸的坎普他汀(SEQ ID NO:15)(圆形)、5-氟代-色氨酸(SEQ ID NO:16)(三角形)、6-氟代色氨酸(SEQ ID NO:17(星形)、5-羟基-色氨酸(SEQ ID NO:27)(六角形)、7-氮杂-色氨酸(SEQ ID NO:28)(菱形)的肽浓度的图。
图2.合成的坎普他汀类似物的活性。补体抑制百分比对Ac-V4W/H9A(SEQ ID NO:5)(正方形)和在第4位(SEQ ID NO:18)(圆形)、第7位(SEQ ID NO:19)(三角形)、第4和7位(SEQ ID NO:20)(菱形)具有5-氟代-L-色氨酸的坎普他汀类似物的肽浓度的图。
图3.另外的合成的坎普他汀类似物的活性。补体抑制百分比对以下肽浓度的图:(A)Ac-V4W/H9A(SEQ ID NO:5)(三角形)与Ac-V4(5f-l-W)/H9A(SEQ ID NO:18)(倒置三角形)、Ac-V4(5-甲基-W)/H9A (SEQ ID NO:22)(圆形)、Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)(菱形)、Ac-V4(2-Nal)/H9A(SEQ ID NO:7)(正方形)相比;(B)Ac-V4W/H9A(SEQ ID NO:5)(三角形)与Ac-V4W/W7(5f-l-W)/H9A(SEQ ID NO:19)(六边形)相比;和(C)野生型坎普他汀(SEQ ID NO:1)(三角形)与Ac-V4(1-甲基-W)/W7(5f-l-W)/H9A(SEQ ID NO:24)(尖端向左的三角形)相比。
图4.另外的坎普他汀类似物与C3相互作用的热力学表征。表明(A)Ac-V4W/H9A(SEQ ID NO:5);(B)Ac-V4(5f-l-W)/H9A(SEQ ID NO:18);(C)Ac-V4(5-甲基-W)/H9A(SEQ ID NO:22);(D)Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23);(E)Ac-V4(2-Nal)/H9A(SEQ ID NO:7)和(F)Ac-V4W/W7(5f-l-W)/H9A(SEQ ID NO:19)与C3结合的ITC数据。该图是通过使校正的原始数据符合Origin 7.0中“one set of sites”模式而获得的。
图5.显示了以log P表示的类似物的疏水性与抑制常数(A)、以-TΔS表示的熵(B)和结合常数(C)之间关系的图。
图6.另外的合成的坎普他汀类似物的活性。补体抑制百分比对Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)(圆形)和Ac-V4(1-甲酰基-W)/H9A(SEQ ID NO:25)(正方形)的肽浓度的图。
图7.聚乙二醇化坎普他汀类似物的活性。补体抑制百分比对Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)(圆形)和Ac-V4(1-甲基-W)/H9A-K-PEG 5000(SEQ ID NO:36)(正方形)的肽浓度的图。
图8.白蛋白结合蛋白-缀合的坎普他汀类似物的活性。补体抑制百分比对Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)(圆形)和融合肽(Ac-ICV(1MeW)QDWGAHRCTRLIEDICLPRWGCLWEDD-NH2)(SEQ ID NO:39) (正方形)的肽浓度的图。
示例实施方案的详细描述
与本发明的方法及其他方面有关的各种术语贯穿说明书和权利要求书使用。这种术语赋予它们在本领域通常的意思,除非另有陈述。其他具体限定的术语以与本文提供的定义一致的方式进行解释。
定义:
说明书和实施例中可以使用下列缩写:Ac,乙酰基;NH2,氨基;MALDI,基质辅助激光解吸离子化;TOF,飞行时间;ITC,等温滴定量热法;HPLC,高效液相色谱法;NA,非活性的;dT,D-苏氨酸;2-Nal,2-萘基丙氨酸;1-Nal,1-萘基丙氨酸;2-Igl,2-茚满基甘氨酸;Dht,二氢色氨酸;Bpa,4-苯甲酰基-L-苯丙氨酸;5f-l-W,5-氟代-L-色氨酸;6f-l-W,6-氟代-L-色氨酸;5-OH-W,5-羟色氨酸;5-甲氧基-W,5-甲氧基色氨酸;5-甲基-W,5-甲基色氨酸;1-甲基-W,1-甲基色氨酸;氨基酸缩写使用标准的三或单字母命名法,例如对于色氨酸,用Trp或W。
本文使用的术语“大约”当涉及可测值,如量、持续时间等时,意思是包括从给定值±20%或±10%,在一些实施方案中±5%,在一些实施方案中±1%,和在一些实施方案中±0.1%的变化,因为这种变化适于作出并用于公开的化合物和组合物。
术语“药物活性的”和“生物活性的”是指本发明化合物结合C3或其片段并抑制补体激活的能力。这种生物活性可以通过几种本领域认可的试验中的一种或多种进行测定,如本文更详细描述的。
本文使用的“烷基”是指具有大约1至大约10个碳原子(和那里碳原子范围和具体数量的所有组合和亚组合)的任选取代的饱和直链、支链或环状烃,大约1至大约7个碳原子是优选的。烷基包括但不限于甲基、乙基、正-丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、环戊基、异戊基、新戊基、正己基、异己基、环己基、环辛基、金刚烷基、3-甲戊基、2,2-二甲丁基和2,3-二甲丁基。术语“低级烷基”是指具有大约1至大约5个碳原子(和那里碳原子范围和具体数量的所有组合和亚组合)的任选取代的饱和直链、支链或环状烃。低级烷基包括但不限于甲基、乙基、正-丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、环戊基、异戊基和新戊基。
本文使用的“卤代”是指F、Cl、Br或I。
本文使用的“烷酰基”可以与“酰基”互换使用,是指具有大约1至大约10个碳原子(和那里碳原子范围和具体数量的所有组合和亚组合)的任选取代的一个直链或支链或脂肪族非环状残基。烷酰基包括但不限于甲酰基、乙酰基、丙酰基、丁酰基、异丁酰基、戊酰基、异戊酰基、2-甲基-丁酰基、2,2-二甲基丙酰基、己酰基、庚酰基、辛酰基等。术语“低级烷酰基”是指具有大约1至大约5个碳原子(和那里碳原子范围和具体数量的所有组合和亚组合)的任选取代的直链或支链脂肪族非环状残基。低级烷酰基包括但不限于甲酰基、乙酰基、正丙酰基、异丙酰基、丁酰基、异丁酰基、戊酰基、异戊酰基等。
本文使用的“芳基”是指具有大约5至大约14个碳原子(和那里碳原子范围和具体数量的所有组合和亚组合)的任选取代的单环或二环芳环系统,大约6至大约10个碳原子是优选的。非限制实例包括例如苯基和萘基。
本文使用的“芳烷基”是指具有大约6至大约20个碳原子(和那里碳原子范围和具体数量的所有组合和亚组合)的含芳基取代基的烷基,大约6至大约12个碳原子是优选的。芳烷基可以任意取代。非限制实例包括例如苯甲基、萘甲基、二苯甲基、三苯甲基、苯乙基和二苯乙基。
本文使用的术语“烷氧基”是指任选取代的烷基-O-基,其中烷基如前定义。示例的烷氧基包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基和庚氧基等等。
本文使用的“羧基”是指-C(=O)OH基。
本文使用的“烷氧羰基”是指-C(=O)O-烷基,其中烷基如前定义。
本文使用的“芳酰基”是指-C(=O)芳基,其中芳基如前定义。示例的芳酰基包括苯甲酰基和萘酰基。
典型地,取代的化学部分包括取代分子上选定位置的氢的一个或多个取代基。示例的取代基包括例如卤代、烷基、环烷基、芳烷基、芳基、巯基、羟基(-OH)、烷氧基、氰基(-CN)、羧基(-COOH)、酰基(烷酰基:-C(=O)R);-C(=O)O-烷基、甲酰胺基(-C(O)NH2)、-N-取代的甲酰胺基(-C(=0)NHR”)、CF3、CF2CF3等。至于上述取代基,每部分R”可以单独例如是H、烷基、环烷基、芳基或芳烷基中的任何。
本文使用的“L-氨基酸”是指蛋白质中正常存在的任何天然存在的左旋α-氨基酸或那些α-氨基酸的烷基酯。术语“D-氨基酸”是指右旋α-氨基酸。除非另外规定,本文提及的全部氨基酸是L-氨基酸。
“疏水”和“非极性”在本文同义使用,是指不以偶极为特征的分子间或分子内任何相互作用。
本文使用的“pi符号”是指坎普他汀参与pi与C3键合的能力。Pi键由两个平行的p轨域侧向重叠产生。
本文使用的“氢键能”是指坎普他汀参与与C3静电吸引的能力,涉及坎普他汀上修饰的色氨酸残基或色氨酸类似物上带负电的部分和C3上的氢原子。这种带负电部分的非限制实例是氟原子。
“聚乙二醇化”是指其中至少一个聚乙二醇(PEG)部分,无论其大小,与蛋白质或肽化学连接形成PEG-肽缀合物的反应。聚乙二醇化意思是至少一个PEG部分,不管其大小,化学连接于肽或蛋白质。术语PEG通常带有数字后缀,其指明PEG聚合物的近似平均分子量;例如PEG-8,000是指平均分子量为大约8,000的聚乙二醇。
本文使用的“药物可接受盐”是指所公开化合物的衍生物,其中母体化合物通过制备其酸或碱盐而被修饰。药物可接受盐的实例包括但不限于碱性残基例如胺类的无机或有机酸盐;酸性残基例如羧酸类的碱性或有机盐等等。因此术语“酸加成盐”是指通过添加酸而制备的母体化合物的相应盐衍生物。药物可接受盐包括由无机或有机酸形成的母体化合物的常规盐或季铵盐。例如,这种常规盐包括但不限于源自无机酸如盐酸、氢溴酸、硫酸、氨基磺酸、磷酸、硝酸等的盐;和由无机酸如醋酸、丙酸、琥珀酸、乙醇酸、硬脂酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、棕榈酸、马来酸、羟基马来酸、苯乙酸、谷氨酸、苯甲酸、水杨酸、磺胺酸、2-乙酸基苯甲酸、反丁烯二酸、甲苯磺酸、甲磺酸、乙烷二磺酸、草酸、羟乙磺酸等制备的盐。本发明的某些酸性或碱性化合物可以以两性离子形式存在。所有形式的化合物,包括游离酸、游离碱和两性离子都包括在本发明范围内。
说明书:
根据本发明,已经使用关于坎普他汀的生物和物理化学特性的信息设计了与亲本坎普他汀肽相比活性显著提高的坎普他汀类似物。在一些实施方案中,该类似物具有坎普他汀的至少50倍或更大的活性。在其他实施方案中,该类似物具有坎普他汀的60、65、70、75、80、85、90、95、100、105、110、115、120、125或130倍或更大的活性。还在其他的实施方案中,使用实施例中描述的试验来比较,该类似物具有坎普他汀的135、140、145、150、155、160、165、170、175、180、185、190、195、200、205、210、215、220、225、230、235、240、245、250、255、260、265倍或更大的活性。
已经表明根据其他方法合成的坎普他汀类似物具有与亲本肽相比稍微提高的活性,即达到大约99倍(Mallik,B.等,2005,见上;WO2004/026328)。根据本发明生产的类似物甚至具有比迄今为止生产的亲本肽或其类似物更大的活性,如本文附图和实施例中所示的体外试验所证明。
表1B显示了坎普他汀和活性显著提高的所选类似物的氨基酸序列和补体抑制活性。选择的类似物涉及与亲本肽坎普他汀(SEQ ID NO:1)和与SEQ NOS:2-14的肽相比指定位置(1-13)的特异性修饰,如表1A所示,其在WO2004/026328中描述。SEQ ID NOS:15-24的肽代表根据本发明进行的修饰,产生了明显更有效的坎普他汀类似物。如下面更详细描述的,很清楚如SEQ ID NOS:2-13所列出的对第4位上的色氨酸进行的某些修饰可以与第7位上的色氨酸类似物置换结合,形成另外的强效坎普他汀类似物。
表1
这个表中使用的缩写如下:
dT=D-苏氨酸
2-Nal=2-萘基丙氨酸
1-Nal=1-萘基丙氨酸
2-Igl=2-茚满基甘氨酸
Dht=二氢色氨酸
Bpa=4-苯甲酰基-L-苯丙氨酸
5f-l-W=5-氟代-L-色氨酸
6f-l-W=6-氟代-L-色氨酸
5-OH-W=5-羟色氨酸
5-甲氧基-W=5-甲氧基色氨酸
5-甲基-W=5-甲基色氨酸
1-甲基-W=1-甲基色氨酸
1-甲酰基-W=1-甲酰基色氨酸
N-末端的修饰。N-末端的乙酰化典型地增加坎普他汀及其类似物的补体抑制活性,如通过SEQ ID NO:1与SEQ ID NO:2比较具体可见。因此,肽的氨基末端添加酰基,包括但不限于N-乙酰化,是本发明的一个优选实施方案,当合成制备肽时特别实用。然而,通过在原核或真核表达系统中表达编码肽的核酸分子,或通过体外转录和翻译而制备肽时,有时也有利。对于这些实施方案,可以使用天然存在的N-末端。适于体外或体内表达坎普他汀类似物的一个实例以SEQ ID NOS:15-17为代表,其中在N-末端乙酰基被未修饰的甘氨酸替代。如下所讨论的在肽内和在C-末端另外包含修饰的SEQ ID NOS:15-17在本文描述的补体抑制试验中比坎普他汀活性高大约45至大约125倍。
肽内修饰。使用为低能量序列分级的计算方法,以前测定了Tyr和Val是第4位上支持肽稳定性和活性最可能的候选者(Klepeis JL等,2003)。WO2004/026328中公开了Trp在第4位,尤其是联合Ala在第9位,产生比亲本肽活性高很多倍的肽(例如,SEQ ID NOS:4、5和6与SEQ ID NOS:2和3的活性比较)。WO2004/026326还公开了发现在第4位包含色氨酸类似物2-萘基丙氨酸(SEQ ID NOS:7、8)、1-萘基丙氨酸(SEQ ID NO:9)、2-茚满基甘氨酸(SEQ ID NOS:10、11)或二氢色氨酸(SEQ ID NO:12)的肽的补体抑制活性全部增加,范围从大于坎普他汀5倍至99倍。此外,在第4位包含苯丙氨酸类似物、4-苯甲酰基-L-丙氨酸的肽(SEQ ID NO:13)活性比坎普他汀大49倍。
根据本发明,在第4位包含5-氟代-L-色氨酸(SEQ ID NO:19)或5-甲氧基、5-甲基或1-甲基色氨酸或者1-甲酰基色氨酸的肽(分别是SEQ ID NOS:21、22、23和25)比坎普他汀活性大31-264倍。1-甲基或1-甲酰基色氨酸的掺入与其他类似物相比增加了活性和结合亲合力最大。人们相信在第4位吲哚‘N’介导的氢键对于坎普他汀的结合和活性不是必要的。通过在第4位用低级烷基、烷酰基或吲哚氮取代氢引起缺乏这个氢键或极性降低使坎普他汀结合和活性增加。不意欲限于任何特定的理论或作用机制,人们相信在第4位的疏水作用或效应加强坎普他汀与C3的相互作用。因此,保持或增强上述疏水作用的在第4位Trp的修饰(例如根据本领域熟知方法改变侧链结构)或Trp类似物的置换包括在本发明中以生产甚至更大活性的坎普他汀类似物。这种类似物是本领域熟知的,包括但不限于本文例证的类似物,以及其未取代或者取代的衍生物。适当的类似物实例可以参考下列出版物和很多其他的出版物而找到:Beene,等(2002)Biochemistry41:10262-10269(尤其描述了单和多卤代Trp类似物);Babitzky&Yanofsky(1995)J.Biol.Chem.270:12452-12456(尤其描述了甲基化和卤代Trp及其他Trp和吲哚类似物)和美国专利6,214,790、6,169,057、5,776,970、4,870,097、4,576,750和4,299,838。Trp类似物通过体外或体内表达,或通过肽合成被引入坎普他汀肽,如本领域已知的和实施例中更详细描述的。
在某些实施方案中,坎普他汀的第4位Trp被类似物替代,该类似物包含1-烷基取代基,更具体地包含如上定义的低级烷基(例如C1-C5)取代基。这些包括但不限于N(α)甲基色氨酸和5-甲基色氨酸。在其他的实施方案中,坎普他汀第4位的Trp被类似物替代,该类似物包含1-烷酰基取代基,更具体地包含如上定义的低级烷酰基(例如C1-C5)取代基。除了例证的类似物之外,这些包括但不限于1-乙酰基-L-色氨酸和L-β-同型色氨酸。
热力学实验表明相对于野生型坎普他汀,在第7位掺入5-氟代-L-色氨酸增加了坎普他汀与C3之间相互作用的焓,而在坎普他汀第4 位掺入5-氟代-色氨酸降低了这种相互作用的焓。不意欲受任何特定机制的束缚,前面的结果表明在坎普他汀第7位Trp残基上用氟原子替代吲哚氢可以加强吲哚环的氢键能,引入新的氢键能,或通过结合交界面的水分子介导与C3的相互作用。(Katragadda M等,2004)。因此,保持或增强上述氢键能,或通过结合交界面的水分子介导与C3的相互作用,在第7位Trp的修饰(例如根据本领域熟知方法改变侧链结构)或Trp类似物的置换包括在本发明中以生产具有甚至更大活性的类似物。在某些实施方案中,其吲哚环具有修饰的Trp类似物可以通过体外或体内表达,或通过肽合成而引入坎普他汀肽的第7位,所述修饰引起氢键能增加或通过结合交界面的水分子介导与C3的相互作用。发现在第7位包含色氨酸类似物5-氟代-色氨酸的肽(SEQ ID NO:19)与坎普他汀相比,活性增加121倍。
在另一个实施方案中,Trp类似物掺入坎普他汀分子的第4和7位,和任选坎普他汀第9位的His被Ala替代。热力学实验表明相对于野生型坎普他汀,在第4和7位掺入5-氟代-色氨酸增加坎普他汀和C3之间相互作用的焓。因此,经由第4位保持或增加上述与C3的疏水性相互作用和经由第7位保持或增加上述与C3的氢键能,或经由第7位通过结合交界面的水分子介导与C3的相互作用,在第4和7位Trp的修饰(例如根据本领域熟知方法改变侧链结构)或Trp类似物的置换包括在本发明中以生产甚至更大活性的坎普他汀类似物。这种修饰的Trp或Trp类似物可以通过体外或体内表达,或通过肽合成被引入坎普他汀肽的第4和7位。发现在第4和7位包含色氨酸类似物5-氟代-色氨酸的肽(SEQ ID NO:16)和包含色氨酸类似物6-氟代色氨酸(SEQ ID NO:17)具有比坎普他汀显著增加的活性,活性增加范围从112倍至264倍。此外,发现在第4位包含色氨酸类似物1-甲基-色氨酸和第7位包含5-氟代-色氨酸的肽(SEQ ID NO:24)相对于坎普他汀活性增加264倍。
羧基末端的修饰。通过合成法生产的肽通常羧基末端被修饰包含氨基而不是酸;这种常见修饰可以参见表1的坎普他汀(SEQ ID NO:1)和几个类似物。的确,在有些情况下,已经测定到含有末端氨基的肽具有比末端含酸的肽更大的活性(例如SEQ ID NOS:5和7分别与SEQ ID NOS:4和8相比)。因此,本发明的一个优选实施方案使用了C末端氨基修饰。然而,一些情况下在C末端使用酸有利。这种情况包括但不限于肽的溶解度考虑和肽从编码肽的核酸分子体外或体内的表达。
坎普他汀的羧基末端残基是苏氨酸。在本发明的一些实施方案中,C末端苏氨酸被一个或多个天然存在的氨基酸或类似物取代。例如,具有SEQ ID NO:6的肽包含D-苏氨酸而不是L-苏氨酸,和在C末端进一步具有COOH。这个肽显示出活性等于C末端包含L-苏氨酸和CONH2的肽SEQ ID NO:5。此外,His替代了第13位的Thr,获得了活性比坎普他汀大21倍的肽。此外SEQ ID NOS:14-17的肽,其C末端包含Asn的肽延伸,或Ala-Asn二肽延伸,以及C末端COOH和非乙酰化N-末端,证明活性比坎普他汀大38至126倍。它们还适于经由原核或真核生物表达系统来生产,如下面更详细描述的。
本发明的坎普他汀类似物可以通过各种肽合成法,根据常规肽合成方法经由一个或多个氨基酸残基的缩合来制备。例如,根据标准固相法合成肽,例如可以根据厂商说明书,在Applied Biosystems Model431A肽合成仪(Applied Biosystems,Foster City,加利福尼亚州)上进行。通过固相法或液相法合成肽或模拟肽的其他方法是本领域技术人员熟知的。在肽合成过程中,支链氨基和羧基可以根据需要被保护/去保护,使用通常已知的保护基。实施例3中列出了适当的肽合成法的实例。利用选择性肽和肽衍生物的保护基进行修饰对本领域技术人员来说是显而易见的。
作为选择,本发明的某些肽可以通过在适当的原核或真核系统表达来生产。例如,DNA构建体可以插入适宜于在细菌细胞(例如大肠杆菌)或酵母细胞(例如酿酒酵母)中表达的质粒载体,或插入用于在昆虫细胞中表达的杆状病毒载体或用于在哺乳动物细胞中表达的病毒载体。这种载体包含DNA在宿主细胞中表达所需的调控元件,其以使得允许DNA在宿主细胞中表达的方式安置。表达所需的这种调控元件包括启动子序列、转录起始序列和任选增强子序列。
SEQ ID NOS:14-17和设计相似的其他肽适于通过体外或体内表达核酸分子来生产。编码肽多连体的DNA构建体可以被引入体内表达系统,多连体的上限取决于所用的表达系统。多连体产生后,通过多肽暴露于肼来实现C末端Asn和下列N末端G之间的切割。
在重组原核或真核系统中表达基因而生产的肽可以根据本领域已知方法来纯化。实施例1和2列出了适于在本发明中使用的方法。在一个实施方案中,可使用市场上可买到的表达/分泌系统,由此重组肽被表达并此后从宿主细胞分泌出来,易于从周围培养基中纯化。
也可以使用基因表达和合成法组合生产坎普他汀类似物。例如,类似物可以通过基因表达来生产和此后经受一个或多个翻译后合成过程,例如修饰N-或C-末端或使分子环化。
坎普他汀的结构是本领域已知的,上述类似物的结构通过类似方法来测定。一旦已经确定了特别期望的短肽构象,设计肽或肽模拟物以适合那个构象的方法就是本领域熟知的。参见例如G.R.Marshall(1993),Tetrahedron,49:3547-3558;Hruby and Nikiforovich(1991),in Molecular Conformation and Biological Interactions,P.Balaram&S.Ramasehan,eds.,Indian Acad,of Sci.,Bangalore,PP.429-455)。与本发明特别相关的是,通过考虑氨基酸残基的各种侧链所起的作用可以进一步改善肽类似物的设计,如上讨论(即对于官能团或对于空间因素的影响)。
本领域技术人员会意识到为了提供与C3结合和抑制补体激活所需的特殊主链构象和侧链官能度,肽模拟物同样可以很好地充当肽。因此,通过使用天然存在的氨基酸、氨基酸衍生物、类似物或非氨基酸分子生产与C3结合、抑制补体的化合物包括在本发明范围内。非肽类似物或包含肽和非肽成分的类似物有时本文称为“肽模拟物”或“等构模拟物”以指明本发明肽的置换或衍生,其具有相同主链构象特征和/或其他官能度,以致它们与示例肽足够相似以抑制补体激活。
模拟肽用于开发高亲合力肽类似物的用途是本领域熟知的(参见例如Zhao B等,1995;Beeley,N.1994;和Hruby,VJ 1993)。假定旋转限制类似于肽内的氨基酸残基,可以分析包含非氨基酸部分的类似物,和通过拉马钱德兰图(Hruby&Nikiforovich 1991),连同其他已知技术来证实它们的构象基序。
本发明的坎普他汀类似物可以通过将聚乙二醇(PEG)成分添加至肽来修饰。如本领域熟知的,聚乙二醇化可以增加治疗肽和蛋白质在体内的半衰期。在一个实施方案中,PEG平均分子量大约1,000至大约50,000。在另一个实施方案中,PEG平均分子量大约1,000至大约20,000。在另一个实施方案中,PEG平均分子量大约1,000至大约10,000。在示例的实施方案中,PEG平均分子量大约5,000。聚乙二醇可以是支链或直链,和优选直链。
本发明的坎普他汀类似物可以经连接基与PEG共价结合。这种方法是本领域熟知的。(在Kozlowski A.等2001中综述;也参见Harris JM and Zalipsky S,eds.Poly(ethylene glycol),Chemistry and Biological Applications,ACS Symposium Series 680(1997))。可接受的连接基的非限制性实例包括酯基、酰胺基、酰亚胺基、氨基甲酸酯基、羧基、羟基、碳水化合物、琥珀酰亚胺基(包括不限于琥珀酰亚胺基琥珀酸盐(SS)、琥珀酰亚胺基丙酸盐(SPA)、琥珀酰亚胺基羧甲酸盐(SCM)、琥珀酰亚胺基琥珀酸胺(SSA)和N-羟基琥珀酰亚胺(NHS))、环氧基、氧羰基咪唑基(包括不限于羰基咪唑基(CDI))、硝苯基(包括不限于硝苯基碳酸盐(NPC)或三氯苯基碳酸盐(TPC))、trysylate基、醛基、异氰酸酯基、乙烯砜基、酪氨酸基、半胱氨酸基、组氨酸基、或伯胺。在某些实施方案中,连接基是琥珀酰亚胺基。在一个实施方案中,连接基是NHS。
本发明的坎普他汀类似物可以选择通过氨基、巯基、羟基或羧基直接与PEG偶联(即无连接基)。在一个实施方案中,PEG与赖氨酸残基连在一起被添加至坎普他汀C末端。
聚乙二醇化是一种增加治疗肽和蛋白质在体内保留时间的方法。通过将肽与某些其他肽连接还可以降低肽的体内清除率。例如,当某些白蛋白结合肽通过静脉内推注给兔子时,显示出罕见的2.3h的长半衰期(Dennis等,2002)。这个类型的肽与抗组织因子-D3H44的Fab融合使Fab能够与白蛋白结合,同时保留Fab结合组织因子的能力(Nguyen等,2006)。这种相互作用白蛋白导致与野生型D3H44Fab相比时,在小鼠和兔子体内清除率显著降低和半衰期延长,可与对于聚乙二醇化的Fab分子、免疫粘附素和白蛋白融合蛋白中所见的相匹敌。如本文实施例11所述,发明人合成了与白蛋白结合肽融合的坎普他汀类似物并证明了该融合蛋白有抑制补体激活的活性。
坎普他汀类似物、模拟肽和缀合物的补体激活抑制活性可以通过本领域已知的各种试验测试。在一个优选实施方案中,使用实施例4中描述的试验。美国专利6,319,897中列出了其他试验的非详尽清单,包括但不限于(1)肽与C3和C3片段结合试验;(2)各种溶血试验;(3)C3转变酶介导的C3切割的测定;和(4)因子D切割因子B的测定。
本文描述的肽和模拟肽具有实用性,用于应用坎普他汀本身的任何目的,如本领域已知的。这种应用包括但不限于:(1)在患者(人或动物)血清、组织或器官中抑制补体激活,其可以促进某些疾病或病情的治疗,包括但不限于年龄相关的黄斑变性、类风湿性关节炎、脊髓损伤、帕金森氏症和阿尔茨海默氏病;(2)抑制使用人造器官或植入物期间发生的补体激活(例如用本发明的肽涂布或否则处理人造器官);(3)抑制生理性流体(血、尿)的体外分流期间发生的补体激活(例如将本发明的肽涂布分流流体的管道);和(4)筛选小分子文库以鉴定其他的坎普他汀激活抑制剂(例如用来测定测试化合物与坎普他汀类似物竞争结合C3或C3片段的能力的液相或固相高通量试验)。
提供下列实施例更详细地描述本发明。它们的意图是说明,而不是限制本发明。使用实施例1-5中列出的材料和方法产生实施例6-11中描述的结果。
实施例1
坎普他汀的细菌表达
具有下列序列NH2-GICVWQDWGAHRCTN-OH(“G(-1)/V4W/H9A/N14”)(SEQ ID NO:15)的坎普他汀类似物以与壳多糖结合域和DnaB内含肽(New England Biolabs,Beverly,MA)融合的形式表达。在肽序列和大肠杆菌的密码子使用的指导下,使用下列遗传密码产生具有下列序列的这个肽的合成基因:
5’ATTTGCGTTTGGCAGGATTGGGGTGCGCACCGTTGCACCAATTAA3’(SEQ ID NO:29)
为将合成基因克隆入pGEM-T载体,设计含有SapI位点的5'侧翼区和含有PstI位点的3'侧翼区。为构建合成基因,使用DnaWorks软件设计如下显示的四个重叠的寡核苷酸并在Invitrogen Inc.(Carlsbad,CA)合成:
5'GGTGGTGCTCTTCCAACGGTATTTGCGTTTGGCAGGA3'(SEQ ID NO:30)
5'TTGGGGTGCGCACCGTTGCACCAATTAACTGCAGG3'(SEQ ID NO:31)
3'CAACGTGGTTAATTGACGTCCGC5'(SEQ ID NO:32)
3'CATAAACGCAAACCGTCCTAACCCCACGCGTGG5'(SEQ ID NO:33)
重叠DNA片段如Stemmer等,1995所述通过PCR组合。使用下列引物扩增所产生的基因:
5'CGCCTGCAGTTAATTGGT3'(SEQ IDNO:34)
5'GGTGGTGCTCTTCCAACG3'(SEQ IDNO:35)
然后将坎普他汀的PCR扩增片段克隆入pGEM-T载体,并用PstI和SapI消化所得克隆。将编码坎普他汀类似物的PstI-SapI片段进一步亚克隆入表达载体pTWIN1,该载体已经预先用PstI和SapI消化;通过DNA测序来证实该克隆的序列。
为表达坎普他汀类似物,用坎普他汀克隆转化的ER2566大肠杆菌细胞在SOB培养基(20g/L胰化蛋白胨、5g/L酵母膏、0.5g/L NaCl、2.5mM KCl、10mM MgCl2)中37℃生长。当OD600达到0.7时,添加IPTG至终浓度0.3mM诱导表达,随后在37℃另外孵育4小时。离心收集细胞和在补充0.2%Tween-20的缓冲液B1(20mM磷酸盐缓冲液,pH 8.5,500mM NaCl和1mM EDTA)中超声裂解。细胞提取物离心,可溶组分填入缓冲液B1预平衡过的壳多糖结合柱(New England Biolabs,Beverly,MA)。用100ml缓冲液B1洗涤柱,随后用3柱体积的缓冲液B2(50mM乙酸铵,pH 7.0)迅速洗涤。柱在室温下孵育20小时,用缓冲液B2洗脱肽,冻干并在C18HPLC柱上进一步纯化。使用MALDI-TOF质谱法鉴定纯化肽。
实施例2
坎普他汀的色氨酸类似物在大肠杆菌中的表达
为表达含有色氨酸衍生物的坎普他汀类似物,pTWTNl-坎普他汀克隆转化Trp 82营养缺陷型ER2566。在补充1mM如上所述的L-色氨酸的M9基本培养基中进行表达。细胞生长至OD6O0为0.8-1.0,然后离心收集并重悬于含有2mM期望的色氨酸类似物的新鲜基本培养基中,色氨酸类似物为:5-氟代-色氨酸,6-氟代-色氨酸、7-氮杂-色氨酸或5-羟色氨酸。表达的坎普他汀类似物进一步如实施例1所述纯化。
实施例3
肽合成
肽合成和纯化如Sahu等,1996;Sahu等,2000;和Mallik等,2005所述进行。简言之,使用Fmoc氨基树脂和标准的侧链保护基在Applied Biosystem肽合成仪(型号431A)中合成肽。肽在22℃与含有5%苯酚、5%苯硫基甲烷、5%水、2.5%乙二硫醇和82.5%三氟乙酸(TFA)的溶剂混合物孵育3小时,将其从树脂切下来。反应混合物通过玻璃烧结的漏斗过滤,用冷乙醚沉淀,溶于含0.1%TFA的50%乙腈,冻干。
将切割后获得的粗肽溶于含0.1%TFA的10%乙腈并使用反相C-18柱纯化(Waters,Milford,MA)。使用试剂三氟醋酸铊(III)通过树脂上环化法实现二硫化物氧化。这个方法删除了稀释溶液氧化的步骤和随后的反相HPLC前通过冻干而浓缩的耗时步骤。使用这个方法,不存在多聚体的形成并获得了高水平(~90%)的愚笨去保护、氧化或环化物质。激光解吸质谱法和HPLC证实了所有肽的同一性和纯度。
对于5-氟代-色氨酸、1-甲基-色氨酸和5-甲基-色氨酸类似物的合成,使用Fmoc-dl-衍生物。肽对映体的分离如Meyers等1978所述进行。每个肽的dl混合物在C18反相HPLC柱上使用含10%乙腈的0.01M乙酸铵,pH 4.1分离为d和l同质异构肽。用V8蛋白酶处理肽,随后使用MALDI-TOF质谱法(MicroMass TOFspec2E)分析,测定洗脱肽的同分异构的同一性。
实施例4
补体抑制试验
坎普他汀及其类似物对补体系统的抑制活性通过测定它们经免疫复合物对补体系统的激活作用来测定。补体激活抑制通过测定抑制正常人血浆中C3固定于卵白蛋白-抗卵白蛋白复合物来估计。Microliter孔用50μl卵白蛋白(10mg/ml)在25℃包被2小时(4℃包被过夜)。用10mg/ml BSA 200μl在25℃浸透该孔1小时,然后添加兔抗卵白蛋白抗体以形成可被补体激活的免疫复合物。各种浓度的三十微升肽直接添加至每孔,随后添加1:80稀释度的人血浆30μl。30分钟孵育后,使用山羊抗人C3HRP-偶联抗体检测结合的C3b/iC3b。添加ABTS过氧化物酶底物显色并在405nm测量光密度。
基于相当于100%补体激活的吸光度,将在405nm获得的吸光度数据转换为抑制%。抑制%对肽浓度作图,和使用Origin 7.0软件使所得数据集符合对数剂量反应函数。引起C3b/iC3b沉淀50%抑制的肽浓度作为IC50并用于比较各种肽的活性。从达到最低卡方值的符合参数中获得IC50值。
实施例5
C3与坎普他汀及其类似物相互作用的等温滴定量热法分析
使用Microcal VP-ITC量热仪(Micrical Inc,Northampton,MA)实施等温滴定量热实验。3.5-5μM的蛋白浓度和80-200μM的肽浓度用来做这个实验。全部滴定在PBS(含150mM NaCl的10mM磷酸盐缓冲液,pH 7.4)中进行。在每一个实验中,靶蛋白、C3输入细胞,和肽输入注射器。全部实验在25℃进行,对于每个实验,2μl肽注射剂进入含蛋白的细胞。在每个实验中,原始等温线通过减去稀释热减去代表进入缓冲液的肽注射剂的等温线进行校正。使所得等温线适于Origin 7.0软件内的各种模式,和卡方值达到最低的模式被认为适于各自的数据集。结合亲合力和熵值针对log P值作图。
实施例6
通过细菌表达的坎普他汀类似物估计的色氨酸在C3-坎普他汀相互作用中的作用
使用内含肽介导的蛋白表达系统将吲哚环化学性质不同的四种不同的色氨酸类似物引入坎普他汀。表达后,一步纯化肽,最终得率2mg/L培养物。还使用MALDI谱指出的ER2566/Trp 82营养缺陷型表达色氨酸类似物5-氟代-色氨酸、6-氟代-色氨酸、7-氮杂-色氨酸和5-羟色氨酸,纯化所得肽至均一。天然坎普他汀和类似物通过二硫键在体内被环化,通过它们不能与PHMB起反应而表明。在反相C18HPLC柱上进一步地纯化所有的肽。
所表达坎普他汀类似物G(-1)/V4W/H9A/N14(SEQ ID NO:15)的活性显示出1.2μM的IC50,这类似于对Ac-V4W/H9A类似物SEQ ID NO:5)观察到的活性。这个发现表明位于所表达肽N-末端的甘氨酸起着类似于位于Ac-V4W/H9A类似物N-末端的乙酰基的作用。
发现除7-氮杂-色氨酸类似物之外的所有表达的坎普他汀类似物在所测试的浓度下都具有活性。然而,肽显示出相对于类似物Ac-V4W/H9A不同水平的活性(图1;表2)。在第9位含6-氟代-色氨酸和5-氟代-色氨酸以及丙氨酸的坎普他汀分别显示出比Ac-V4W/H9A类似物高2.8和2.5倍的活性。
表⒉所表达肽的抑制活性
*相对于肽H-I(CVVQDWGHHRC)T-NH2(坎普他汀,SEQ ID NO:1)的活性c代表羟基
b代表合成肽
不受任何特定机制的限制,人们相信添加氟原子通过增加吲哚环的疏水性而增加肽的活性。也研究了掺入疏水性较低的色氨酸类似物5-羟色氨酸和7-氮杂-色氨酸。与5-氟代和6-氟代类似物的结果相比,含5-羟色氨酸的坎普他汀类似物与Ac-V4W/H9A类似物(SEQ ID NO:5)相比显示出活性降低27.5倍,和含7-氮杂-色氨酸的肽在所测试浓度下根本未显示出活性。7-氮杂-色氨酸在分子结构上类似于色氨酸,除了它在吲哚环的第7位具有一个氮原子,而不是碳原子。7-氮杂-色氨酸置换时观察到的活性降低显示出这个碳原子的相对重要性。
实施例7
各个色氨酸在C3-坎普他汀相互作用中的作用
使用固相肽合成产生在第4位、第7位或第4和第7两个位置选择性掺入含5-氟代-色氨酸,在第9位掺入丙氨酸的坎普他汀类似物。使用Fmoc-5-氟代-dL-色氨酸进行合成。这个反应产生含有5-氟代-d-色氨酸和5-氟代-L-色氨酸的肽的对映异构混合物。合成三种不同的肽:两种肽在第4或第7位独立地具有单个置换,和一个肽在第4和第7位含有置换。在单个置换的情况下可以产生5-氟代-L-色氨酸和5-氟代-d-色氨酸类似物的混合物,而在双置换的情况下就可能产生四个对映异构体组合的混合物。每种肽混合物进一步经受反相HPLC以分离肽对映异构体。对映异构体的鉴定通过用V8蛋白酶消化肽,随后使用MALDI分析消化的产品来进行。只有当其后是一个l-氨基酸时,V8蛋白酶才在Asp残基的C末端侧切开。质谱法鉴定切割产物表明首先洗脱的l-对映异构肽后面跟随d-形式,这种情况下没有检测到切割片段。
测试含5-氟代-L-色氨酸或5-氟代-d-色氨酸或两者的所有肽的补体抑制活性。在两个位置都被5-氟代-L-色氨酸置换的合成肽显示出比Ac-V4W/H9A(SEQ ID NO:5)高2.5倍的活性(表3)。
表⒊合成的含5-氟代-L-色氨酸的坎普他汀类似物的补体抑制活性
*相对于肽H-I(CVVQDWGHHRC)T-NH2(坎普他汀,SEQ ID NO:1)的活性
补体抑制试验(图2;表3)表明单独第4位的5-氟代-L-色氨酸的置换致使肽活性比Ac-V4W/H9A(SEQ ID NO:5)至少降低1.5倍。单独第7位的5-氟代-L-色氨酸的置换与Ac-V4W/H9A相比活性增加2.7倍。在第4和第7位5-氟代-L-色氨酸的同时置换也导致活性相对于Ac-V4W/H9A(SEQ ID NO:5)增加2.5倍。在第4或7位,或在两个位置5-氟代-d-色氨酸的置换致使肽无活性。
实施例8
色氨酸介导的C3识别坎普他汀的热力学基础
使用等温滴定量热法研究肽与C3的结合和研究它们的活性的热力学基础。使对于所有肽与C3的相互作用获得的量热数据适于化学计算接近于1的一组位点模式。据信这些肽与C3的结合以1:1的比例发生。由这些拟合产生的热动力参数在表4中示出。如Kd值显然可知,在第7位含5-氟代-L-色氨酸单个置换和在第4和7位含双置换显示出比Ac-V4W/H9A(SEQ ID NO:5)和Ac-V4(5f-l-W)/H9A(SEQ ID NO:18)类似物更强的结合。这个发现与补体抑制试验(表3)中观察到的相对活性一致,表明存在结合-活性相关性。
与C3结合的所有的肽具有负焓和正熵。这种结合是坎普他汀与C3相互作用的特征。在研究的所有肽之中,第7位置换的Ac-V4W/W7(5f-l-W)/H9A类似物(SEQ ID NO:19)显示出比野生型副本更高的结合焓(ΔH=-21.83,ΔΔH=-3.69)。与C3结合的第4位置换的Ac-V4(5f-l-W)/H9A类似物(SEQ ID NO:18)的焓是-16.69kcal/mole,显示比其野生型副本低1.45kcal/mole。
第4位掺入5-氟代-色氨酸引起焓相对于这个位置是色氨酸的肽减少1.45kcal/mole(表4)。由于色氨酸和5-氟代-色氨酸之间唯一的差异是吲哚C5的氟原子,因此这种焓减少可以归于氢被氟取代。
表4.合成的含5-氟代-L-色氨酸的坎普他汀类似物和C3相互作用的热动力参数
第7位掺入5-氟代-色氨酸,焓相对于野生型增加3.69kcal/mole(表4)。不受任何特定机制的限制,据信第7位色氨酸参与利于焓的相互作用,例如氢键。用氟原子取代一个吲哚氢可以加强吲哚NH的氢键特性,这是由于pKa下降。或者,氟形成氢键是它的给电子性质的结果,如十四碳(3-氟代酪氨酰)谷胱甘肽转移酶的结构所证明。
所观察到焓增加的另一种解释是水分子桥接氟原子和C3上氢受体之间的相互作用,这种情况下需要形成两个氢键(相当于大约4kcal/mole能量)。这个学说的支持来自所观察到的第7位置换的Ac-V4W/W7(5fW)/H9A类似物(SEQ ID NO:19)相对于野生型类似物的相互作用(表4)的熵减少,这种减少可以通过交界面上另外的水分子的结合而产生。在其他系统种已经观察到水介导的氟原子及其他氢键受体之间的相互作用。
双置换的类似物与C3的结合导致-19.85kcal/mole的焓变,-9.35kcal/mole的熵变和-10.5kcal/mole的自由能变化。相信在两个位置同时掺入5-氟代-色氨酸取消了单个置换的影响。
实施例9
另外的坎普他汀类似物
在第4位掺入色氨酸类似物。实施例5和6表明在坎普他汀的第4位用色氨酸置换缬氨酸使其活性增加45倍。为进一步研究坎普他汀与C3结合过程中第4位残基介导的相互作用的性质,用色氨酸类似物和2-萘基丙氨酸置换第4位的色氨酸。
使用基于ELISA的试验测试了第4位和第9位丙氨酸上含色氨酸类似物的所有肽类似物的活性。虽然用1-甲基-色氨酸(Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)和2-萘基丙氨酸(Ac-V4(2-Nal)/H9A)(SEQ ID NO:7)置换,活性比坎普他汀分别增加264和99倍,但是5-氟代-色氨酸(Ac-V4(5f-l-W)/W7/H9A)(SEQ ID NO:18和5-甲基色氨酸(Ac-V4(5-甲基-W)/H9A)(SEQ ID NO:22)的置换导致较低的活性;比野生型肽显示的活性高31和67倍(表5)。图3显示了描绘活性的抑制曲线和表5显示了由曲线计算的IC50值和肽与原坎普他汀的活性相比的相对活性。图5显示了针对色氨酸类似物和2-萘基丙氨酸的log P值描绘的抑制常数(IC50)。
表5.坎普他汀类似物的补体抑制活性
*相对于OfH-I(CVVQDWGHHRC)T-NH2(坎普他汀,SEQ ID NO:1)的活性。
也使用等温滴定量热法研究了坎普他汀肽的结合。使对于所有肽与C3的相互作用获得的量热数据适于化学计算接近于1的一组位点模式(图4)。这个结果提示这些肽与C3的结合以1:1的比例发生。由这些拟合产生的热动力参数在表6中示出。从Kd值显然可知,Ac-V4(1-甲基-W)/H9A与第4位具有单个置换的所有其他肽相比显示出更高的结合亲和力(Kd=0.015μM)。针对类似物的log P描绘这些值表明色氨酸类似物和2-萘基丙氨酸的结合亲合力和疏水性之间存在相关性。对于每个相关性,结合亲合力随着第4位掺入的类似物疏水性的增加而增加。这个观察结果与log P和抑制常数之间显示的相关性一致。
表6.合成的含5-氟代-L-色氨酸的坎普他汀类似物和C3相互作用的热动力参数
与C3结合的所有肽具有负焓和正熵,提示这种结合是焓驱动的。这种结合是坎普他汀与C3相互作用的特征。然而,这些肽结合的特征是焓变低于野生型,和熵变朝着有利端变动。图5B显示了log P对-TΔS的图,其表明随着第4位掺入的类似物的疏水性增加,熵更有利由此使得对自由能变化产生积极影响。
在第7位掺入色氨酸类似物。实施例7提出第7位色氨酸与C3上残基形成氢键。为进一步研究这个可能性,用类似于第4位取代的色氨酸类似物取代第7位色氨酸,以阐明在这个位置由色氨酸引起的相互作用的性质。具有5-氟代-色氨酸的置换(Ac-V4W/W7(5f-l-W)/H9A)(Seq ID NO:19)产生高活性121倍的肽。(图3,表5)。第7位用类似物5-甲基-色氨酸或1-甲基-色氨酸置换致使坎普他汀无活性(未显示数据)。因此,观察到色氨酸类似物的活性和疏水性之间无相关性。
通过量热法同时研究了不同的色氨酸7-类似物的热力学特性。(表6)。由于没有检测到在第7位含5-甲基-色氨酸或者1-甲基色氨酸的肽的结合,因此不存在结合参数。仅肽Ac-V4W/W7(5f-l-W)/H9A(SEQ ID NO:19)与C3结合。结合亲合力是0.035uM,这高于对于在第4位含有色氨酸类似物的肽所观察到的,除了肽Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)。与第4位具有色氨酸类似物的肽相比,与C3结合的Ac-V4W/W7(5f-l-W)/H9A(SEQ ID NO:19)具有很有利的结合焓变(ΔH=-21.83,ΔΔH=-3.69)和不利的熵变(-TΔS=11.56,-TΔΔS=2.77),提示进一步有利的极性的非共价相互作用。
该结果表明第7位掺入5-氟代-色氨酸导致坎普他汀活性增加,而类似物5-甲基-色氨酸和1-甲基-色氨酸的掺入致使坎普他汀无活性。1-甲基-色氨酸掺入时坎普他汀活性丧失支持色氨酸7的N-H介导的氢键对于坎普他汀与C3相互作用很重要的推论。此外,5-甲基-色氨酸掺入时坎普他汀活性完全丧失提示第7位不甚允许疏水性氨基酸。
在第4和第7两个位置掺入色氨酸类似物。由于用1-甲基-色氨酸置换第4位色氨酸和用5-氟代-色氨酸置换第7位色氨酸产生活性急剧增加的坎普他汀类似物,因此产生在第4和7位含置换的坎普他汀类似物。所得肽(Ac-V4(1-甲基-W)/W7(5f-l-W)/H9A)(SEQ ID NO:24)产生类似于用1-甲基-色氨酸(Ac-V4(1-甲基-W)/H9A)(SEQ ID NO:23)单个置换的抑制曲线(图3,表5)。量热器中观察到这个肽的结合亲合力(Kd=0.017)也类似于Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)。这些观察结果提示在这些实验条件下,在第4位1-甲基-色氨酸存在下第7位5-氟代-色氨酸没有作用。
在第4位掺入另一个色氨酸类似物。为进一步研究坎普他汀与C3结合过程中,第4位残基介导的相互作用的性质,用色氨酸类似物1-甲酰基-色氨酸取代第4位的色氨酸。
图6显示了补体抑制百分比vs.Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)(圆形)和Ac-V4(1-甲酰基-W)/H9A(SEQ ID NO:25)的肽浓度的比较。如所见,1-甲酰基-W类似物与1-甲基-W类似物的补体抑制活性基本上相同。
实施例10
坎普他汀类似物的聚乙二醇化
坎普他汀半衰期延长对于它在慢性治疗中的使用是有利的。通过聚乙二醇化,在几种情况下已经实现测试的治疗性肽半衰期延长(参见Veronese等,2001),因为PEG具有延迟生物分子被各种机制从循环中除去的能力,包括降低肾清除率、蛋白水解和免疫原性。聚乙二醇化包括PEG聚合物与大分子,优选与赖氨酸的伯胺的共价连接。
这个实施例描述了聚乙二醇化坎普他汀类似物Ac-V4(1-甲基-W)/H9A-K-PEG 5000(SEQ ID NO:36)的制备方法,并评价了该化合物抑制补体激活的能力。
Fmoc-NH-NHS-5000PEG购自Nektar transforming therapeutics,490discovery Dr,Huntsville,AL 35806。
化合物Ac-V4(1-甲基-W)/H9A-K-PEG 5000(SEQ ID NO:36)由Fmoc固相肽化学根据改进的标准规约进行化学合成。简言之,PEG溶于3ml二氯甲烷中,人工添加1ml 2M DIEA,和PEG混合5分钟。
然后将PEG转入容器中,使之偶联过夜。接着用20%哌啶对PEG去保护20分钟。
然后根据标准方案进行合成,为了使PEG与分子侧链连接,赖氨酸插入该分子C末端。
用Reagent D(TFA:H2O:TIS:苯酚,87.5:5:2.5:5)(4mL)在25℃90分钟实现肽的最终切割,以提供期望的产物。然后在C18反相HPLC柱上纯化肽、冻干和MALDI-TOF表征。
使用实施例4描述的体外试验测试聚乙二醇化的坎普他汀类似物的补体抑制活性。如图7所示,聚乙二醇化的类似物具有抑制补体激活的活性,然而,比非聚乙二醇化的类似物Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)达到相同抑制所需缀合物要多7倍。
实施例11
坎普他汀类似物的白蛋白结合蛋白缀合物
Dennis等(2002)鉴定了一系列具有核心序列DICLPRWGCLW(SEQ ID NO:37)的肽,它们以高亲合力特异性结合来自多个物种的血清白蛋白。化学计量法测定这些肽与白蛋白以1:1结合,位点不同于已知的小分子结合部位。肽SA21(AcRLIEDICLPRWGCLWEDDNH2;SEQ ID NO:38)当通过静脉内推注注射给兔子时,具有罕见的2.3h的长半衰期。如详细说明书中提及,与D3H44的抗组织因子Fab融合的相关序列使Fab能够以与SA21相似的亲合力结合白蛋白,同时保留Fab结合组织因子1的能力(Nguyen等2006)。在小鼠和兔子中,与白蛋白的这种相互作用导致体内清除率与野生型D3H44Fab相比降低25和58倍。在兔子中,半衰期延长37倍达32.4小时,和在小鼠中延长26倍达10.4小时,在这些动物中达到白蛋白半衰期的25-43%。超过Fab 2的这些半衰期可与聚乙二醇化的Fab分子、免疫粘附素和白蛋白融合物中所见的情形相匹敌。
这个实施例描述了与白蛋白结合肽融合的坎普他汀类似物的合成,和它在体外试验中抑制补体的活性。
化合物4(1MeW)-ABP由Fmoc固相肽化学根据标准规约进行化学合成。肽的N-和C末端用乙酰基和酰胺基保护。进一步在C18反相HPLC柱上纯化肽、冻干和MALDI质谱法TOF表征。
对于环化,肽-树脂(根据氨基酸分析0.10mmol/g填载)在二氯甲烷(DCM)(2mL)中溶胀5分钟、过滤和用94:1:5的DCM/TFA/TIS(5mL)25℃处理3次×2分钟,每次选择性去保护S-Mmt保护基,除去溶剂N2压力。用CH2Cl2、DMF和NMP(各5次×2分钟,2mL)洗涤这些二(硫醇)、二(Acm)-肽-树脂中间物、在NMP(2mL)中进一步溶胀5分钟,然后用含Et3N(2当量)的NMP 25℃处理4小时。然后用DMF和CH2Cl2洗涤肽-树脂(各5次×2分钟,2mL)。树脂结合形式的第一个环形成后,再次用DMF洗涤肽-树脂(5次×2分钟,2mL)并在DMF(2mL)中溶胀5分钟、过滤并用含T1(tfa)3(1.5当量)的DMF-苯甲醚(4mL)处理以环化第二个二硫环。25℃和缓搅搅拌4小时后,用DMF除去铊试剂(8次x 2分钟,2mL)和用CH2Cl2进一步洗涤肽-树脂(5次x 2分钟,2mL)。用Reagent D(TFA:H2O:TIS:苯酚,87.5:5:2.5:5)(4mL)在25℃90分钟完成双环肽的最终切割,以提供期望的产物。
生成的缀合肽(SEQ ID NO:39)如下所示。
使用实施例4描述的体外试验测试了白蛋白结合肽-坎普他汀的补体抑制活性。如图8所示,该缀合物具有抑制补体激活的活性,然而,比非缀合类似物Ac-V4(1-甲基-W)/H9A(SEQ ID NO:23)达到相同量抑制所需的缀合物要多7倍。
本发明涉及以下实施方式:
1.一种抑制补体激活的化合物,包含具有下述序列的肽:Xaa1-Cys-Val-Xaa2-Gln-Asp-Xaa3-Gly-Xaa4-His-Arg-Cys-Xaa5(SEQ ID NO:26);
其中:
Xaa1是Ile、Val、Leu、Ac-Ile、Ac-Val、Ac-Leu或包含Gly-Ile的二肽;
Xaa2是Trp或Trp的类似物,其中Trp的类似物与Trp相比疏水特性增加,条件是,如果Xaa3是Trp,则Xaa2是Trp的类似物;
Xaa3是Trp或在其吲哚环上包含化学修饰的Trp的类似物,其中化学修饰增加吲哚环的氢键能;
Xaa4是His、Ala、Phe或Trp;
Xaa5是L-Thr、D-Thr、Ile、Val、Gly、包含Thr-Asn或Thr-Ala的二肽、或包含Thr-Ala-Asn的三肽,其中L-Thr、D-Thr、Ile、Val、Gly或Asn中任何一个的羧基末端-OH任选被-NH2替代;并且
两个Cys残基通过二硫键相连。
2.实施方式1的化合物,其中Xaa2参与与C3的非极性相互作用。
3.实施方式1的化合物,其中Xaa2参与与C3的非极性相互作用,和Xaa3参与与C3的氢键键合。
4.实施方式1的化合物,其中Xaa3参与与C3的氢键键合。
5.实施方式1的化合物,其中Xaa2的色氨酸类似物包含卤代色氨酸。
6.实施方式5的化合物,其中卤代色氨酸是5-氟代-L-色氨酸或6-氟代-L-色氨酸。
7.实施方式1的化合物,其中Xaa2的色氨酸类似物在色氨酸第5位包含低级烷氧基或低级烷基取代基。
8.实施方式7的化合物,其中Xaa2的色氨酸类似物是5-甲氧基色氨酸或5-甲基色氨酸或1-甲基色氨酸或1-甲酰基色氨酸。
9.实施方式1的化合物,其中Xaa2的色氨酸类似物在色氨酸第1位包含低级烷酰基或低级烷基取代基。
10.实施方式9的化合物,其中Xaa2的色氨酸类似物是1-甲基色氨酸或1-甲酰基色氨酸。
11.实施方式1的化合物,其中Xaa3的色氨酸类似物包含卤代色氨酸。
12.实施方式11的化合物,其中卤代色氨酸是5-氟代-L-色氨酸或6-氟代-L-色氨酸。
13.实施方式1的化合物,其中Xaa4是Ala。
14.实施方式1的化合物,其中Xaa2在色氨酸第1位包含低级烷酰基或低级烷基取代基,Xaa3任选包含卤代色氨酸和Xaa4包含丙氨酸。
15.实施方式14的化合物,其中Xaa2是1-甲基色氨酸或1-甲酰基色氨酸和Xaa3任选包含5-氟代-L-色氨酸。
16.实施方式1的化合物,包含SEQ ID NOS:15-25任何序列。
17.实施方式1的化合物,其包括由编码该肽的多核苷酸表达而生产的肽。
18.实施方式1的化合物,其中该化合物至少部分由肽合成法生产。
19.实施方式1的化合物,包含SEQ ID NOS:15-25中的任何一个。
20.实施方式1的化合物,其中该化合物是聚乙二醇化的。
21.实施方式20的化合物,包含SEQ ID NO:36。
22.实施方式1的化合物,进一步包含延长该化合物体内保留时间的另外的肽成分。
23.实施方式22的化合物,其中另外的肽成分是白蛋白结合肽。
24.实施方式23的化合物,包含SEQ ID NO:39。
25.一种抑制补体激活的化合物,包含SEQ ID NO:26的非肽或部分肽模拟物,其中该化合物结合C3并抑制补体激活,在同等试验条件下,其活性比包含SEQ ID NO:1的肽大至少100倍。
参考文献:
Babitzke P,and Yanofsky C.(1995)Structural features of L-tryptophan required for activation of TRAP,the trp RNA-binding attenuation protein of Bacillus subtilis.J.Biol.Chem.270:12452-6.
Beeley N.(1994)Peptidomimetics and small-molecule drug design:towards improved bioavailability and in vivo stability.Trends Biotechnol.12:213-6.
Beene DL,Brandt GS,Zhong W,Zacharias NM,Lester HA,and Dougherty DA.(2002)Cation-pi interactions in ligand recognition by serotonergic(5-HT3A)and nicotinic acetylcholine receptors:the anomalous binding properties of nicotine.Biochemistry.41:10262-9.
Dennis MS,Zhang M,Meng YG,Kadkhodayan M,Kirchhofer D,Combs D,Damico LA.(2002)Albumin binding as a general strategy for improving the pharmacokinetics of proteins.J Biol Chem.277:35035-43
Fiane AE,Mollnes TE,Videm V,Hovig T,Hogasen K,Mellbye OJ,Spruce L,Moore WT,Sahu A,and Lambris JD.(1999a)Prolongation of ex vivo-perfused pig xenograft survival by the complement inhibitor Compstatin.Transplant.Proc.31:934-5.
Fiane AE,Mollnes TE,Videm V,Hovig T,Hogasen K,Mellbye OJ,Spruce L,Moore WT,Sahu A,and Lambris JD.(1999b)Compstatin,a peptide inhibitor of C3,prolongs survival of ex vivo perfused pig xenografts.Xenotransplantation.6:52-65.
Fiane AE,Videm V,Lambris JD,Geiran OR,Svennevig JL,and Mollnes TE.(2000)Modulation of fluid-phase complement activation inhibits hyperacute rejection in a porcine-to-human xenograft model.Transplant.Proc.32:899-900.
Furlong ST,Dutta AS,Coath MM,Gormley JJ,Hubbs SJ,Lloyd D,Mauger RC,Strimpler AM,Sylvester MA,Scott CW,and Edwards PD.(2000)C3 activation is inhibited by analogs of compstatin but not by serine protease inhibitors or peptidyl alpha-ketoheterocycles.
Immunopharmacology.48:199-212.
Hruby VJ.(1993)Conformational and topographical considerations in the design of biologically active peptides.Biopolymers.33:1073-82.
Kalli KR,Hsu P,Fearon and DT.(1994)Therapeutic uses of recombinant complement protein inhibitors.Springer Semin.Immunopathol.15:417-31.
Katragadda M,Morikis D,and Lambris JD.(2004)Thermodynamic studies on the interaction of the third complement component and its inhibitor,compstatin.J.Biol.Chem.279:54987-95.
Klepeis JL,Floudas CA,Morikis D,Tsokos CG,Argyropoulos E,Spruce L,and Lambris JD.(2003)Integrated computational and experimental approach for lead optimization and design of compstatin variants with improved activity.J.Am.Chem.Soc.125:8422-3.
Kozlowski A,Charles SA,and Harris JM.(2001)Development of pegylated interferons for the treatment of chronic hepatitis C.BioDrugs.15:419-29.
Mallik B,Katragadda M,Spruce LA,Carafides C,Tsokos CG,Morikis D,and Lambris JD(2005)Design and NMR Characterization of Active Analogs of Compstatin Containing Non-Natural Amino Acids.J.Med.Chem.48:274-286.
Morikis D,Assa-Munt N,Sahu A,and Lambris JD.(1998)Solution structure of Compstatin,a potent complement inhibitor.Protein Sci.7:619-27.
Nguyen A,Reyes AE 2nd,Zhang M,McDonald P,Wong WL,Damico LA,Dennis MS.(2006)The pharmacokinetics of an albumin-binding Fab(AB.Fab)can be modulated as a function of affinity for albumin.Protein Eng Des Sel.19:291-7.
Nilsson B,Larsson R,Hong J,Elgue G,Ekdahl KN,Sahu,A,and Lambris JD.(1998)Compstatin inhibits complement and cellular activation in whole blood in two models of extracorporeal circulation.Blood.92:1661-7.
Sahu A,Kay BK,and Lambris JD.(1996)Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library.J.Immunol.157:884-91.
Soulika AM,Khan MM,Hattori T,Bowen FW,Richardson BA,Hack CE,Sahu A,Edmunds LH Jr,and Lambris JD.(2000)Inhibition of heparin/protamine complex-induced complement activation by Compstatin in baboons.Clin.Immunol.96:212-21.
Spruce L.,E.Argyropoulos,D.Mastellos,G.Sfyroera,and J.D.Lambris(2002)Chemical synthesis of small complement proteins and protein modules.International Immunopharmacology.2:1320-1321.
Veronese FM.(2001)Peptide and protein PEGylation:a review of problems and solutions.
Biomaterials 22:405-417.
Wang Y,Hu Q,Madri JA,Rollins SA,Chodera A,and Matis LA.(1996)Amelioration of lupus-like autoimmune disease in NZB/WF1 mice after treatment with a blocking monoclonal antibody specific for complement component C5.Proc.Natl.Acad.Sci.USA.93:8563-8.
Zhao B,Helms LR,DesJarlais RL,Abdel-Meguid SS,and Wetzel R.(1995)A paradigm for drug discovery using a conformation from the crystal structure of a presentation scaffold.Nat.Struct.Biol.2:1131-7.
本发明不限于上面描述和举例说明的实施方案,而是能够在所附权利要求范围内进行变化和改进。
- 还没有人留言评论。精彩留言会获得点赞!