2-多取代芳环-嘧啶类衍生物及制备和医药用途的制作方法
本发明涉及药物领域,具体涉及2-多取代芳环-嘧啶类衍生物及其光学异构体或其药学上可接受的盐或溶剂合物、含其的药物组合物及其在抗肿瘤领域的应用。
背景技术:
随着人类生存环境的变化及人口老龄化,恶性肿瘤正严重威胁着人类的生命,在我国恶性肿瘤已成为第一大致死疾病。传统的癌症的治疗方法主要有手术治疗、放射治疗和药物化疗等,其中以药物化疗最为重要。近年来,随着肿瘤分子靶标被逐渐阐释,不少靶向抗肿瘤药物己进入临床应用,但由于肿瘤的复杂性、遗传的多样性,单一靶向药物并不足以治愈肿瘤。传统的化疗药物大多为DNA损伤药物,其通过直接干扰肿瘤细胞的DNA合成,调控DNA的转录、修复等过程诱导肿瘤细胞凋亡,延长癌症患者的生存期。但因其选择性差,会引起多种毒副作用,且治疗过程中会产生明显的耐药性。因此,根据DNA损伤药物的作用特点,开发本身毒性低的药物,使之与DNA损伤药物联合用药,在降低DNA损伤药物用药剂量的同时,增强DNA损伤药物的治疗效果,可降低毒副作用及多药耐药发生的风险。其中,针对细胞周期相关药物的研发及其与DNA损伤药物联合用药的策略近年来引起了药物研究人员的极大兴趣和广泛关注。
真核细胞具有自身调控机制。在受到放、化疗等外界刺激时,可暂时被阻滞在G1,S或G2/M期进行DNA修复,完成修复后进入下一时相。细胞中大量的蛋白激酶相互作用于同一或不同的信号通路,构成了错综复杂的信号网络,共同调节着细胞的生长、增殖、血管生成、转移、凋亡等生命活动。其中,肿瘤基因抑制蛋白p53主要负责G1期检查点的调控,而S和G2/M期则主要由细胞周期检查点激酶1(Checkpoint kinase 1)调控。大部分肿瘤细胞因p53功能的缺失而更加依赖S或G2/M期的阻滞作为DNA损伤诱导凋亡的防御机制。针对p53缺失的肿瘤细胞,抑制Chk1蛋白可清除细胞周期阻滞,直接诱导肿瘤细胞凋亡,而正常细胞由于存在完整的p53调控机制,会被暂时阻滞在G1期而不受影响。因此,Chk1抑制剂可作为辅助治疗药物,选择性地增强肿瘤细胞对放疗或化疗的敏感性,改善治疗效果。
此外,在特定基因缺陷的背景下,如固有DNA损伤过高而造成较大复制压力存在的情况下,Chk1抑制剂也可单独使用,通过“合成致死”的机制杀死肿瘤细胞,达到治疗的目的。基于这一治疗策略,Chk1抑制剂可单用于B细胞淋巴癌、白血病、成神经细胞瘤及一些乳腺癌和肺癌等原癌基因高表达的敏感肿瘤的治疗。
过去的二十年中,不同结构类型的小分子化合物已经作为Chk1抑制剂被发现,这些化合物在临床前体内外评价中表现出了较强的抗肿瘤作用。目前,已有11个小分子Chk1抑制剂进入了临床研究,更加验证了Chk1作为肿瘤治疗靶点的正确性。
技术实现要素:
本发明的目的是提供一种2-多取代芳环-嘧啶类衍生物及其光学异构体或其药学上可接受的盐或溶剂合物。是一种抗癌作用强,具有Chk1抑制作用的新型2-多取代芳环-嘧啶类衍生物。
本发明所提供的2-多取代芳环-嘧啶类衍生物具有通式(Ⅰ)结构:
及其光学异构体或其药学上可接受的盐或溶剂合物,其中:
环A选自取代或无取代的五元或六元芳基、包含1~3个选自O、N和S的五元或六元杂环芳基,所述取代的取代基选自和R5基团;其中环A优选下列五元~六元的含氮芳杂环:
B选自-NH、其中B1自H、C1-4烷基、卤代的C1-4烷基、C1-4烷氧基、卤代的C1-4烷氧基;
R1选自卤原子、C1-6烷基、卤代的C1-6烷基、C3-6环烷基、卤代的C3-6环烷基、C1-6烷氧基、卤代的C1-6烷氧基、C2-6烯基、C2-6羟基取代的烯基、C2-6炔基、C2-6羟基取代的炔基、无取代或取代的五元或六元芳环或芳杂环,所述的芳杂环包含1~3个选自O、N和S的杂原子,取代为单取代、双取代或三取代,所述的取代基选自Ra基团;作为优选,所述的芳基或者杂环芳基选自苯环、呋喃、噻吩、吡唑、噻唑、嘧啶;
Ra任选自H、卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、-C(=O)ORb、-C(=O)NHRb、-NHRb、-ORb、-NHCORb;Rb任选自H、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-7烷胺基;
R2选自H、-NHRc、-N(Rc)2、-ORc、-SRc;Rc选自C1-7烷基、卤代的C1-7烷基、C1-7羟烷基、C1-7胺烷基;
R3选自卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-3烷胺基、卤代的C1-3烷胺基;
L1选自O、S、NH或者缺失;
m=0~2;
R4选自C1-7烷基、卤代的C1-7烷基、羟基取代的C1-7烷基、C1-7烷胺基、卤代的C1-7烷胺基、C1-7烷氧基、卤代的C1-7烷氧基、五元~八元含氮脂杂环;
R5选自卤素、硝基、氰基、三氟甲基、C1-3烷基、C1-3烷氧基、酰胺基、取代烷基酰胺。
进一步地,本发明优选的化合物具有通式(Ⅱ)或通式(III)的结构:
及其光学异构体或其药学上可接受的盐或溶剂合物,其中:
W、X、Y和Z相同或不同,分别独立选自N、C和O;
B选自-NH、其中B1选自H、C1-4烷基、卤代的C1-4烷基、C1-4烷氧基、卤代的C1-4烷氧基;
R1选自卤原子、C1-6烷基、卤代的C1-6烷基、C3-6环烷基、卤代的C3-6环烷基、C1-6烷氧基、卤代的C1-6烷氧基、C2-6烯基、C2-6羟基取代的烯基、C2-6炔基、C2-6羟基取代的炔基,无取代或取代的五元或六元芳环或芳杂环,所述的杂环包含1~3个选自O、N和S的杂原子,取代为单取代、双取代或三取代,所述的取代基选自Ra基团;
Ra任选自H、卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、-C(=O)ORb、-C(=O)NHRb、-NHRb、-ORb、-NHCORb;Rb任选自H、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-7烷胺基;
R2选自H、-NHRc、-N(Rc)2、-ORc、-SRc;Rc选自C1-7烷基、卤代的C1-7烷基、C1-7羟烷基、C1-7胺烷基、C1-7烷氧基;
R3选自卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-3烷胺基、卤代的C1-3烷胺基;
L1选自O、S、NH或者缺失;
m=0~2;
R4选自H、C1-7烷基、卤代的C1-7烷基、羟基取代的C1-7烷基、C1-7烷胺基、卤代的C1-7烷胺基、C1-7烷氧基、卤代的C1-7烷氧基、五元~八元含氮脂杂环;
R5选自卤素、硝基、氰基、三氟甲基、C1-3烷基、C1-3烷氧基、酰胺基、取代烷基酰胺。
及其光学异构体或其药学上可接受的盐或溶剂合物,其中:
W、X、Y和Z相同或不同,分别独立选自N或C;
B选自-NH、其中B1选自H、C1-4烷基、卤代的C1-4烷基、C1-4烷氧基、卤代的C1-4烷氧基;
R1选自卤原子、C1-6烷基、卤代的C1-6烷基、C3-6环烷基、卤代的C3-6环烷基、C1-6烷氧基、卤代的C1-6烷氧基、C2-6烯基、C2-6羟基取代的烯基、C2-6炔基、C2-6羟基取代的炔基、无取代或取代的五元或六元芳环或芳杂环,所述的杂环包含1~3个选自O、N和S的杂原子,取代为单取代、双取代或三取代,所述的取代基选自Ra基团;
Ra任选自H、卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、-C(=O)ORb、-C(=O)NHRb、-NHRb、-ORb、-NHCORb;Rb任选自H、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-7烷胺基;
R2选自H、-NHRc、-N(Rc)2、-ORc、-SRc;Rc选自C1-7烷基、C1-7羟烷基、C1-7胺烷基;
R3选自卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-3烷胺基、卤代的C1-3烷胺基;
L1选自O、S、NH或者缺失;
m=0~2;
R4选自H、C1-7烷基、卤代的C1-7烷基、羟基取代的C1-7烷基、C1-7烷胺基、卤代的C1-7烷胺基、C1-7烷氧基、卤代的C1-7烷氧基、五元~八元含氮脂杂环;
R5选自卤素、硝基、氰基、三氟甲基、C1-3烷基、C1-3烷氧基、酰胺基、取代烷基酰胺。
更进一步地,本发明优选的化合物具有通式(Ⅳ)或通式(V)的结构:
及其光学异构体或其药学上可接受的盐或溶剂合物,其中:
W、X、Y和Z相同或不同,分别独立选自N、C和O;
R1选自卤原子、C1-6烷基、卤代的C1-6烷基、C3-6环烷基、卤代的C3-6环烷基、C1-6烷氧基、卤代的C1-6烷氧基、C2-6烯基、C2-6羟基取代的烯基、C2-6炔基、C2-6羟基取代的炔基,无取代或取代的五元或六元芳环或芳杂环,所述的芳杂环包含1~3个选自O、N和S的杂原子,取代为单取代、双取代或三取代,所述的取代基选自Ra基团;
Ra任选自H、卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、-C(=O)ORb、-C(=O)NHRb、-NHRb、-ORb、-NHCORb;Rb任选自H、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-7烷胺基;
R2选自H、-NHRc、-N(Rc)2、-ORc、-SRc;Rc选自C1-7烷基、C1-7羟烷基、C1-7胺烷基;
R3选自卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-3烷胺基、卤代的C1-3烷胺基;
L1选自O、S、NH或者缺失;
m=0~2
R4选自H、C1-7烷基、卤代的C1-7烷基、羟基取代的C1-7烷基、C1-7烷胺基、卤代的C1-7烷胺基、C1-7烷氧基、卤代的C1-7烷氧基、五元~八元含氮脂杂环;
R5选自卤素、硝基、氰基、三氟甲基、C1-3烷基、C1-3烷氧基、酰胺基、取代烷基酰胺。
更具体地,本发明通式(Ⅳ)结构的优选化合物选自:
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(1-甲基-5-(哌啶-4-氧基)吡唑-3-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(3-(哌啶-4-氧基)异恶唑-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(1-甲基-2-(哌啶-4-氧基)咪唑-4-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)异噻唑-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)-4,5-2H-恶唑啉-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)噻唑-5-基)-2,4-二氨基嘧啶
进一步地,本发明优选的化合物具有通式(V)结构
及其光学异构体或其药学上可接受的盐或溶剂合物,其中:
W、X、Y和Z相同或不同,分别独立选自N或C;
R1选自卤原子、C1-6烷基、卤代的C1-6烷基、C3-6环烷基、卤代的C3-6环烷基、C1-6烷氧基、卤代的C1-6烷氧基、C2-6烯基、C2-6羟基取代的烯基、C2-6炔基、C2-6羟基取代的炔基、无取代或取代的五元或六元芳环或芳杂环,所述的芳杂环包含1~3个选自O、N和S的杂原子,取代为单取代、双取代或三取代,所述的取代基选自Ra基团;
Ra任选自H、卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、-C(=O)ORb、-C(=O)NHRb、-NHRb、-ORb、-NHCORb;Rb任选自H、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-7烷胺基;
R2选自H、-NHRc、-N(Rc)2、-ORc、-SRc;Rc选自C1-7烷基、C1-7羟烷基、C1-7胺烷基;
R3选自卤素、硝基、氰基、C1-3烷基、卤代的C1-3烷基、C1-3烷氧基、卤代的C1-3烷氧基、C1-3烷胺基、卤代的C1-3烷胺基;
L1选自O、S、NH或者缺失;
m=0~2;
R4选自H、C1-7烷基、卤代的C1-7烷基、羟基取代的C1-7烷基、C1-7烷胺基、卤代的C1-7烷胺基、C1-7烷氧基、卤代的C1-7烷氧基、五元~八元含氮脂杂环;
R5选自卤素、硝基、氰基、三氟甲基、C1-3烷基、C1-3烷氧基、酰胺基、取代烷基酰胺。
更具体地,本发明通通式(V)结构的优选化合物选自:
5-苯基-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(3-氟苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(4-氟苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(3-甲氧基苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(4-甲氧基苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(吡啶-3-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(吡啶-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(噻吩-2-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-(呋喃-2-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
5-三氟甲基-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
(R)-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
(S)-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶
4-甲氧基-5-(3-氟苯基)-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶
4-甲氧基-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶
4-甲氧基-5-(3-氟苯基)-N-(2-氰基-3-(2-二甲氨基乙氧基)吡啶-5-基)-2-氨基嘧啶
4-甲氧基-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(2-二甲氨基乙氧基)吡啶-5-基)-2-氨基嘧啶
4-甲氧基-5-三氟甲基-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶
N4-甲基-5-苯基-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(3-氟苯基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(4-氟苯基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(2-氟苯基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(3-甲氧基苯基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(4-甲氧基苯基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(2,4-二甲氧基苯基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(吡啶-3-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(吡啶-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(噻吩-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(呋喃-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(5-氯-呋喃-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(5-甲氧羰基噻吩-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-5-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-三氟甲基-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2,4-二氨基嘧啶
(R)-N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
(S)-N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
(R)-N4-甲基-5-三氟甲基-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
(S)-N4-甲基-5-三氟甲基-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
(R)-N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(吡咯-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
(S)-N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(吡咯-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-4-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(2-二甲氨基乙氧基)吡啶-5-基)-2,4-二氨基嘧啶
(R)-N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(1-二甲氨基丙基-2-氧基)吡啶-5-基)-2,4-二氨基嘧啶
(S)-N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(1-二甲氨基丙基-2-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(N-甲基哌啶-4-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-三氟甲基-N2-(2-氰基-3-(N-甲基哌啶-4-氧基)吡啶-5-基)-2,4-二氨基嘧啶
N4-甲基-5-(4-甲基噻唑-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶
及其上述化合物的药学上可接受的盐或溶剂合物。
本发明采用本领域技术人员所熟知的方法可以制备本发明所述的2-取代嘧啶类化合物的盐。所述的盐可以是有机酸盐、无机酸盐等,所述的有机酸盐包括枸橼酸盐、富马酸盐、草酸盐、苹果酸盐、L-苹果酸盐、D-苹果酸盐、乳酸盐、樟脑磺酸盐、对甲苯磺酸盐、甲磺酸盐、苯甲酸盐等;所述的无机酸盐包括氢卤酸盐、硫酸盐、磷酸盐、硝酸盐等。例如,与低级烷基磺酸,如甲磺酸,三氟甲磺酸等可形成甲磺酸盐、三氟甲磺酸盐;与芳基磺酸,如苯磺酸或对甲苯磺酸等可形成对甲苯磺酸盐、苯磺酸盐;与有机羧酸,如乙酸,富马酸,酒石酸盐、L-酒石酸,D-酒石酸草酸,马来酸,苹果酸盐、L-苹果酸,D-苹果酸,琥珀酸或柠檬酸等可形成相应的盐;与氨基酸,如谷氨酸或天冬氨酸可形成谷氨酸盐或天冬氨酸盐。与无机酸,如氢卤酸(如氢氟酸、氢溴酸、氢碘酸、氢氯酸),硝酸,碳酸,硫酸或磷酸等也可形成相应的盐。
本发明的第二个目的是提供一种药物组合物,所述药物组合物包含至少一种活性组分以及一种或多种药学上可接受的载体或赋形剂,所述的活性组分可以是本发明的2-取代嘧啶类化合物、所述化合物的光学异构体、所述化合物或其光学异构体在药学上可接受的盐、所述化合物或其光学异构体的溶剂合物中的任意一种或任意多种。
所述载体包括药学领域的常规稀释剂,赋形剂,填充剂,粘合剂,湿润剂,崩解剂,吸收促进剂,表面活性剂,吸附载体,润滑剂等,必要时还可以加入香味剂,甜味剂等。本发明药物可以制成片剂,粉剂,粒剂,胶囊,口服液及注射用药等多种形式,上述各剂型的药物均可以按照药学领域的常规方法制备。
本发明还提供通式(I)~通式(V)所述的化合物、及其光学异构体或其药学上可接受的盐或溶剂合物、及其光学异构体或其药学上可接受的盐或溶剂合物单独和/或与放疗、其他药物联合使用在制备Chk1抑制剂中的应用,特别是在制备治疗细胞增生疾病中的应用。所述的细胞增生疾病包括肿瘤,所述的肿瘤为乳腺癌、卵巢癌、肉瘤、肺癌、前列腺癌、结肠癌、直肠癌、肾癌、胰腺癌、血癌、淋巴癌、成神经细胞瘤、神经胶质瘤、头癌、颈癌、甲状腺癌、肝癌、外阴癌、子宫颈癌、子宫内膜癌、睾丸癌、膀胱癌、食管癌、胃癌、鼻咽癌、颊癌、口腔癌、胃肠道间质瘤、皮肤癌、多发性骨髓瘤。能和本发明所提供的化合物或其可药用盐联合使用的抗肿瘤药包括但并非限定至少一种以下种类:抗代谢药物(吉西他滨,5-氟尿嘧啶,羟基脲,培美曲赛);烷基化试剂(如顺铂、卡铂);拓扑异构酶抑制剂(伊立替康、多柔比星);小分子抑制剂(MEK抑制剂,PARP抑制剂,Scr家族激酶抑制剂,mTOR抑制剂,法尼基转移酶抑制剂等)。
本发明的另一个目的是提供上述目标化合物的制备方法,通过以下步骤实现:
方法一:
(1)5-溴-2-取代-3-硝基吡啶(或5-溴-2-氰基-3-硝基吡啶)在碱性条件(NaH存在的条件)下,与不同的脂肪醇经取代反应得到不同取代的吡啶片段;
(2)以5-溴-2,4-二氯嘧啶为起始原料,依次经甲醚化(或甲胺化)和氨化得到5-溴-2,4-二取代嘧啶或以2-氨基嘧啶为原料经溴代后得到5-溴-2-氨基嘧啶,先经Suzuki偶联,再与上述吡啶片段发生钯催化的Buchwald-Hartwig交叉偶联反应,最后脱Boc保护基得到目标化合物;也可以采用2-氨基嘧啶中间体先与取代的5-溴吡啶经Buchwald-Hartwig交叉偶联,然后经Suzuki偶联反应,酸性条件下脱Boc保护基得到目标分子;
化合物1-6按照下列合成路线制备:
化合物7-16按照下列合成路线制备:
化合物17-43按照下列合成路线制备:
化合物44按照下列合成路线制备:
方法二:
(1)5-溴-2-氰基-3-硝基吡啶在碱性条件(NaH存在的条件)下,与不同的脂肪醇经取代反应得到不同取代的吡啶片段;
(2)以2,4-二氯-5-三氟甲基嘧啶为起始原料,依次经甲醚化(或甲胺化)和氨化得到2,4-二取代-5-三氟甲基嘧啶或以2-氯-5-三氟甲基嘧啶为原料经氨化,再与上述吡啶片段发生钯催化的Buchwal-Hartwig交叉偶联反应,最后脱Boc保护基得到目标化合物。
化合物45合成路线如下:
化合物46-50合成路线如下:
本发明发明人通过实验证实,本发明中的大部分化合物具有中等到强的Chk1激酶抑制活性,可应用于治疗人或动物细胞增殖性相关的实体瘤或血癌的药物中。本发明以通过基于结构的虚拟筛选得到的2-氨基嘧啶为先导化合物,设计并合成了一系列全新的小分子Chk1抑制剂,并对该类化合物进行了分子水平的Chk1激酶抑制活性测试。结果表明,大部分化合物表现出中等到强的Chk1抑制活性,是有前景的Chk1抑制剂,为癌症治疗提供了新的药物。
附图说明
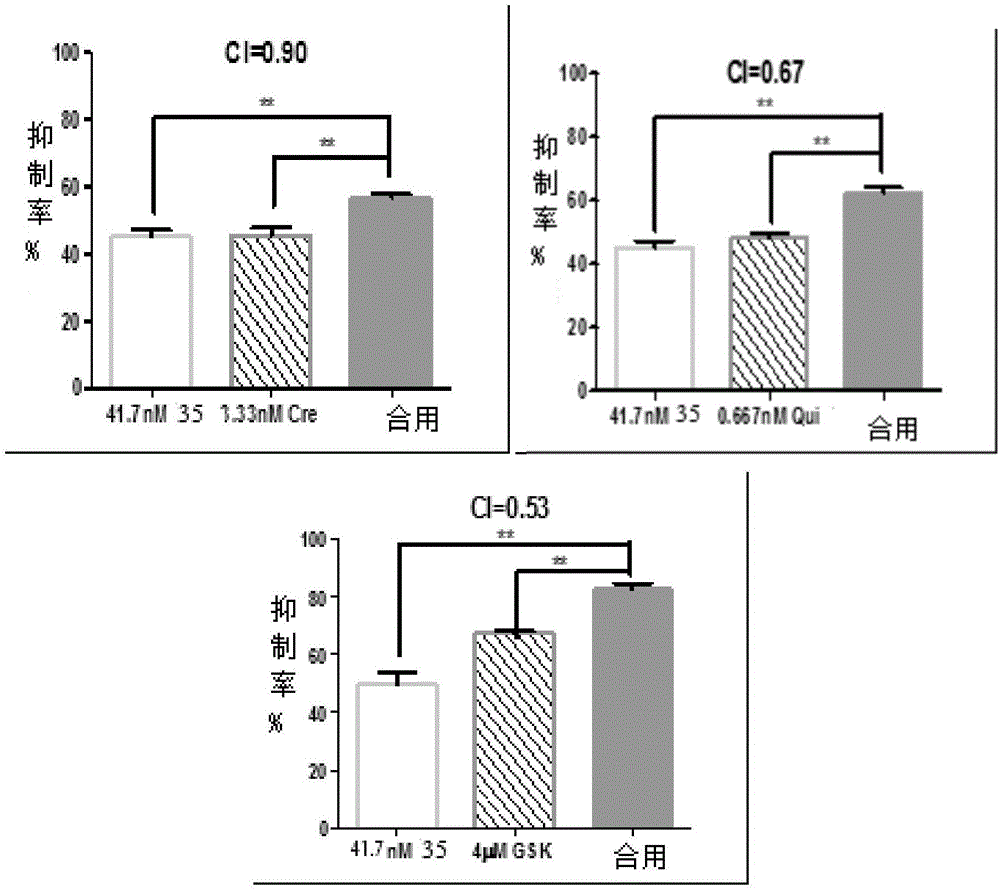
图1是CHK1抑制剂与其他药物在MV 4-11细胞上联用的活性。
具体实施方式
本发明结合实施例作进一步的说明,以下实施例仅是说明本发明,而不是以任何方式限制本发明。
制备实施例1.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(1-甲基-5-(哌啶-4-氧基)吡唑-3-基)-2,4-二氨基嘧啶(化合物1)
步骤1.N-叔丁氧羰基-4-((3-溴-1-甲基-1H-吡唑-5-基)氧基)哌啶(中间体1-2)合成
将N-叔丁氧羰基-4-羟基哌啶(460mg,2.29mmol)溶于无水THF(7.2mL)中,冰浴冷却下,分批加入60%氢化钠(108mg,4.5mmol),室温下搅拌10min。升温至35℃,搅拌10min。将3-溴-1-甲基-5-硝基吡唑(360mg,1.75mmol)置于50mL三颈瓶中,氮气保护下加入无水THF(4.8mL),将上述制得的钠盐缓慢滴加入反应液中(约20min滴加完毕),室温下搅拌1h。加入饱和氯化铵溶液淬灭反应,减压回收溶剂。用硅胶柱层析纯化,以PE:EtOAc(50:9)为洗脱剂,得金黄色油状物。Yield:79%;LCMS:m/z=361[M+1]+。
步骤2. 4-甲氨基-2-氯-5-溴嘧啶(中间体1-4)合成
将5-溴-2,4-二氯嘧啶(5g,22mmol)溶于甲醇(42mL)中,冰浴冷却下滴加33%的甲胺乙醇溶液(6.75mL),室温下搅拌30min。减压回收溶剂,用硅胶柱层析纯化,以PE:EtOAc(5:1)为洗脱剂,得白色固体。Yield:88%;mp:139-141℃;1H NMR(500MHz,DMSO-d6):δ8.85(s,Ar-H,1H),7.75(br,NH,1H),2.85(d,J=3.9Hz,CH3,3H);ESI-MS:m/z=222[M+1]+。
步骤3. 5-溴-N4-甲基-2,4-二氨基嘧啶(中间体1-5)合成
将化合物1-4(275mg,1.23mmol)置于封管中,加入氨气饱和的乙醇溶液(20mL),100℃下搅拌24h。冷却至室温,减压回收溶剂得残余物。用硅胶柱层析纯化,以PE:EtOAc(2:1)为洗脱剂,得白色固体。Yield:78%;1H NMR(500MHz,CDCl3):δ7.86(s,Ar-H,1H),5.22(br,NH,1H),4.85(br,NH2,2H),2.99(d,J=6.0Hz,CH3,3H);ESI-MS:m/z=204[M+1]+。
步骤4. 5-(1-甲基-1H-吡唑-4-基)-N4-甲基-2,4-二氨基嘧啶(中间体1-6)合成
氮气保护下,向化合物1-5(290mg,1.43mmol),1-甲基-1H-吡唑-4-硼酸频哪醇酯(358mg,1.72mmol),Pd(dppf)Cl2(54mg,0.07mmol)的混合物中加入乙二醇二甲醚(14mL),1N Na2CO3水溶液(2.8mL),回流搅拌过夜。减压回收溶剂得残余物。用硅胶柱层析纯化,以CH2Cl2:EtOH(25:1)为洗脱剂,得白色固体。Yield:75%;mp:226-228℃;1H NMR(500MHz,DMSO-d6):δ7.88(s,Ar-H,1H),7.49(s,Ar-H,1H),7.35(s,Ar-H,1H),6.22(br,NH2,2H),5.96(q,J=4.5Hz,NH,1H),3.51(s,CH3,3H),2.86(d,J=4.5Hz,CH3,3H);ESI-MS:m/z=205[M+1]+。
步骤5.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(1-甲基-5-(哌啶-4-氧基)吡唑-3-基)-2,4-二氨基嘧啶(化合物1)合成
氮气保护下,向化合物5a(169mg,0.986mmol),2a(376mg,0.986mmol),三(二亚苄基丙酮)二钯(9mg,0.00986mmol),4,5-双二苯基膦-9,9-二甲基氧杂蒽(15mg,0.026mmol),碳酸铯(450mg,1.38mmol)的混合物中加入无水二氧六环(6mL),回流搅拌过夜。抽滤,减压回收溶剂得残余物。用硅胶柱层析纯化,以CH2Cl2:EtOH(30:1)为洗脱剂,得白色固体。用三氟乙酸脱Boc保护基得白色固体。Yield:65%;LCMS:m/z=385[M+1]+。
制备实施例2.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(3-(哌啶-4-氧基)异恶唑-5-基)-2,4-二氨基嘧啶(化合物2)
步骤1.N-叔丁氧羰基-4-((5-溴-异恶唑-3-基)氧基)哌啶(中间体1-8)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,从5-溴-3-硝基吡唑(化合物1-7)制备化合物1-8。Yield:63%;LCMS:m/z=348[M+1]+。
步骤2.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(3-(哌啶-4-氧基)异恶唑-5-基)-2,4-二氨基嘧啶(化合物2)合成
合成步骤参考实施案例1步骤5.用类似于化合物1的合成方法,以中间体1-8和1-6为原料,制备得到化合物2。Yield:65%;LCMS:m/z=371[M+1]+。
制备实施例3.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(1-甲基-2-(哌啶-4-氧基)咪唑-4-基)-2,4-二氨基嘧啶(化合物3)
步骤1.N-叔丁氧羰基-4-((4-溴-1-甲基-1H-咪唑-2-基)氧基)哌啶(中间体1-10)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,从4-溴-1-甲基-2-硝基咪唑(化合物1-9)制备化合物1-10。Yield:67%;LCMS:m/z=361[M+1]+。
步骤2.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(1-甲基-2-(哌啶-3-氧基)咪唑-4-基)-2,4-二氨基嘧啶(化合物3)合成
合成步骤参考实施案例1步骤5.用类似于化合物1的合成方法,以中间体1-10和1-6为原料,制备得到化合物3。Yield:65%;LCMS:m/z=384[M+1]+。
制备实施例4.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)异噻唑-5-基)-2,4-二氨基嘧啶(化合物4)
步骤1.N-叔丁氧羰基-4-((5-溴-异噻唑-3-基)氧基)哌啶(中间体1-12)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,从5-溴-3-硝基咪唑(化合物1-11)制备化合物1-12。Yield:66%;LCMS:m/z=364[M+1]+。
步骤2.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)异噻唑-5-基)-2,4-二氨基嘧啶(化合物4)合成
合成步骤参考实施案例1步骤5.用类似于化合物1的合成方法,以中间体1-12和1-6为原料,制备得到化合物4。Yield:65%;LCMS:m/z=387[M+1]+。
制备实施例5.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)-4,5-2H-恶唑啉-5-基)-2,4-二氨基嘧啶(化合物5)
步骤1.N-叔丁氧羰基-4-((5-溴--4,5-2H-恶唑啉-2-基)氧基)哌啶(中间体1-14)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,从5-溴-2-硝基-4,5-2H恶唑啉化合物1-13)制备化合物1-14。Yield:67%;LCMS:m/z=361[M+1]+。
步骤2.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)-4,5-2H-恶唑啉-5-基)-2,4-二氨基嘧啶(化合物5)合成
合成步骤参考实施案例1步骤5.用类似于化合物1的合成方法,以中间体1-14和1-6为原料,制备得到化合物5。Yield:65%;LCMS:m/z=373[M+1]+。
制备实施例6.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-(哌啶-4-氧基)噻唑-5-基)-2,4-二氨基嘧啶(化合物6)
步骤1.N-叔丁氧羰基-4-((5-溴-噻唑-2-基)氧基)哌啶(中间体1-16)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,从5-溴-2-硝基噻唑(化合物1-15)制备化合物1-16。Yield:63%;LCMS:m/z=364[M+1]+。
步骤2.N4-甲基-5-(1-甲基-1H-咪唑-4-基)-N2-(1-甲基-2-(哌啶-4-氧基)吡唑-4-基)-2,4-二氨基嘧啶(化合物3)合成
合成步骤参考实施案例1步骤5.用类似于化合物1的合成方法,以中间体1-16和1-6为原料,制备得到化合物6。Yield:65%;LCMS:m/z=387[M+1]+。
制备实施例7. 5-苯基-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物7)
步骤1. 2-氰基-5-溴-3-(N-叔丁氧羰基哌啶-3-氧基)吡啶(中间体2-2)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,以5-溴-3-硝基-2-氰基吡啶(化合物2-1)和N-叔丁氧羰基-3-羟基哌啶为原料制备化合物2-2。Yield:79%;1H NMR(500MHz,DMSO-d6):δ8.26(s,Ar-H,1H),7.53(s,Ar-H,1H),4.38(br,CH,1H),3.63(br,CH,1H),3.51(br,CH,1H),3.38(br,CH,2H),1.98-1.96(m,CH,1H),1.91-1.85(m,CH,2H),1.51(br,CH,1H),1.36(s,CH3×3,9H);ESI-MS:m/z=382[M+1]+。
步骤2. 5-溴-2-氨基嘧啶(中间体2-4)合成
将2-氨基嘧啶(2.5g,26.29mmol)溶于乙腈(25mL)中,冰浴冷却下加入N-溴代丁二酰亚胺(4.6g,27.9mmol),室温下避光搅拌过夜。减压回收溶剂,用水(100mL)洗涤,抽滤,真空干燥得白色固体。Yield:97%;mp:241-243℃。
步骤3. 2-氨基-5-苯基嘧啶(中间体2-5)合成
合成步骤参考实施案例1步骤4.以中间体2-4和苯硼酸为原料制备化合物2-5。Yield:86%;mp:159-161℃;1H NMR(400MHz,DMSO-d6):δ8.57(s,Ar-H,2H),7.62(d,J=7.6Hz,Ar-H,2H),7.45(t,J=7.2Hz,Ar-H,2H),7.34(t,J=7.2Hz,Ar-H,1H),6.77(s,NH,2H);ESI-MS:m/z=172[M+1]+。
步骤4. 5-苯基-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物7)合成
合成步骤参考实施案例1步骤5.以中间体2-5和2-2为原料制备化合物7。Yield:65%;mp:84-86℃;1H NMR(500MHz,DMSO-d6):δ10.55(s,NH,1H),8.99(s,Ar-H,2H),8.65(d,J=2.0Hz,Ar-H,1H),8.39(d,J=2.0Hz,Ar-H,1H),7.78(d,J=7.0Hz,Ar-H,2H),7.52(t,J=7.5Hz,Ar-H,2H),7.43(t,J=7.0Hz,Ar-H,1H),4.42-4.37(m,CH,1H),3.19(d,J=12.0Hz,2.0Hz,CH2,1H),2.81-2.77(m,CH2,1H),2.66-2.62(m,CH2,1H),2.56-2.53(m,CH2,1H),2.14-2.11(m,CH2,1H),1.76-1.71(m,CH2,1H),1.65-1.58(m,CH2,1H),1.54-1.46(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ160.22,158.43,157.73,153.05,142.82,137.60,133.83,129.37,116.42,113.77,111.72,106.92,104.53,86.99,57.06,45.54,38.63,28.17;ESI-MS:m/z=373[M+1]+。
制备实施例8. 5-(3-氟苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物8)
步骤1. 2-氨基-5-(3-氟苯基)嘧啶(中间体2-6)合成
合成步骤参考实施案例1步骤4.以中间体2-4和3-氟苯硼酸为原料制备化合物2-6,得白色固体。Yield:83%;mp:168-170℃;1H NMR(400MHz,DMSO-d6):δ8.62(s,Ar-H,2H),7.53-7.43(m,Ar-H,3H),7.16-7.11(m,Ar-H,1H),6.86(s,NH2,2H);ESI-MS:m/z=190[M+1]+。
步骤2. 5-(3-氟苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物8)合成
合成步骤参考实施案例1步骤5.以中间体2-6和2-2为原料制备化合物8,得白色固体。Yield:72%;mp:107-109℃;1H NMR(500MHz,DMSO-d6):δ10.59(br,NH,1H),9.03(s,Ar-H,2H),8.65(d,J=1.5Hz,Ar-H,1H),8.37(s,Ar-H,1H),7.70(d,J=10.5Hz,Ar-H,1H),7.65(d,J=8.0Hz,Ar-H,1H),7.56(dd,J=14.0Hz,8.0Hz,Ar-H,1H),7.26(td,J=8.5Hz,2.5Hz,Ar-H,1H),4.42-4.37(m,CH,1H),3.19(dd,J=12.0Hz,2.0Hz,CH2,1H),2.82-2.78(m,CH2,1H),2.67-2.63(m,CH2,1H),2.57-2.52(m,CH2,1H),2.14-2.11(m,CH2,1H),1.77-1.71(m,CH2,1H),1.66-1.59(m,CH2,1H),1.54-1.47(m,CH2,1H);ESI-MS:m/z=391[M+1]+。
制备实施例9. 5-(4-氟苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物9)
步骤1. 2-氨基-5-(4-氟苯基)嘧啶(中间体2-7)合成
合成步骤参考实施案例1步骤4.以中间体2-4和4-氟苯硼酸为原料制备化合物2-7,得白色固体。Yield:85%;mp:172-174℃;1H NMR(400MHz,DMSO-d6):δ8.58(s,Ar-H,2H),7.68-7.64(m,Ar-H,2H),7.29(t,J=8.8Hz,Ar-H,2H),6.78(s,NH2,2H);ESI-MS:m/z=190[M+1]+。
步骤2. 5-(4-氟苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物9)合成
合成步骤参考实施案例1步骤5.以中间体2-7和2-2为原料制备化合物9,得白色固体。Yield:78%;mp:182-184℃;1H NMR(500MHz,DMSO-d6):δ10.54(br,NH,1H),8.96(s,Ar-H,2H),8.65(d,J=2.0Hz,Ar-H,1H),8.36(s,Ar-H,1H),7.83(dd,J=8.5Hz,5.5Hz,Ar-H,2H),7.35(t,J=8.0Hz,Ar-H,2H),4.41-4.36(m,CH,1H),3.20(dd,J=12.0Hz,2.5Hz,CH2,1H),2.82-2.78(m,CH2,1H),2.67-2.63(m,CH2,1H),2.57-2.52(m,CH2,1H),2.14-2.11(m,CH2,1H),1.77-1.71(m,CH2,1H),1.65-1.58(m,CH2,1H),1.54-1.46(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ163.09,161.14,158.27,157.25,155.90,141.82,134.15,130.59,128.17,128.11,125.15,116.21,116.08,115.91,113.84,109.21,74.69,49.65,45.28,29.82,24.27;ESI-MS:m/z=391[M+1]+。
制备实施例10. 5-(3-甲氧基苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物10)
步骤1. 2-氨基-5-(3-甲氧基苯基)嘧啶(中间体2-8)合成
合成步骤参考实施案例1步骤4.以中间体2-4和3-甲氧基苯硼酸为原料制备化合物2-8,得白色固体。Yield:87%;mp:133-135℃;1H NMR(400MHz,DMSO-d6):δ8.58(s,Ar-H,2H),7.36(t,J=8.0Hz,Ar-H,1H),7.18(d,J=7.6Hz,Ar-H,2H),6.90(dd,J=8.4Hz,1.6Hz,Ar-H,1H),6.78(s,NH2,2H),3.81(s,CH3,3H);ESI-MS:m/z=202[M+1]+。
步骤2. 5-(3-甲氧基苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物10)合成
合成步骤参考实施案例1步骤5.以中间体2-8和2-2为原料制备化合物10,得白色固体。Yield:74%;mp:102-104℃;1H NMR(500MHz,DMSO-d6):δ10.54(br,NH,1H),8.98(s,Ar-H,2H),8.65(d,J=2.0Hz,Ar-H,1H),8.37(d,J=2.0Hz,Ar-H,1H),7.42(t,J=8.0Hz,Ar-H,1H),7.33-7.32(m,Ar-H,2H),6.99-6.97(m,Ar-H,1H),4.42-4.37(m,CH,1H),3.85(s,CH3,3H),3.20(dd,J=12.0Hz,2.0Hz,CH2,1H),2.82-2.78(m,CH2,1H),2.68-2.64(m,CH2,1H),2.57-2.53(m,CH2,1H),2.14-2.11(m,CH2,1H),1.77-1.71(m,CH2,1H),1.66-1.59(m,CH2,1H),1.55-1.47(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ159.92,158.41,157.25,156.03,141.83,135.43,134.18,130.23,125.86,118.19,116.23,113.84,113.65,111.37,109.21,74.66,55.22,49.63,45.29,29.81,24.24;ESI-MS:m/z=403[M+1]+。
制备实施例11. 5-(4-甲氧基苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物11)
步骤1. 2-氨基-5-(4-甲氧基苯基)嘧啶(中间体2-9)合成
合成步骤参考实施案例1步骤4.以中间体2-4和4-甲氧基苯硼酸为原料制备化合物2-9,得白色固体。Yield:82%;mp:165-167℃;1H NMR(400MHz,DMSO-d6):δ8.51(s,Ar-H,2H),7.55(d,J=8.8Hz,Ar-H,2H),7.01(d,J=8.8Hz,Ar-H,2H),6.67(s,NH2,2H),3.78(s,CH3,3H);ESI-MS:m/z=202[M+1]+。
步骤2. 5-(4-甲氧基苯基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物11)合成
合成步骤参考实施案例1步骤5.以中间体2-9和2-2为原料制备化合物11,得白色固体。Yield:75%;mp:218-220℃;1H NMR(400MHz,DMSO-d6):δ10.47(s,NH,1H),8.91(s,Ar-H,2H),8.64(s,Ar-H,1H),8.36(s,Ar-H,1H),7.71(d,J=8.8Hz,Ar-H,2H),7.06(d,J=8.8Hz,Ar-H,2H),4.40-4.36(m,CH,1H),3.81(s,CH3,3H),3.20(d,J=12.0Hz,CH2,1H),2.81-2.78(m,CH2,1H),2.67-2.62(m,CH2,1H),2.56-2.51(m,CH2,1H),2.14-2.12(m,CH2,1H),1.76-1.72(m,CH2,1H),1.66-1.57(m,CH2,1H),1.54-1.46(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ159.25,157.86,157.26,155.34,141.92,134.08,127.20,126.29,125.85,116.24,114.61,113.65,109.00,74.69,55.20,49.69,45.31,29.84,24.31;ESI-MS:m/z=403[M+1]+。
制备实施例12. 5-(吡啶-3-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合12)
步骤1. 2-氨基-5-(吡啶-3-基)嘧啶(中间体2-10)合成
合成步骤参考实施案例1步骤4.以中间体2-4和吡啶-3-硼酸为原料制备化合物2-10,得白色固体。Yield:79%;mp:183-185℃;1H NMR(400MHz,DMSO-d6):δ8.87(s,Ar-H,1H),8.64(s,Ar-H,2H),8.53(br,Ar-H,1H),8.05(d,J=7.6Hz,Ar-H,1H),7.46(d,J=4.0Hz,Ar-H,1H),6.89(s,NH2,2H);ESI-MS:m/z=173[M+1]+。
步骤2. 5-(吡啶-3-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物12)合成
合成步骤参考实施案例1步骤5.以中间体2-10和2-2为原料制备化合物12,得白色固体。Yield:75%;mp:221-223℃;1H NMR(500MHz,DMSO-d6):δ10.61(br,NH,1H),9.06(s,Ar-H,2H),9.00(d,J=2.5Hz,Ar-H,1H),8.67(d,J=1.5Hz,Ar-H,1H),8.62(dd,J=4.5Hz,1.5Hz,Ar-H,1H),8.37(d,J=2.0Hz,Ar-H,1H),8.21(td,J=8.0Hz,2.0Hz,Ar-H,1H),7.54(dd,J=8.0Hz,4.0Hz,Ar-H,1H),4.43-4.39(m,CH,1H),3.20(dd,J=12.0Hz,2.0Hz,CH2,1H),2.83-2.78(m,CH2,1H),2.69-2.65(m,CH2,1H),2.59-2.54(m,CH2,1H),2.15-2.12(m,CH2,1H),1.78-1.72(m,CH2,1H),1.67-1.60(m,CH2,1H),1.55-1.47(m,CH2,1H);ESI-MS:m/z=374[M+1]+。
制备实施例13. 5-(噻吩-2-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合13)
步骤1. 2-氨基-5-(噻吩-2-基)嘧啶(中间体2-11)合成
合成步骤参考实施案例1步骤4.以中间体2-4和噻吩-2-硼酸为原料制备化合物2-11,得白色固体。Yield:84%;mp:156-158℃;1H NMR(400MHz,DMSO-d6):δ8.53(s,Ar-H,2H),7.49(d,J=4.8Hz,Ar-H,1H),7.38(d,J=2.8Hz,Ar-H,1H),7.12(t,J=4.4Hz,Ar-H,1H),6.87(s,NH2,2H):ESI-MS:m/z=178[M+1]+。
步骤2. 5-(噻吩-2-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物13)合成
合成步骤参考实施案例1步骤5.以中间体2-11和2-2为原料制备化合物13,得白色固体。Yield:74%;mp:212-214℃;1H NMR(500MHz,DMSO-d6):δ10.58(br,NH,1H),8.93(s,Ar-H,2H),8.61(d,J=2.0Hz,Ar-H,1H),8.35(d,J=2.0Hz,Ar-H,1H),7.63-7.61(m,Ar-H,2H),7.20(dd,J=5.0Hz,3.5Hz,Ar-H,1H),4.42-4.37(m,CH,1H),3.19(dd,J=12.0Hz,2.0Hz,CH2,1H),2.82-2.78(m,CH2,1H),2.67-2.63(m,CH2,1H),2.57-2.52(m,CH2,1H),2.14-2.10(m,CH2,1H),1.77-1.71(m,CH2,1H),1.66-1.59(m,CH2,1H),1.54-1.46(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ158.00,157.25,154.63,141.67,136.46,134.18,128.55,126.15,124.27,121.13,116.20,113.91,109.24,74.64,49.63,45.30,29.81,24.26;ESI-MS:m/z=379[M+1]+。
制备实施例14. 5-(呋喃-2-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合14)
步骤1. 2-氨基-5-(呋喃-2-基)嘧啶(中间体2-12)合成
合成步骤参考实施案例1步骤4.以中间体2-4和呋喃-2-硼酸为原料制备化合物2-12,得白色固体。Yield:83%;mp:156-158℃;1H NMR(400MHz,DMSO-d6):δ8.57(s,Ar-H,2H),7.69(s,Ar-H,1H),6.88(s,NH,2H),6.78(d,J=2.0Hz,Ar-H,1H),6.56(s,Ar-H,1H);ESI-MS:m/z=162[M+1]+。
步骤2. 5-(呋喃-2-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物14)合成
合成步骤参考实施案例1步骤5.以中间体2-12和2-2为原料制备化合物14,得白色固体。Yield:70%;mp:200-202℃;1H NMR(400MHz,DMSO-d6):δ10.58(br,NH,1H),8.95(s,Ar-H,2H),8.59(d,J=1.2Hz,Ar-H,1H),8.36(s,Ar-H,1H),7.81(s,Ar-H,1H),7.05(d,J=2.8Hz,Ar-H,1H),6.65(t,J=1.6Hz,Ar-H,1H),4.41-4.37(m,CH,1H),3.20(d,J=12Hz,CH2,1H),2.82-2.79(m,CH2,1H),2.67-2.62(m,CH2,1H),2.57-2.55(m,CH2,1H),2.14-2.12(m,CH2,1H),1.76-1.73(m,CH2,1H),1.67-1.58(m,CH2,1H),1.54-1.49(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ157.76,157.23,153.18,148.23,143.35,141.62,134.17,117.90,116.19,113.89,112.08,109.23,106.19,74.66,49.63,45.30,29.81,24.27;ESI-MS:m/z=363[M+1]+。
制备实施例15.(R)-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合15)
步骤1.(R)-2-氰基-5-溴-3-(N-叔丁氧羰基哌啶-3-氧基)吡啶(中间体2-13)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,以5-溴-3-硝基-2-氰基吡啶(化合物2-1)和(R)-N-叔丁氧羰基-3-羟基哌啶为原料制备化合物2-13,得金黄色油状物。Yield:76%;1H NMR(500MHz,DMSO-d6):δ8.26(s,Ar-H,1H),7.53(s,Ar-H,1H),4.38(br,CH,1H),3.63(br,CH,1H),3.51(br,CH,1H),3.38(br,CH,2H),1.98-1.96(m,CH,1H),1.91-1.85(m,CH,2H),1.51(br,CH,1H),1.36(s,CH3×3,9H);ESI-MS:m/z=382[M+1]+。
步骤2. 2-氨基-5-(1-甲基-1H-吡唑-4-基)嘧啶(中间体2-14)合成
合成步骤参考实施案例1步骤4.以中间体2-4和1-甲基吡唑-4-硼酸频哪醇酯为原料制备化合物2-14,得白色固体。Yield:85%;mp:174-176℃;1H NMR(400MHz,DMSO-d6):δ8.46(s,Ar-H,2H),8.03(s,Ar-H,1H),7.78(s,Ar-H,1H),6.57(s,NH2,2H),3.84(s,CH3,3H);ESI-MS:m/z=176[M+1]+。
步骤3.(R)-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物15)合成
合成步骤参考实施案例1步骤5.以中间体2-14和2-13为原料制备化合物15,得白色固体。Yield:73%;mp:230℃(分解);1H NMR(500MHz,DMSO-d6):δ10.42(br,NH,1H),8.87(s,Ar-H,2H),8.59(d,J=1.5Hz,Ar-H,1H),8.35(d,J=2.0Hz,Ar-H,1H),8.23(s,Ar-H,1H),7.97(s,Ar-H,1H),4.41-4.36(m,CH,1H),3.89(s,CH3,3H),3.19(dd,J=12.0Hz,2.0Hz,CH2,1H),2.82-2.78(m,CH2,1H),2.68-2.64(m,CH2,1H),2.58-2.53(m,CH2,1H),2.14-2.11(m,CH2,1H),1.78-1.72(m,CH2,1H),1.65-1.58(m,CH2,1H),1.54-1.46(m,CH2,1H);13C NMR(125MHz,DMSO-d6):δ157.28,154.26,141.98,135.75,134.02,127.58,119.77,116.28,115.46,113.46,108.76,74.59,49.62,45.27,38.74,29.78,24.19;ESI-MS:m/z=377[M+1]+。
制备实施例16.(R)-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合16)
步骤1.(S)-2-氰基-5-溴-3-(N-叔丁氧羰基哌啶-3-氧基)吡啶(中间体2-15)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,以5-溴-3-硝基-2-氰基吡啶(化合物2-1)和(S)-N-叔丁氧羰基-3-羟基哌啶为原料制备化合物2-15,得金黄色油状物。Yield:78%;1H NMR(500MHz,DMSO-d6):δ8.26(s,Ar-H,1H),7.53(s,Ar-H,1H),4.38(br,CH,1H),3.63(br,CH,1H),3.51(br,CH,1H),3.38(br,CH,2H),1.98-1.96(m,CH,1H),1.91-1.85(m,CH,2H),1.51(br,CH,1H),1.36(s,CH3×3,9H);ESI-MS:m/z=382[M+1]+。
步骤2.(S)-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物16)合成
合成步骤参考实施案例1步骤5.以中间体2-14和2-15为原料制备化合物16,得白色固体。Yield:72%;mp:230℃(分解);1H NMR(500MHz,DMSO-d6):δ10.42(br,NH,1H),8.87(s,Ar-H,2H),8.59(d,J=1.5Hz,Ar-H,1H),8.35(d,J=2.0Hz,Ar-H,1H),8.23(s,Ar-H,1H),7.97(s,Ar-H,1H),4.41-4.36(m,CH,1H),3.89(s,CH3,3H),3.19(dd,J=12.0Hz,2.0Hz,CH2,1H),2.82-2.78(m,CH2,1H),2.68-2.64(m,CH2,1H),2.58-2.53(m,CH2,1H),2.14-2.11(m,CH2,1H),1.78-1.72(m,CH2,1H),1.65-1.58(m,CH2,1H),1.54-1.46(m,CH2,1H);13C NMR(125MHz,DMSO-d6):δ157.28,154.26,141.98,135.75,134.02,127.58,119.77,116.28,115.46,113.46,108.76,74.59,49.62,45.27,38.74,29.78,24.19;ESI-MS:m/z=377[M+1]+。
制备实施例17. 4-甲氧基-5-(3-氟苯基)-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶(化合17)
步骤1. 2-氰基-5-溴-3-(N-叔丁氧羰基哌啶-4-甲基)氧基吡啶(中间体2-16)合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,以5-溴-3-硝基-2-氰基吡啶(化合物2-1)和N-叔丁氧羰基哌啶-4-甲醇为原料制备化合物2-16,得金黄色油状物。Yield:80%;1H NMR(500MHz,CDCl3):δ8.34(s,Ar-H,1H),7.51(s,Ar-H,1H),4.20(d,J=11.5Hz,CH2,2H),3.95(br,CH2,2H),2.80(br,CH2,2H),2.14-2.05(m,CH,1H),1.89(br,CH2,2H),1.47(s,CH3×3,9H),1.33-1.26(m,CH2,2H);ESI-MS:m/z=206[M+1]+。
步骤2. 4-甲氧基-2-氯-5-溴嘧啶(中间体2-17)合成
将5-溴-2,4-二氯嘧啶(500mg,2.194mmol)溶于无水甲醇(5mL)中,氮气保护下,滴加甲醇钠(钠56mg,2.42mmol)的甲醇溶液(1.85mL),室温下搅拌过夜。加入饱和氯化铵溶液淬灭反应,减压回收溶剂,加入CH2Cl2(30mL),用水(30mL)洗涤,减压回收溶剂。用硅胶柱层析纯化,以PE:EtOAc(4:1)为洗脱剂,得白色固体。Yield:90%;1H NMR(400MHz,CDCl3):δ8.44(s,Ar-H,1H),4.11(s,CH3,3H);ESI-MS:m/z=223[M+1]+。
步骤3. 4-甲氧基-5-溴-2-氨基嘧啶(中间体2-18)合成
合成步骤参考实施案例1步骤3.用类似于化合物1-5的合成方法,中间体2-17为原料制备化合物2-18,得白色固体。Yield:75%;1H NMR(500MHz,CDCl3):δ8.13(s,Ar-H,1H),5.41(s,NH2,2H),3.91(s,CH3,3H);ESI-MS:m/z=205[M+1]+。
步骤4. 4-甲氧基-5-(3-氟苯基)-2-氨基嘧啶(中间体2-19)合成
合成步骤参考实施案例1步骤4.以中间体2-18和3-氟苯硼酸为原料制备化合物2-19,得白色固体。Yield:88%;mp:127-128℃;1H NMR(500MHz,CDCl3):δ7.98(s,Ar-H,1H),7.46-7.42(m,Ar-H,1H),7.18(d,J=7.5Hz,Ar-H,1H),7.12-7.08(m,Ar-H,2H),5.19(s,NH2,2H),4.10(s,NH2,2H),3.96(s,CH3,3H);ESI-MS:m/z=220[M+1]+。
步骤4. 4-甲氧基-5-(3-氟苯基)-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶(化合物17)合成
合成步骤参考实施案例1步骤5.以中间体2-16和2-19为原料制备化合物17,得白色固体。Yield:79%;mp:139-141℃;1H NMR(500MHz,CDCl3):δ8.36(d,J=2.0Hz,Ar-H,1H),8.18(s,Ar-H,1H),8.08(d,J=2.0Hz,Ar-H,1H),7.58-7.54(m,Ar-H,1H),7.24-7.21(m,Ar-H,2H),7.15-7.12(m,Ar-H,1H),7.04(br,NH,1H),4.05(s,CH3,3H),3.97(d,J=6.5Hz,CH2,2H),3.20-3.18(m,CH2,2H),2.73-2.72(m,CH2,2H),2.13-2.06(m,CH,1H),1.93-1.91(m,CH2,2H),1.40-1.32(m,CH2,2H);13C NMR(125MHz,DMSO-d6):δ163.85,163.46,161.52,158.92,158.03,157.93,141.22,136.25,135.99,135.92,131.07,131.01,125.40,116.22,116.05,115.84,114.98,114.82,114.64,114.14,111.54,72.92,55.50,44.11,34.25,27.19;ESI-MS:m/z=435[M+1]+。
制备实施例18. 4-甲氧基-5-(3-氟苯基)-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶(化合18)
步骤1. 4-甲氧基-5-(1-甲基-1H-吡唑-4-基)-2-氨基嘧啶(中间体2-20)合成
合成步骤参考实施案例1步骤4.以中间体2-18和1-甲基吡唑-4-硼酸频那醇酯为原料制备化合物2-20,得白色固体。Yield:85%;1H NMR(500MHz,CDCl3):δ7.96(s,Ar-H,1H),7.58(s,Ar-H,1H),7.47(s,Ar-H,1H),5.18(s,NH2,2H),3.97(s,CH3,3H),3.94(s,CH3,3H);ESI-MS:m/z=206[M+1]+。
步骤2. 4-甲氧基-5-(1-甲基-1H-吡唑-4-基)-N-(2-氰基-3-(哌啶-4-甲基)氧基吡啶-5-基)-2-氨基嘧啶(化合物18)合成
合成步骤参考实施案例1步骤5.以中间体2-16和2-20为原料制备化合物18,得白色固体。Yield:82%;mp:213-215℃;1H NMR(500MHz,DMSO-d6):δ9.02(s,Ar-H,1H),8.69(d,J=2.0Hz,Ar-H,1H),8.25(s,Ar-H,1H),8.24(d,J=2.0Hz,Ar-H,1H),8.05(s,NH,1H),7.70(s,Ar-H,1H),5.33(s,NH,1H),4.05(d,J=6.5Hz,CH2,2H),3.92(s,CH3,3H),3.91(s,CH3,3H),3.31-3.28(m,CH2,2H),2.93-2.87(m,CH2,2H),2.19-2.11(m,CH,1H),1.93-1.91(m,CH2,2H),1.55-1.47(m,CH2,2H);ESI-MS:m/z=421[M+1]+。
制备实施例19.化合物19~31的制备
步骤1.中间体2-21~2-33合成
合成步骤参考实施案例1步骤4.以中间体1-5和相应的硼酸为原料制备化合物2-21~2-33,得白色固体。
步骤2.化合物19-31合成
合成步骤参考实施案例1步骤5.以中间体2-21~2-33和2-2为原料制备化合物19-31,得白色固体。
其中化合物30的合成方法如下:
合成步骤参考实施案例1步骤5.以中间体2-31和2-2为原料制备化合物30的中间体,将Boc保护的中间体(100mg,0.203mmol)溶于DMF/THF(1:1)的混合溶液(2mL),滴加N-氯代丁二酰亚胺(27mg,0.203mmol),60℃下搅拌5小时,减压回收溶剂,加入乙酸乙酯,用水洗除DMF,合并有机层,用无水硫酸钠干燥,减压回收溶剂的残余物,用三氟乙酸脱保护基得黄色固体。
制备实施例20.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶(化合物32)
步骤1.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶(化合物32)合成
合成步骤参考实施案例1步骤5.以中间体1-6和2-2为原料制备化合物32,得白色固体。Yield:79%;mp:207-209℃;1H NMR(500MHz,DMSO-d6):δ9.92(s,NH,1H),8.51(s,Ar-H,2H),7.91(s,Ar-H,1H),7.90(s,Ar-H,1H),7.61(s,Ar-H,1H),6.74(q,J=4.5Hz,NH,1H),4.40-4.35(m,CH,1H),3.89(s,CH3,3H),3.15(dd,J=12.0Hz,2.0Hz,CH2,1H),2.94(d,J=5.0Hz,CH3,3H),2.79-2.74(m,CH2,1H),2.63-2.59(m,CH2,1H),2.54-2.48(m,CH2,1H),2.11-2.07(m,CH2,1H),1.74-1.68(m,CH2,1H),1.62-1.55(m,CH2,1H),1.45-1.37(m,CH2,1H);ESI-MS:m/z=406[M+1]+。
制备实施例21.N4-甲基-5-(1-甲基-1H-吡唑-5-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶(化合物33)
步骤1. 5-(1-甲基-1H-吡唑-5-基)-N4-甲基-2,4-二氨基嘧啶(中间体2-35)合成
合成步骤参考实施案例1步骤4.以中间体1-5和1-甲基-1H-吡唑-5-硼酸频哪醇酯为原料制备化合物2-35,得白色固体。Yield:65%;1H NMR(500MHz,DMSO-d6):δ7.54(s,Ar-H,1H),7.47(d,J=1.5Hz,Ar-H,1H),6.24(br,NH2,2H),6.20(d,J=1.5Hz,Ar-H,1H),6.07(q,J=4.5Hz,NH,1H),3.61(s,CH3,3H),2.75(d,J=4.5Hz,CH3,3H);ESI-MS:m/z=205[M+1]+。
步骤2.N4-甲基-5-(1-甲基-1H-吡唑-4-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶(化合物33)合成
合成步骤参考实施案例1步骤5.以中间体2-35和2-2为原料制备化合物33,得白色固体。Yield:74%;1H NMR(400MHz,DMSO-d6):δ10.07(br,NH,1H),8.55(s,Ar-H,1H),8.51(s,Ar-H,1H),7.87(s,Ar-H,1H),7.54(d,J=1.6Hz,Ar-H,1H),6.78(q,J=4.0Hz,NH,1H),6.32(d,J=1.6Hz,Ar-H,1H),4.41-4.36(m,CH,1H),3.67(s,CH3,3H),3.16(dd,J=9.6Hz,2.0Hz,CH2,1H),2.93(d,J=3.6Hz,CH3,3H),2.79-2.75(m,CH2,1H),2.64-2.59(m,CH2,1H),2.54-2.49(m,CH2,1H),2.11-2.07(m,CH2,1H),1.74-1.68(m,CH2,1H),1.63-1.56(m,CH2,1H),1.45-1.37(m,CH2,1H);13C NMR(100MHz,DMSO-d6):δ160.55,159.15,157.30,155.63,142.41,138.23,135.67,134.32,116.34,113.22,108.60,107.38,101.53,74.30,49.78,45.27,36.50,29.89,28.14,24.20;ESI-MS:m/z=406[M+1]+。
制备实施例22.化合物34~43的制备
步骤1.中间体2-36~2-42合成
合成步骤参考实施案例1步骤1.用类似于化合物1-2的合成方法,以5-溴-3-硝基-2-氰基吡啶(化合物2-1)和相应的醇为原料制备化合物2-36~2-42,得产物。
步骤2.化合物34-43合成
合成步骤参考实施案例1步骤5.分别以2-13、2-15、2-16、2-36~2-42和中间体2-34为原料制备化合物34-43,得白色固体。
制备实施例23.N4-甲基-5-(4-甲基噻唑-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶(化合物44)
步骤1.锂(4-甲基噻唑-2-基)硼酸三异丙酯(中间体2-44)合成
氮气保护下,将4-甲基噻唑(1g,10.09mmol),硼酸三异丙酯(2.35mL,10.09mmol)溶于无水甲苯和THF的混合溶液(32mL,v/v,4:1)中。冷却至-78℃,缓慢滴加正丁基锂(3.83mL,2.5mol/L,9.58mmol),约85min滴完,搅拌135min。缓慢升温至0℃(约1.5h),加入异丙醇(2.84mL),搅拌过夜。减压回收溶剂,加入无水丙酮(17mL),减压旋蒸至均相,冷却至室温。在氮气保护下抽滤,用55℃的乙腈洗涤,真空干燥得白色固体,无需纯化,直接进行下一步反应。
步骤2.N4-甲基-5-溴-N2-(2-氰基-3-(N-叔丁氧羰基哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧(中间体2-45)合成
合成步骤参考实施案例1步骤5.以中间体1-5和2-2为原料制备中间体2-45,得白色固体。Yield:70%;1H NMR(400MHz,DMSO-d6):δ10.05(s,NH,1H),8.46(s,Ar-H,1H),8.41(s,Ar-H,1H),8.13(s,Ar-H,1H),7.32(q,J=4.5Hz,NH,1H),4.59(br,CH,1H),3.96-3.93(m,CH2,1H),3.77-3.70(m,CH2,1H),3.54-3.43(m,CH2,1H),3.07-2.96(m,CH2,1H),2.93(d,J=4.5Hz,CH3,3H),1.94(br,CH2,2H),1.85-1.80(m,CH2,1H),1.48-1.46(m,CH2,1H),1.38(s,CH3×3,9H);ESI-MS:m/z=504[M+1]+。
步骤3.N4-甲基-5-(4-甲基噻唑-2-基)-N2-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2,4-二氨基嘧啶(化合物44)合成
氮气保护下,向化合物2-45(85.3mg,0.211mmol),2-44(124mg,0.422mmol),Pd(dppf)Cl2(7.7mg,0.011mmol),CuCl(2.1mg,0.021mmol),ZnCl2(28.8mg,0.211mmol),Cs2CO3(137.5mg,0.422mmol)的混合物中加入无水DMF(10mL),加热至100℃搅拌过夜。抽滤,减压回收溶剂得残余物。用硅胶柱层析纯化,以CH2Cl2:EtOH(30:1)为洗脱剂,得白色固体。将得到的白色固体溶于二氯甲烷(3mL),冰浴下滴加三氟乙酸(3mL),搅拌30分钟,室温下搅拌4.5小时,用饱和碳酸氢钠溶液中和pH至9,加入乙酸乙酯(40mL),用饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压回收溶剂得残余物。用硅胶柱层析纯化,以CH2Cl2/EtOH(NH3)(100:3)为洗脱剂,得白色固体。Yield:40%;1H NMR(400MHz,DMSO-d6):δ10.05(s,NH,1H),8.46(s,Ar-H,1H),8.41(s,Ar-H,1H),8.13(s,Ar-H,1H),7.32(q,J=4.5Hz,NH,1H),4.59(br,CH,1H),3.96-3.93(m,CH2,1H),3.77-3.70(m,CH2,1H),3.54-3.43(m,CH2,1H),3.07-2.96(m,CH2,1H),2.93(d,J=4.5Hz,CH3,3H),1.94(br,CH2,2H),1.85-1.80(m,CH2,1H),1.48-1.46(m,CH2,1H),1.38(s,CH3×3,9H);ESI-MS:m/z=504[M+1]+。
制备实施例24. 5-三氟甲基-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物45)
步骤1. 5-三氟甲基-二氨基嘧啶(中间体2-47)合成
将化合物2-46(225mg,1.23mmol)置于封管中,加入氨水(10mL),N-甲基吡咯烷酮(10mL),120℃下搅拌24h。冷却至室温,减压回收溶剂得残余物。用硅胶柱层析纯化,以PE:EtOAc(2:1)为洗脱剂,得白色固体。LC-MS:m/z=164[M+1]+。
步骤2. 5-三氟甲基-N-(2-氰基-3-(哌啶-3-氧基)吡啶-5-基)-2-氨基嘧啶(化合物45)合成
合成步骤参考实施案例1步骤5.以中间体2-47和2-2为原料制备化合物45,得白色固体。LC-MS:m/z=365[M+1]+。
制备实施例25.化合物46-50的制备
步骤1.中间体2-49/2-51合成
合成步骤分别参考实施案例17步骤2、实施案例1步骤2,得化合物2-49/2-51,质谱数据分别为LC-MS:m/z=194[M+1]+;LC-MS:m/z=193[M+1]+。
步骤2.中间体2-50/2-52合成
合成步骤参考实施案例24步骤1.分别以2-49/2-51为原料制备化合物2-50/2-52,得白色固体。质谱数据分别为LC-MS:m/z=194[M+1]+;LC-MS:m/z=193[M+1]+。
步骤3.化合物46-50合成
合成步骤参考实施案例1步骤5.分别以2-16、2-13、2-35、2-44和中间体2-50/2-52为原料制备化合物46-50,得白色固体。质谱数据分别为LC-MS:m/z=409[M+1]+;LC-MS:m/z=394[M+1]+;LC-MS:m/z=394[M+1]+;LC-MS:m/z=394[M+1]+;LC-MS:m/z=393[M+1]+。
中间体2-21~2-33、2-36~2-42和化合物19-31、34-43的核磁及质谱数据见表1-1、1-2、1-3、1-4。
表1-1
表1-2
表1-3
表1-4
本发明公开的化合物的Chk1抑制作用
以Staurosporine为阳性对照,采用ADP-Glo试剂盒评价Chk1酶抑制活性(IC50)。化合物作用于Chk1蛋白激酶,抑制其磷酸化底物Cdc25C,磷酸化过程需消耗ATP,反应结束后利用ADP-GloTM Reagent消耗掉剩余ATP,反应过程中所产生的ADP可被ADP-Glo Detection Reagent转化为ATP,ATP可作为Ultra-GloTM萤光素酶催化反应的底物,产生光信号。将待测化合物溶于DMSO中制成10mM的储备液,并按一定比例稀释至12个不同浓度以备测试。于384孔板中,每孔加入待测化合物1μL,2.5X Chk1激酶2μL,对照组加入1X缓冲液2μL,室温下孵育10min,加入2.5X底物2μL,在37℃下孵育1h,加入ADP-GloTM Reagent 5μL终止反应,在37℃下孵育1h。加入ADP-Glo Detection Reagent 10μL,在37℃下孵育30min,并且每个样品设置三个平行孔,采用luminescence荧光检测酶标仪测定吸光度,利用GraphPad Prism 5软件处理数据,计算IC50值。
本发明公开的化合物对Chk1激酶的抑制活性
表2化合物对Chk1激酶的IC50(μM)
从表中数据可以看出,大部分化合物是Chk1蛋白激酶的高效抑制剂,有24个化合物的Chk1抑制活性与阳性化合物Staurosporine相当,5个化合物优于阳性对照Staurosporine。因此,本发明所涉及的可用作Chk1抑制剂的2-取代嘧啶类衍生物具有广阔的抗肿瘤应用前景。
本发明公开的化合物对各种肿瘤细胞的增殖抑制活性
细胞株:人多发性骨髓瘤细胞RPMI 8226、人套细胞淋巴瘤细胞Mino、Jeko-1、人淋巴瘤细胞Romas、人急性单核细胞白血病细胞MV-4-11、人乳腺癌细胞MCF-7、人肺癌细胞A549、人前列腺癌细胞LnCAP、人胃癌细胞BGC-823、人结肠癌细胞HCT116、Colo205、人卵巢癌细胞OVCAR-8实验方法:MTS法测定化合物对不同的肿瘤细胞株的体外增殖抑制活性(IC50)。
将处于对数生长期的细胞用胰酶消化,计数,以1×104细胞/孔的密度接种在96孔板中,每孔100μL,置于含5%CO2的37℃培养箱中过夜培养,每一化合物设六个浓度梯度,每一浓度设三组复孔,加入后,培养72小时,加入20μL MTS。37℃下孵育2小时后,用SpectraMAX 340酶标仪测490nm(L1)下的光吸收值,参考波长690nm(L2),将(L1-L2)值对抑制剂的不同浓度作图,经公式拟合得半数抑制浓度IC50。
表3-1化合物对各肿瘤细胞株的增殖抑制作用
表3-2
aIC50:三次实验平均值;b未测。
本发明公开的化合物与其他药物联用活性
MV 4-11细胞以5000/孔接种至96孔板。联用时,药物按两药IC50之比确定药物的配比,各药浓度选择范围为IC20~IC80(或者1/8,1/4,1/2,1,2and 4of IC50)。72小时后,加入MTS试剂检测细胞活力,以未加药组为100%计算出抑制率Fa。采用Chou-Talalay method将抑制率Fa及相应药物浓度输入CompuSyn软件进行分析,得出单一浓度药物联用CI值及Fa-CI曲线。CI(combination index)的计算公式是CI=DA/ICX,A+DB/ICX,B(A,B代表两种不同药物,ICX,A和ICX,B是两种药物单独使用时生长抑制率达X时的药物浓度,DA和DB是两药联用时生长抑制率达X时两种药物的浓度)。结果见图1。图中:CHK1抑制剂(35);FLT3抑制剂Crenolanib(Cre)、Quizartinib(Qui);Akt抑制剂GSK2141795(GSK);CI=联用指数,根据Soriano等的判断方法,0.9≤CI≤1.1为叠加作用,0.8≤CI<0.9为低度协同作用,0.6≤CI<0.8为中度协同作用,0.4≤CI<0.6为高度协同作用,0.2≤CI<0.4为强协同作用。
- 还没有人留言评论。精彩留言会获得点赞!