一种TCR‑/PD‑1‑双阴性T细胞及其构建方法与流程
本发明属于生物医药领域,具体地说涉及一种TCR-/PD-1-双阴性T细胞及其构建方法。
背景技术:
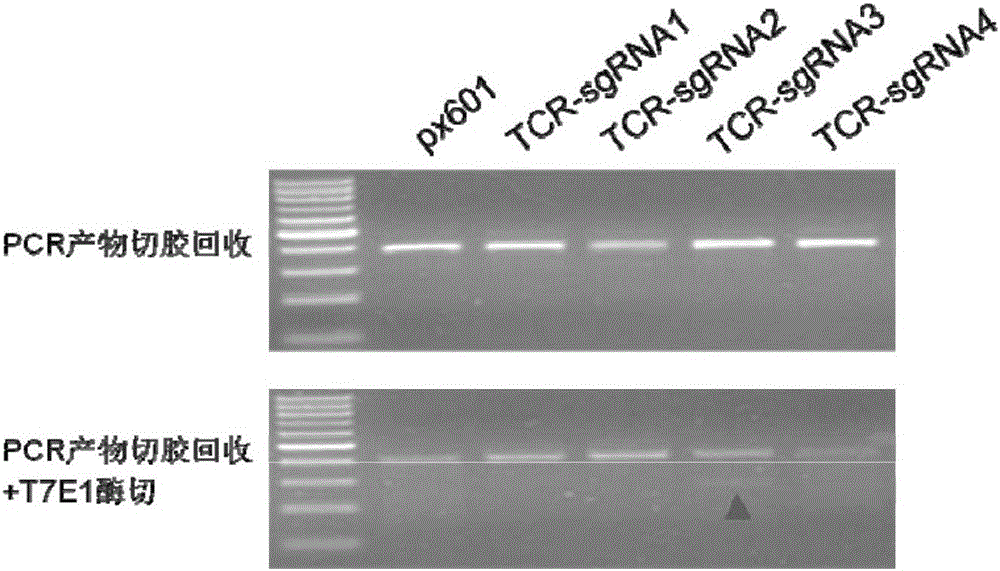
:随着免疫学的发展,肿瘤免疫治疗取得巨大进步。肿瘤免疫治疗通过激发或调动患者自身的免疫系统,增强肿瘤微环境抗肿瘤免疫力,从而控制和杀伤肿瘤细胞,被认为是继手术、放疗、化疗后第4大肿瘤治疗技术。近20年来,以过继细胞免疫疗法(adoptivecelltherapy,ACT)和免疫检查点疗法(immunechenkpointtherapy)疗法为代表的肿瘤免疫治疗取得了突破性进展,显示出良好的应用前景,标志着肿瘤治疗新时代的开启。过继细胞免疫疗法(ACT)是将活化的具有杀伤性的免疫细胞输给肿瘤病人,使其获得抗肿瘤免疫力,以达到治疗肿瘤的目的。过继细胞免疫疗法主要包括非特异性的淋巴因子激活的杀伤细胞(lymphokineactivatedkiller,LAK)疗法、细胞因子介导的杀伤细胞(cytokine-inducedkiller,CIK)疗法和特异性的肿瘤浸润淋巴细胞(tumorinfiltratinglymphocytes,TIL)疗法、细胞毒性T细胞(cytotoxicT-lymphocyte,CTL)疗法、T细胞受体(Tcellreceptors,TCR)疗法(TCRT)、嵌合抗原受体(Chimericantigenreceptors,CAR)修饰的T细胞疗法(CART)。其中CART是研究的主要重点。LAK细胞是利用白细胞介素-2(IL-2)刺激外周血单核细胞而得到的多种淋巴细胞的混合细胞。由于LAK需要使用大剂量的IL-2,毒副作用大,而且LAK细胞体外扩增能力较低。不能特异性聚集在肿瘤细胞表面,体内杀瘤活性不高,限制了临床应用。CIK是在体外将人外周血单核细胞利用多种细胞因子(如CD3单抗、IL-2、IFN-γ等)诱导而成的一群异质细胞,由于该种细胞同时表达CD3+和CD56+两种膜蛋白分子,故又被称为NK细胞样T淋巴细胞,理论上兼具有T淋巴细胞强大的抗瘤活性和NK细胞的非MHC限制性杀瘤优点。但是,目前CIK疗法仅对很少的病人具有一定的效果,总体疗效不好。TIL是从病人体内取出肿瘤组织,分离出其中的淋巴细胞经IL-2诱导扩增,然后回输病人体内增强免疫应答。TIL具有较高的特异性,杀瘤效果高于LAK和CIK。但是,TIL疗法面临2大难题:首先,病人在接受治疗之前,需要等待4-6周以扩增细胞;其次,需要从新鲜肿瘤组织分离,限制了其临床应用。CTL是在体外将人外周血单核细胞利用靶细胞抗原和淋巴因子的诱导、分化、扩增成具有强大杀伤能力的细胞,然后回输体内达到特异性清除病毒和杀伤肿瘤细胞的作用。但是肿瘤微环境会诱导CD8+T细胞升高抑制性受体PD-1的表达。肿瘤通过细胞表面PD-L1与之结合,显著抑制CD8+CTL清除肿瘤细胞的能力。大量的基础研究和临床实验表明抗PD-1或抗PDL-1的抗体能显著增强机体清除肿瘤的效率,因此通过敲除CTL的PD-1、PDL-1基因可能是提高CTL免疫治疗效果一个有效途径。TCRT是通过基因工程修饰后,使来自病人的T细胞表达能特异识别肿瘤细胞表面抗原-HLA复合物的T细胞受体,从而成为肿瘤特异性杀伤细胞。目前,TCRT已经表明能使一些黑色素瘤、结肠直肠癌和滑膜肉瘤患者的肿瘤减小。但是克隆与患者的免疫类型相匹配、与肿瘤细胞表面抗原高亲和力结合的TCR难度大。CART是分离肿瘤患者T细胞,通过基因工程嵌入特定的抗原受体(CAR),使得T细胞的靶向性、杀伤活性和持久性增强,并且对肿瘤细胞表面抗原的识别不依赖MHC的限制性。CARs由胞外抗原结合区、跨膜区、以及胞内的T细胞受体的信号转导区(如CD3ζ和CD28)组成。胞外抗原结合区由单克隆抗体的轻链(VL)和重链(VH)组成,中间由铰链连接形成单链抗体(singlechainfragmentvariable,scFv),能够识别特定的肿瘤抗原。有相关临床试验表明,CAR在其它治疗方法无效的淋巴癌患者身上具有较好的治疗效果。美国宾夕法尼亚大学CarlJune进行的CART-19研究表明75名白血病患者(包括成人和儿童患者),有45人经过CART细胞治疗之后病情完全缓解(completeremission)。除了CART会导致细胞因子风暴等副作用外,还存在3大问题:首先,对于一些因淋巴细胞数量较低或质量较差的晚期患者失去CART治疗的机会;其次,CART疗法在实体瘤中的疗效仍不显著,可能因为免疫抑制检验点信号通路的影响,导致免疫细胞在肿瘤组织内的存活率较差、活性不高;最后,由于CART是个体化治疗,成本昂贵,增加了病人负担。因此开发同种异体来源的通用CAR-T细胞能够推广其应用。Cellectis公司通过TALEN技术开发的异体CAR-T疗法UCART19定向敲除TCR-α基因(降低GVHD)和CD52基因(使细胞对alemtuzumab耐药)已经成功治愈了一例复发性急性淋巴细胞白血病(ALL)患儿。但是Cellectis通过TALEN敲除TCR需要繁琐的构建过程和大规模测序,并且脱靶率高。当前,规律成簇间隔短回文重复系统(clusteredregularlyinterspacedshortpalindromicrepeat;CRISPR-associated,CRISPR-Cas9)通过识别特定的DNA序列实现对基因的编辑,并且比TALEN更简单、高效。免疫检验点疗法是一类通过调节T细胞活性来提高抗肿瘤免疫反应的治疗方法。T细胞的激活需要双信号:一是MHC-多肽的信号;另一个是共刺激分子信号,主要有正向共刺激CD28、CD137等通路,以及调节T细胞不被过度刺激的负向共刺激CTLA4、PD1/PDL1通路。在共刺激信号激活之后,T细胞获得了有效的功能,可以到达肿瘤附近发挥杀伤作用。激活后的T细胞如何进入肿瘤微环境中又是一项难题。即使T细胞成功进入了微环境,还需要克服物理屏障,表皮细胞的阻挡,调节性T细胞的抑制作用,抑制性细胞因子等来发挥功能。其中T细胞负向共刺激分子的抑制性通路会被肿瘤利用来对抗免疫系统,使其逃脱机体免疫系统的监控和攻击。美国FDA批准了3种免疫检验点疗法药物,特异性结合T细胞表面CTLA-4受体的抗体类药物ipilimumab(Yervoy)以及特异性结合T细胞表面PD-1受体的抗体类药物pembrolizumab(Keytruda)和Opdivo(nivolumab)。2011年批准ipilimumab(Yervoy)用于治疗晚期黑色素瘤;2014年批准pembrolizumab(Keytruda)用于治疗对其它不再反应的晚期或不可切除黑色素瘤、Opdivo(nivolumab)用于治疗不再对其它药物响应的不可切除或转移性黑色素瘤患者;2015年批准用于Opdivo(nivolumab)治疗以铂类为基础化疗或化疗后疾病进展的晚期(转移性)非小细胞肺癌患者。相关临床试验显示,PD1/PDL1单抗比CTLA4单抗有更强的抗肿瘤作用。但是免疫检验点单抗会导致T细胞的过度激活和扩增等有关不良反应,一些患者的器官会发生临床上可观测到的自身免疫损伤。此外免疫检验点单抗研发困难、需要巨大的前期投入,阻断作用只是暂时性,需要长期用药,导致治疗费用昂贵。对于晚期癌症患者,上面介绍的免疫疗法为他们带来一丝希望。但是现有利用过继细胞免疫治疗或利用PD1抗体进行免疫检测阻断治疗肿瘤技术仍然存在一些问题:(1)对于一些因淋巴细胞数量较低或质量较差的晚期患者失去CART治疗的机会;(2)可能因为免疫抑制检验点信号通路的影响,CART疗法在实体瘤中的疗效仍不显著;(3)无论是过继细胞免疫治疗还是免疫检验点疗法都是个体化治疗,费用昂贵;(4)PD1抗体由于全身用药,导致自身免疫损伤。技术实现要素:本发明的目的在于克服现有技术在免疫治疗存在的以上问题,提供一种TCR-/PD-1-双阴性T细胞的构建方法,通过CRISPR/Cas9基因编辑技术敲除TCR和PD-1,并通过磁珠分选出TCR-/PD-1-双阴性T细胞,用于肿瘤的过继细胞免疫治疗等方面。本发明的目的在于提供一种TCR-/PD-1-双阴性T细胞。本发明的另一目的在于提供一种TCR-/PD-1-双阴性T细胞的构建方法。本发明的再一目的在于提供上述TCR-/PD-1-双阴性T细胞的应用。本发明所采取的技术方案是:一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列选自SEQIDNO:1~15中的任意一种,靶向人PD-1基因的sgRNA序列选自SEQIDNO:16~33中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列选自SEQIDNO:1~4中的任意一种,靶向人PD-1基因的sgRNA序列选自SEQIDNO:16~19中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列的反向互补DNA为SEQIDNO:34~48中的任意一种,靶向人PD-1基因的sgRNA序列的反向互补DNA为SEQIDNO:49~66中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列的反向互补DNA为SEQIDNO:34~37中的任意一种,靶向人PD-1基因的sgRNA序列的反向互补DNA为SEQIDNO:49~52中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的DNA寡核苷酸,靶向人TCR-α基因的sgRNA所对应的DNA寡核苷酸选自由SEQIDNO:67和68互补配对形成的双链DNA寡核苷酸、由SEQIDNO:69和70互补配对形成的双链DNA寡核苷酸、由SEQIDNO:71和72互补配对形成的双链DNA寡核苷酸、由SEQIDNO:73和74互补配对形成的双链DNA寡核苷酸中的任意一种;靶向人PD-1基因的sgRNA所对应的DNA寡核苷酸选自由SEQIDNO:75和76互补配对形成的双链DNA寡核苷酸、由SEQIDNO:77和78互补配对形成的双链DNA寡核苷酸、由SEQIDNO:79和80互补配对形成的双链DNA寡核苷酸、由SEQIDNO:81和82互补配对形成的双链DNA寡核苷酸中的任意一种。一种TCR-/PD-1-双阴性T细胞的构建方法,包括以下步骤:1)分别构建CRISPR/Cas9-TCR-sgRNA和CRISPR/Cas9-PD1-sgRNA载体CRISPR/Cas9-TCR-sgRNA载体的构建:将权利要求5所述的靶向人TCR-α基因的sgRNA所对应的DNA寡核苷酸与线性化的骨架载体进行连接,重组载体CRISPR/Cas9-TCR-sgRNA;CRISPR/Cas9-PD1-sgRNA载体的构建:将权利要求5所述的靶向人PD-1基因的sgRNA所对应的DNA寡核苷酸与线性化的骨架载体进行连接,重组载体CRISPR/Cas9-PD1-sgRNA;2)TCR-/PD-1-双阴性T细胞的获得:将分离的外周血单核细胞激活成T细胞,然后将上步制得的载体CRISPR/Cas9-TCR-sgRNA和CRISPR/Cas9-PD1-sgRNA共同转染T细胞,对转染后T细胞进行分离纯化,即可获得TCR-/PD-1-双阴性T细胞。进一步的,所述骨架载体选自px601-AAV-CMV、px602-AAV-CMV、px603-AAV-CMV、px552中的一种。TCR-/PD-1-双阴性T细胞在制备抗肿瘤药物中的应用。TCR-/PD-1-双阴性T细胞在制备防治病毒或细菌引起的感染性疾病药物中的应用。进一步的,所述病毒或细菌引起的感染性疾病包括乙肝病毒引起的乙型肝炎、HIV病毒引起的艾滋病。本发明的有益效果是:1)本发明通过CRISPR/Cas9基因编辑技术敲除TCR,不仅可以为淋巴细胞数量较低或质量较差的患者提供同种异体供体来源的CAR-T细胞,而且该T细胞是一种能携带针对不同肿瘤抗原的CAR的通用型CART细胞(UCART)。2)本发明使得所有同种肿瘤抗原的患者可以使用提前制备的同一种UCART,有利于CAR-T治疗的标准化,并且可以提前大规模制备,降低治疗成本。3)本发明通过CRISPR/Cas9基因编辑技术敲除PD-1基因,可以实现永久的效果,降低PD-1抗体导致的免疫损伤。4)由于肿瘤的异质性,单一的疗法(如CART或PD1抗体)可能并不能达到理想的治疗效果,然而本发明制备的TCR-/PD-1-双阴性T细胞具有杀瘤效果,可用于构建CART,相当于过继细胞免疫治疗和PD1抗体的联合治疗,达到意想不到的效果。本发明制备的TCR-/PD-1-双阴性T细胞同时还可以用于病毒或细菌引起的感染性疾病的治疗,如于乙肝病毒(HBV)引起的乙型肝炎(CHB)、HIV病毒引起的艾滋病等。附图说明图1为px601-AAV-CMV载体图谱;图2T7E1酶切检测本发明CRISPR/Cas9-TCR-sgRNA介导的TCR-α基因特异性切割;图中TCR-sgRNA1~4均为CRISPR/Cas9-TCR-sgRNA1~4的简写;图3为PCR产物测序验证TCR-α基因已经被敲除;图中TCR-sgRNA3为CRISPR/Cas9-TCR-sgRNA3的简写;图4为T7E1酶切检测本发明CRISPR/Cas9-PD1-sgRNA介导的PD-1基因特异性切割;图中PD1-sgRNA1~4均为CRISPR/Cas9-PD1-sgRNA1~4的简写;图5为PCR产物测序验证PD-1基因已经被敲除;图中PD1-sgRNA2~4均为CRISPR/Cas9-PD1-sgRNA2~4的简写;图6为TCR-/PD-1-双阴性T细胞体外杀瘤实验结果。具体实施方式一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列选自SEQIDNO:1~15中的任意一种,靶向人PD-1基因的sgRNA序列选自SEQIDNO:16~33中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列选自SEQIDNO:1~4中的任意一种,靶向人PD-1基因的sgRNA序列选自SEQIDNO:16~19中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列的反向互补DNA为SEQIDNO:34~48中的任意一种,靶向人PD-1基因的sgRNA序列的反向互补DNA为SEQIDNO:49~66中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNA,靶向人TCR-α基因的sgRNA序列的反向互补DNA为SEQIDNO:34~37中的任意一种,靶向人PD-1基因的sgRNA序列的反向互补DNA为SEQIDNO:49~52中的任意一种。一种基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的DNA寡核苷酸,靶向人TCR-α基因的sgRNA所对应的DNA寡核苷酸选自由SEQIDNO:67和68互补配对形成的双链DNA寡核苷酸、由SEQIDNO:69和70互补配对形成的双链DNA寡核苷酸、由SEQIDNO:71和72互补配对形成的双链DNA寡核苷酸、由SEQIDNO:73和74互补配对形成的双链DNA寡核苷酸中的任意一种;靶向人PD-1基因的sgRNA所对应的DNA寡核苷酸选自由SEQIDNO:75和76互补配对形成的双链DNA寡核苷酸、由SEQIDNO:77和78互补配对形成的双链DNA寡核苷酸、由SEQIDNO:79和80互补配对形成的双链DNA寡核苷酸、由SEQIDNO:81和82互补配对形成的双链DNA寡核苷酸中的任意一种。一种TCR-/PD-1-双阴性T细胞的构建方法,包括以下步骤:1)分别构建CRISPR/Cas9-TCR-sgRNA和CRISPR/Cas9-PD1-sgRNA载体CRISPR/Cas9-TCR-sgRNA载体的构建:将权利要求5所述的靶向人TCR-α基因的sgRNA所对应的DNA寡核苷酸与线性化的骨架载体进行连接,重组载体CRISPR/Cas9-TCR-sgRNA;CRISPR/Cas9-PD1-sgRNA载体的构建:将权利要求5所述的靶向人PD-1基因的sgRNA所对应的DNA寡核苷酸与线性化的骨架载体进行连接,重组载体CRISPR/Cas9-PD1-sgRNA;2)TCR-/PD-1-双阴性T细胞的获得:将分离的外周血单核细胞激活成T细胞,然后将上步制得的载体CRISPR/Cas9-TCR-sgRNA和CRISPR/Cas9-PD1-sgRNA共同转染T细胞,对转染后T细胞进行分离纯化,即可获得TCR-/PD-1-双阴性T细胞。优选的,所述骨架载体选自px601-AAV-CMV、px602-AAV-CMV、px603-AAV-CMV、px552中的一种。优选的,在转染T细胞之前,先在293FT细胞中验证所构建的载体CRISPR/Cas9-TCR-sgRNA和/或CRISPR/Cas9-PD1-sgRNA是否具有敲除活性。优选的,CRISPR/Cas9-TCR-sgRNA和/或CRISPR/Cas9-PD1-sgRNA转染293FT细胞的转染方法选自脂质体转染法、电穿孔转染法。优选的,步骤2)所述CRISPR/Cas9-TCR-sgRNA和CRISPR/Cas9-PD1-sgRNA转染T细胞的转染方法选自电穿孔转染法、病毒感染法。优选的,在对转染后T细胞进行分离纯化之前,先对转染后T细胞进行T7E1酶切和/或测序验证,验证T细胞中的TCR-α和PD-1基因是否被敲除。优选的,步骤2)所述分离纯化的具体操作为:通过使用TCR-α抗体和PD-1抗体经磁珠分选出高纯度的TCR-/PD-1-双阴性T细胞。优选的,所述抗体为生物素标记的抗体,所述磁珠分选系统为MIdiMACSStartingKit。优选的,所述Cas9蛋白是SaCas9,来源于金黄色葡萄球菌(Streptococcuspyogenes)。TCR-/PD-1-双阴性T细胞在制备抗肿瘤药物中的应用。TCR-/PD-1-双阴性T细胞在制备防治病毒或细菌引起的感染性疾病药物中的应用。优选的,所述病毒或细菌引起的感染性疾病包括乙肝病毒引起的乙型肝炎、HIV病毒引起的艾滋病。下面结合具体实施例对本发明作进一步的说明,但并不局限于此。实施例1基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的sgRNAPD-1是T细胞表面的一种抑制性受体,TCR(T细胞受体)是T细胞特异性识别和结合抗原肽-MHC分子的分子结构,通常与CD3分子呈复合物形式存在于T细胞表面;大多数T细胞的TCR由α和β肽链组成。为了构建TCR-/PD-1-双阴性T细胞,为了尽可能的破坏靶基因的读码框,本发明目的在于获得对靶基因TCR-α和PD1具有敲除活性的sgRNA。本发明获得了15种对靶向基因TCR-α的sgRNA,和18种靶向基因PD-1的sgRNA。其中,靶向人TCR-α基因的sgRNA序列选自SEQIDNO:1~15中的任意一种;序列SEQIDNO:1~15的反向互补DNA序列如SEQIDNO:34~48所示。靶向人PD-1基因的sgRNA序列选自SEQIDNO:16~33中的任意一种;序列选自SEQIDNO:16~33的反向互补DNA序列如SEQIDNO:49~66所示。下文中分别选取了4个TCR-α的sgRNA(SEQIDNO:1~4)和4个PD-1的sgRNA(SEQIDNO:16~19)进行后续试验。实施例2基于CRISPR/Cas9系统构建TCR-/PD-1-双阴性T细胞的DNA寡核苷酸根据上述实施例1设计的sgRNA合成相应的DNA寡核苷酸,在正向寡核苷酸5’加上CACC,在反向寡核苷酸5’加上AAAC;上述TCR-α的sgRNA序列SEQIDNO:1~4所对应的正向寡核苷酸的序列分别为SEQIDNO:67、69、71、73,所对应的反向寡核苷酸的序列分别为SEQIDNO:68、70、72、74;PD-1的sgRNA序列SEQIDNO:16~19所对应的正向寡核苷酸的序列分别为SEQIDNO:75、77、79、81,所对应的反向寡核苷酸的序列分别为SEQIDNO:76、78、80、82。将上述合成的正向寡核苷酸序列和反向寡核苷酸序列分别配对退火,形成双链DNA寡聚核苷酸,TCR-α和PD-1的sgRNA所对应的DNA寡核苷酸的具体配对情况如下:靶向人TCR-α基因的sgRNA所对应的DNA寡核苷酸有:由SEQIDNO:67和68互补配对形成的双链DNA寡核苷酸(命名为TCR-DNAOligos-1)、由SEQIDNO:69和70互补配对形成的双链DNA寡核苷酸(命名为TCR-DNAOligos-2)、由SEQIDNO:71和72互补配对形成的双链DNA寡核苷酸(命名为TCR-DNAOligos-3)、由SEQIDNO:73和74互补配对形成的双链DNA寡核苷酸中(命名为TCR-DNAOligos-4)。靶向人PD-1基因的sgRNA所对应的DNA寡核苷酸有:由SEQIDNO:75和76互补配对形成的双链DNA寡核苷酸(命名为PD1-DNAOligos-1)、由SEQIDNO:77和78互补配对形成的双链DNA寡核苷酸(命名为PD1-DNAOligos-2)、由SEQIDNO:79和80互补配对形成的双链DNA寡核苷酸(命名为PD1-DNAOligos-3)、由SEQIDNO:81和82互补配对形成的双链DNA寡核苷酸(命名为PD1-DNAOligos-4)。实施例3CRISPR/Cas9-TCR-sgRNA质粒的构建方法1)将px601-AAV-CMV质粒(图谱如图1所示,下文均简写为px601)进行酶切,得线性化px601质粒;所述酶切体系如下:1μgpx601质粒;2μl10×FastDigest®buffer;1μlFastDigest®BsaI(ThermoScientific);补水至20μl,37℃孵育1小时,然后切胶回收。2)连接将实施2所得的双链DNA寡核苷酸(TCR-DNAOligos-1~4)分别与线性化的px601进行连接,连接体系如下:2.5μlpx601质粒;2.5μl退火的双链sgRNA;5μlSolutionI(Takara);16℃孵育1小时。3)转化:将上述连接产物转化到大肠杆菌DH5α感受态细胞,37℃孵育16-18h,并挑取阳性克隆。4)验证:用Omega的Endo-freeplasmidMiniKitII-fast抽提阳性克隆菌中无内毒素的CRISPR/Cas9-TCR-sgRNA质粒,用如序列表SEQIDNO:83所示的引物U6,用常规测序的方法鉴定获得阳性克隆质粒;测序列正确的质粒即为CRISPR/Cas9-TCR-sgRNA质粒;TCR-DNAOligos-1~4所对应的重组质粒分别命名为CRISPR/Cas9-TCR-sgRNA1、CRISPR/Cas9-TCR-sgRNA2、CRISPR/Cas9-TCR-sgRNA3、CRISPR/Cas9-TCR-sgRNA4。实施例4CRISPR/Cas9-TCR-sgRNA的活性筛选一、细胞传代与转染转染前一天每孔按约2x105个细胞的密度将HEK293FT细胞接种到24孔板,加DMEM培养基至400μL,培养过夜。一共转染5组:其中px601作为后续T7E1酶切实验阴性对照,CRISPR/Cas9-TCR-sgRNA1、CRISPR/Cas9-TCR-sgRNA2、CRISPR/Cas9-TCR-sgRNA3、CRISPR/Cas9-TCR-sgRNA4作为后续T7E1酶切实验组,然后按下面步骤进行转染:1)对于每个孔的细胞,在无菌的1.5ml离心管加入0.8μg的DNA至50μl没有血清的培养基OptiMEM®IMedium中,室温下放置5min。2)对于每个孔的细胞,在无菌的1.5ml离心管加入2μl的Lipofectamine2000至50μl没有血清的培养基OptiMEM®IMedium中,在室温下放置5min。3)5min之后,分别将含有质粒(CRISPR/Cas9-TCR-sgRNA1、CRISPR/Cas9-TCR-sgRNA2、CRISPR/Cas9-TCR-sgRNA3、CRISPR/Cas9-TCR-sgRNA4)和脂质体的OptiMEM®Medium混合(这种混合必须在30min之内完成,过长时间则会导致活性下降),轻轻地混匀,然后在室温下放置20min使之形成质粒-脂质体2000复合体。4)将100μl质粒-脂质体2000复合体直接加入到300μl不含抗生素的DMEM培养基中,然后将其加入到含有细胞的孔中,轻轻地混匀进行转染,转染后6-12h换液,72h后分别收集转染了px601、CRISPR/Cas9-TCR-sgRNA1、CRISPR/Cas9-TCR-sgRNA2、CRISPR/Cas9-TCR-sgRNA3、CRISPR/Cas9-TCR-sgRNA4的细胞并提取基因组DNA(使用TransEasyPureGenomicDNAKit试剂盒)。二、T7E1酶切检测敲除1)PCR扩增目的片段分别以提取转染px601、CRISPR/Cas9-TCR-sgRNA1、CRISPR/Cas9-TCR-sgRNA2、CRISPR/Cas9-TCR-sgRNA3、CRISPR/Cas9-TCR-sgRNA4质粒的HEK293FT细胞的基因组DNA为模板,以TCR-seq-F(如SEQIDNO:84所示)和TCR-seq-R(如SEQIDNO:85所示)为引物对扩增出包含各sgRNA靶序列的目的片段,使用AxyPrepDNA凝胶回收试剂盒纯化PCR产物。2)PCR产物变性退火各取200ngPCR产物按下列体系(19.5μL)变性退火:成分用量DNA200ng10×NEBBuffer22μlddH2O加到19.5μL变性退火程序如下:95℃,5min;95–85℃,-2℃/s;85–25℃,-0.1℃/s;4℃,∞。3)T7E1酶切反应向上述变性退火后的体系中加入0.5μlT7E1,37℃反应20min后,加入1.5μl0.25MEDTA,2%凝胶电泳检测。电泳检测结果如图2所示,通过琼脂糖胶电泳可以发现:CRISPR/Cas9-TCR-sgRNA3有T7E1切割条带(红色箭头),说明CRISPR/Cas9-TCR-sgRNA3敲掉了TCR-α。三、PCR产物测序验证TCR-α基因是否被敲除将上述PCR产物不进行变性退火,直接切胶回收测序,测序结果如图3所示,sgRNA靶序列以红色方框标出,PAM序列以红线标出,从CRISPR/Cas9-TCR-sgRNA3的靶序列(红色方框)开始出现套峰,说明CRISPR/Cas9-TCR-sgRNA3引起了TCR-α基因突变,即成功敲除了TCR-α基因。实施例5CRISPR/Cas9-PD1-sgRNA质粒的构建方法CRISPR/Cas9-PD1-sgRNA质粒的构建方法同实施例3中所述的CRISPR/Cas9-TCR-sgRNA质粒的构建方法。最终PD1-DNAOligos-1~4对应所获得的重组质粒分别命名为CRISPR/Cas9-PD1-sgRNA1、CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4。实施例6CRISPR/Cas9-PD1-sgRNA的活性筛选一、细胞传代与转染转染前一天每孔按约2x105个细胞的密度将HEK293FT细胞接种到24孔板,加DMEM培养基至400μl,培养过夜。一共转染5组:其中px601作为后续T7E1酶切实验阴性对照,CRISPR/Cas9-PD1-sgRNA1、CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4作为后续T7E1酶切实验组,然后按下面步骤进行转染:1)对于每个孔的细胞,在无菌的1.5ml离心管加入0.8μg的DNA至50μl没有血清的培养基OptiMEM®IMedium中,室温下放置5min。2)对于每个孔的细胞,在无菌的1.5ml离心管加入2μl的Lipofectamine2000至50μl没有血清的培养基OptiMEM®IMedium中,在室温下放置5min。3)5min之后,分别将含有质粒(CRISPR/Cas9-PD1-sgRNA1、CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4)和脂质体的OptiMEM®Medium混合(这种混合必须在30min之内完成,过长时间则会导致活性下降),轻轻地混匀,然后在室温下放置20min使之形成质粒-脂质体2000复合体。(4)将100μl质粒-脂质体2000复合体直接加入到300μl不含抗生素的DMEM培养基中,然后将其加入到含有细胞的孔中,轻轻地混匀进行转染;转染后6-12h换液,72h后收集转染了px601、CRISPR/Cas9-PD1-sgRNA1、CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4的细胞并提取基因组DNA(使用TransEasyPureGenomicDNAKit试剂盒)。二、T7E1酶切检测敲除1)PCR扩增目的片段分别以提取转染了px601、CRISPR/Cas9-PD1-sgRNA1、CRISPR/Cas9-PD1-sgRNA4质粒的HEK293FT细胞的基因组DNA为模板,以PD1-seq-F14(如SEQIDNO:86所示)和PD1-seq-R14(如SEQIDNO:87所示)为引物对扩增出包含各sgRNA靶序列的目的片段;分别以提取转染px601、CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3质粒的HEK293FT细胞的基因组DNA为模板,以PD1-seq-F23(如SEQIDNO:88所示)和PD1-seq-R23(如SEQIDNO:89所示)为引物对扩增出包含各sgRNA靶序列的目的片段。使用AxyPrepDNA凝胶回收试剂盒纯化PCR产物。2)PCR产物变性退火各取200ngPCR产物按下列体系(19.5μl)变性退火:变性退火程序如下::95℃,5min;95–85℃,-2℃/s;85–25℃,-0.1℃/s;4℃,∞。3)T7E1酶切反应向上述变性退火后的体系中加入0.5μlT7E1,37℃反应20min后,加入1.5μl0.25MEDTA,2%凝胶电泳检测。电泳检测结果如图4所示,CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4都有T7E1切割条带(红色),说明CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4都能敲除PD-1基因。三、PCR产物测序验证PD-1基因是否被敲除将上述PCR产物不进行变性退火,直接切胶回收测序,测序结果如图5所示,sgRNA靶序列以红色方框标出,PAM序列以红线标出,PD1-sgRNA2、PD1-sgRNA3、PD1-sgRNA4的靶序列开始都出现套峰,说明CRISPR/Cas9-PD1-sgRNA2、CRISPR/Cas9-PD1-sgRNA3、CRISPR/Cas9-PD1-sgRNA4都引起了PD-1基因突变,即成功敲除了PD-1基因。实施例7TCR-/PD-1-双阴性T细胞的构建方法1)T细胞的制备分离外周血单核细胞(PBMC)并用CD3、CD28单抗激活,IL-2(终浓度300IU/ml)持续培养,即得激活的T细胞。2)转染将上步分离培养72h后的T细胞,通过电穿孔转染法将筛选出的具有敲除活性的CRISPR/Cas9-TCR-sgRNA(构建方法如实施例3所示)和CRISPR/Cas9-PD1-sgRNA(构建方法如实施例5所示)质粒共同转染到T细胞。3)T7E1酶切和测序检测敲除效果转染72h后,提取细胞基因组DNA,然后用T7E1酶检测敲除效率,并将PCR产物通过TA克隆测序进一步验证TCR-α和PD-1基因是否被敲除。4)磁珠分选出TCR-/PD-1-双阴性T细胞将检测具有敲除效果的转染细胞用生物素标记的TCR抗体和生物素标记的PD-1抗体经磁珠(磁珠分选系统为MIdiMACSStartingKit)分选出高纯度的TCR-/PD-1-双阴性T细胞。实施例8TCR-/PD-1-双阴性T细胞体外杀瘤实验LDH释放法测定细胞杀伤活性(碧云天乳酸脱氢酶细胞毒性检测试剂盒C1007):接种100μl1×104/孔靶细胞AGS(PDL1阳性)到96孔细胞培养板。靶细胞自然释放孔(阴性对照)和最大释放孔(阳性对照)加100μl培养液;各实验孔加100μl实施例7制备的TCR-/PD-1-双阴性T细胞(效应细胞),设置按不同的效靶比(E/T值1:1、5:1、10:1、20:1),每组设三个复孔。37℃,5%CO2,培养24h。在最大释放孔中加入试剂盒提供的LDH释放试剂,加入量为原有培养液体积的10%(20μl)。加入LDH释放试剂后,反复吹打数次混匀,然后继续在细胞培养箱中孵育1h。到达预定时间后,将细胞培养板用多孔板离心机400g离心5min。分别取各孔的上清液120μl,加入到一新的96孔板相应孔中,然后各孔分别加入60μlLDH检测工作液。混匀,室温(约25℃)避光孵育30min(可用铝箔包裹后置于水平摇床或侧摆摇床上缓慢摇动)。然后在490nm处测定吸光度。实验过程中,同时用患者自体来源的T细胞(效应细胞),进行上述相同的实验,作为对照比较。根据以下公式计算细胞杀伤活性:杀伤活性(%)=[(实验组A值-总自然释放A值)/(最大释放组A值-总自然释放A值)]×100%。检测结果如图6所示,从中可以看出,当E/T值大于1时,本发明制备的TCR-/PD-1-双阴性T细胞的肿瘤杀伤能力明显强于患者自体来源的T细胞,尤其是当E/T值为20时,本发明TCR-/PD-1-双阴性T细胞的肿瘤杀伤能力达50%,而患者自体来源T细胞的肿瘤杀伤能力仅约为28%。实施例8TCR-/PD-1-双阴性T细胞在病毒或细菌引起的感染性疾病中的应用PD-1/PDL-1信号通路的免疫抑制作用对多种病毒或细菌引起的感染性疾病的发生、发展具有重要作用。在人HIV、HBV、HCV感染患者体内发现PD-1在病毒特异性T细胞过量表达,抑制T细胞对病毒的杀伤作用,造成病毒慢性持续性感染。研究发现HIV特异性CD8+T细胞上PD-1分子高水平表达,阻断PD-1信号能够提高T细胞的增殖和和效应能力(Dayetal.,2006)。HIV病程的严重程度与HIV特异性CD8+T细胞上PD-1表达水平和PD-1表达的阳性率之间有显著相关性,高水平表达的PD-1也与体外抗原刺激后特异性CD8+T细胞低下的增殖能力有关系(Dayetal.,2006;Trautmannetal.,2006)。这些发现显示PD-1与T细胞功能衰竭有关。此外,HCV和HBV也利用PD-1/PDL-1通路抑制机体免疫反应。在急性和HBV和HCV感染早期,病毒特异性CTL细胞PD-1分子高水平表达,随着急性感染者的康复,PD-1的表达明显下降。而HBV和HCV慢性感染者特异性CTL细胞PD-1分子仍然维持高水平表达,并伴随着功能低下;体外阻断PD-1/PDL-1相互作用能够恢复这些CTL细胞因子释放和增殖能力。因此将实施例7制备的TCR-/PD-1-双阴性T细胞利用靶抗原和淋巴因子的诱导、分化、扩增成CD8+CTL,然后回输给患者可能具有较好的治疗效果。综上所述,从理论上来说,本发明TCR-/PD-1-双阴性T细胞对相关的病毒或细菌引起的感染性疾病也具有一定的治疗效果,所述病毒或细菌引起的感染性疾病包括但不限于乙肝病毒(HBV)引起的乙型肝炎(CHB)、HIV病毒引起的艾滋病等。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。<110>广东华南联合疫苗开发院有限公司<120>一种TCR-/PD-1-双阴性T细胞及其构建方法<130><160>89<170>PatentInversion3.5<210>1<211>20<212>RNA<213>人工序列<400>1gucucucagcugguacacgg20<210>2<211>21<212>RNA<213>人工序列<400>2gcagcugguacacggcagggu21<210>3<211>21<212>RNA<213>人工序列<400>3gauaggcagacagacuuguca21<210>4<211>21<212>RNA<213>人工序列<400>4guacacggcagggucaggguu21<210>5<211>20<212>RNA<213>人工序列<400>5guacuuggcuggacagcaag20<210>6<211>20<212>RNA<213>人工序列<400>6gacagacuugucacuggauu20<210>7<211>21<212>RNA<213>人工序列<400>7guuguuugagaaucaaaaucg21<210>8<211>21<212>RNA<213>人工序列<400>8gacuuugugacacauuuguuu21<210>9<211>21<212>RNA<213>人工序列<400>9gaaacaaaugugucacaaagu21<210>10<211>21<212>RNA<213>人工序列<400>10guuugucugugauauacacau21<210>11<211>21<212>RNA<213>人工序列<400>11gcuggggaagaaggugucuuc21<210>12<211>21<212>RNA<213>人工序列<400>12gcuuucaaaaccugucaguga21<210>13<211>21<212>RNA<213>人工序列<400>13gaaccugucagugauuggguu21<210>14<211>21<212>RNA<213>人工序列<400>14gaauccuccuccugaaagugg21<210>15<211>21<212>RNA<213>人工序列<400>15gaacccggccacuuucaggag21<210>16<211>20<212>RNA<213>人工序列<400>16gagagccugcgggcagagcu20<210>17<211>20<212>RNA<213>人工序列<400>17gcuacaacugggcuggcggc20<210>18<211>21<212>RNA<213>人工序列<400>18gacgacuggccagggcgccug21<210>19<211>21<212>RNA<213>人工序列<400>19gcagggcuggggagaaggugg21<210>20<211>20<212>RNA<213>人工序列<400>20gggguuccagggccugucug20<210>21<211>20<212>RNA<213>人工序列<400>21ggcuggccugggugaggggc20<210>22<211>21<212>RNA<213>人工序列<400>22guuuggaacuggccggcuggc21<210>23<211>21<212>RNA<213>人工序列<400>23gccgcccacgacaccaaccac21<210>24<211>20<212>RNA<213>人工序列<400>24ggcagccuggugcugcuagu20<210>25<211>21<212>RNA<213>人工序列<400>25gagagaacacaggcacggcug21<210>26<211>21<212>RNA<213>人工序列<400>26gucucuguggacuauggggag21<210>27<211>21<212>RNA<213>人工序列<400>27gacacagggcacggggggcuc21<210>28<211>20<212>RNA<213>人工序列<400>28gcccugugucccugagcaga20<210>29<211>20<212>RNA<213>人工序列<400>29gccaccauugucuuuccuag20<210>30<211>20<212>RNA<213>人工序列<400>30gccaccauugucuuuccuag20<210>31<211>21<212>RNA<213>人工序列<400>31gcagcugagccccugcgggcg21<210>32<211>20<212>RNA<213>人工序列<400>32ggggcucagcugacggcccu20<210>33<211>20<212>RNA<213>人工序列<400>33gugcccagccacugaggccu20<210>34<211>20<212>DNA<213>人工序列<400>34ccgtgtaccagctgagagac20<210>35<211>21<212>DNA<213>人工序列<400>35accctgccgtgtaccagctgc21<210>36<211>21<212>DNA<213>人工序列<400>36tgacaagtctgtctgcctatc21<210>37<211>21<212>DNA<213>人工序列<400>37aaccctgaccctgccgtgtac21<210>38<211>20<212>DNA<213>人工序列<400>38cttgctgtccagccaagtac20<210>39<211>20<212>DNA<213>人工序列<400>39aatccagtgacaagtctgtc20<210>40<211>21<212>DNA<213>人工序列<400>40cgattttgattctcaaacaac21<210>41<211>21<212>DNA<213>人工序列<400>41aaacaaatgtgtcacaaagtc21<210>42<211>21<212>DNA<213>人工序列<400>42actttgtgacacatttgtttc21<210>43<211>21<212>DNA<213>人工序列<400>43atgtgtatatcacagacaaac21<210>44<211>21<212>DNA<213>人工序列<400>44gaagacaccttcttccccagc21<210>45<211>21<212>DNA<213>人工序列<400>45tcactgacaggttttgaaagc21<210>46<211>21<212>DNA<213>人工序列<400>46aacccaatcactgacaggttc21<210>47<211>21<212>DNA<213>人工序列<400>47ccactttcaggaggaggattc21<210>48<211>21<212>DNA<213>人工序列<400>48ctcctgaaagtggccgggttc21<210>49<211>20<212>DNA<213>人工序列<400>49agctctgcccgcaggctctc20<210>50<211>20<212>DNA<213>人工序列<400>50gccgccagcccagttgtagc20<210>51<211>21<212>DNA<213>人工序列<400>51caggcgccctggccagtcgtc21<210>52<211>21<212>DNA<213>人工序列<400>52ccaccttctccccagccctgc21<210>53<211>20<212>DNA<213>人工序列<400>53cagacaggccctggaacccc20<210>54<211>20<212>DNA<213>人工序列<400>54gcccctcacccaggccagcc20<210>55<211>21<212>DNA<213>人工序列<400>55gccagccggccagttccaaac21<210>56<211>21<212>DNA<213>人工序列<400>56gtggttggtgtcgtgggcggc21<210>57<211>20<212>DNA<213>人工序列<400>57actagcagcaccaggctgcc20<210>58<211>21<212>DNA<213>人工序列<400>58cagccgtgcctgtgttctctc21<210>59<211>21<212>DNA<213>人工序列<400>59ctccccatagtccacagagac21<210>60<211>21<212>DNA<213>人工序列<400>60gagccccccgtgccctgtgtc21<210>61<211>20<212>DNA<213>人工序列<400>61tctgctcagggacacagggc20<210>62<211>20<212>DNA<213>人工序列<400>62ctaggaaagacaatggtggc20<210>63<211>20<212>DNA<213>人工序列<400>63ctaggaaagacaatggtggc20<210>64<211>21<212>DNA<213>人工序列<400>64cgcccgcaggggctcagctgc21<210>65<211>20<212>DNA<213>人工序列<400>65agggccgtcagctgagcccc20<210>66<211>20<212>DNA<213>人工序列<400>66aggcctcagtggctgggcac20<210>67<211>24<212>DNA<213>人工序列<400>67caccgtctctcagctggtacacgg24<210>68<211>24<212>DNA<213>人工序列<400>68aaacccgtgtaccagctgagagac24<210>69<211>25<212>DNA<213>人工序列<400>69caccgcagctggtacacggcagggt25<210>70<211>25<212>DNA<213>人工序列<400>70aaacaccctgccgtgtaccagctgc25<210>71<211>25<212>DNA<213>人工序列<400>71caccgataggcagacagacttgtca25<210>72<211>25<212>DNA<213>人工序列<400>72aaactgacaagtctgtctgcctatc25<210>73<211>25<212>DNA<213>人工序列<400>73caccgtacacggcagggtcagggtt25<210>74<211>25<212>DNA<213>人工序列<400>74aaacaaccctgaccctgccgtgtac25<210>75<211>24<212>DNA<213>人工序列<400>75caccgagagcctgcgggcagagct24<210>76<211>24<212>DNA<213>人工序列<400>76aaacagctctgcccgcaggctctc24<210>77<211>24<212>DNA<213>人工序列<400>77caccgctacaactgggctggcggc24<210>78<211>24<212>DNA<213>人工序列<400>78aaacgccgccagcccagttgtagc24<210>79<211>25<212>DNA<213>人工序列<400>79caccgacgactggccagggcgcctg25<210>80<211>25<212>DNA<213>人工序列<400>80aaaccaggcgccctggccagtcgtc25<210>81<211>25<212>DNA<213>人工序列<400>81caccgcagggctggggagaaggtgg25<210>82<211>25<212>DNA<213>人工序列<400>82aaacccaccttctccccagccctgc25<210>83<211>21<212>DNA<213>人工序列<400>83gcctatttcccatgattcctt21<210>84<211>20<212>DNA<213>人工序列<400>84catcactggcatctggactc20<210>85<211>19<212>DNA<213>人工序列<400>85cctgaagcaaggaaacagc19<210>86<211>21<212>DNA<213>人工序列<400>86gcagagacttctcaatgacat21<210>87<211>19<212>DNA<213>人工序列<400>87cttccagagctagaggaca19<210>88<211>19<212>DNA<213>人工序列<400>88cgtggatgtggaggaagag19<210>89<211>18<212>DNA<213>人工序列<400>89ggccaggggaggagttgg18当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1