一种含有双芘基团的多肽纳米材料及其制备方法和应用与流程
本发明属于纳米材料技术领域,涉及一种含有双芘基团的多肽纳米材料及其制备方法和应用。
背景技术:
肿瘤转移是恶性肿瘤的重要生物学特征,也是临床绝大多数肿瘤患者治疗失败和死亡的主要原因。恶性肿瘤对患者健康的威胁越来越大,防止肿瘤转移是改善肿瘤治疗的关键。据2015年1月国家卫生计生委关于全国死因登记报告信息系统统计分析报告显示,所有31个省份中,恶性肿瘤在6个省份中(占46.2%)排名第一位,从死因顺位及死亡率的分析结果来看,影响我国居民健康的前两位死因为恶性肿瘤和脑血管病。目前国内外已研发的抗肿瘤药物众多,1953年至今FDA批准的抗肿瘤药物约210种,但由于肿瘤的浸润和转移是一个非常复杂的过程,目前高效抑制肿瘤转移的药物却未见批准。伴有细胞外基质破坏的肿瘤细胞浸润是肿瘤转移这一复杂机制的最主要的过程。目前在临床肿瘤学中有一个紧急战略目标就是试图抑制MMP(基质金属蛋白酶)的活性以阻止肿瘤的转移。然而,大多数肿瘤疾病一旦发现已进入晚期阶段,肿瘤细胞的浸润和转移已经发生,因此抑制MMP的活性来达到控制肿瘤发展的做法收效甚微。
因此,在本领域迫切需要开辟一种新的策略用于抑制肿瘤的侵袭和转移。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种含有双芘基团的多肽纳米材料及其制备方法和应用,所述的多肽纳米材料可在活体肿瘤区域原位构筑人造细胞外基质,从而在肿瘤迁移破坏细胞外基质(ECM)的过程中,形成转移屏障,能够高效的抑制肿瘤的浸润和转移。
为达到此发明目的,本发明采用以下技术方案:
一方面,本发明提供一种含有双芘基团的多肽纳米材料,所述纳米材料具有式I所示的结构:
其中,R1为具有分子内多重氢键的多肽序列,R2和R3为肿瘤部位靶向肽,R1、R2和R3之间由酰胺键相连。
本发明所述结构的多肽纳米材料包括双芘荧光分子,强氢键作用力的自组装多肽单元和结合癌细胞表面受体的靶向肽,该材料可仿造生物体内层粘连蛋白纤维化过程,靶向性结合癌细胞表面的受体蛋白,在肌动蛋白和材料本身氢键作用力下可发生形貌转变自组装成纳米纤维网状结构,在细胞外基质降解过程中可作为一个长期的屏障,高效抑制肿瘤的侵袭和转移。
优选地,所述含有双芘基团的多肽纳米材料中双芘基团的供体为具有式II所示结构的双芘化合物:
在本发明所述含有双芘基团的多肽纳米材料中双芘基团与R1通过形成酰胺键而连接在一起。
优选地,R1为
其中表示基团连接位点。
在本发明中,R1的供体结构为:即KLVFFK(Lys-Leu-Val-Phe-Phe-Lys);
或即KLPFFDK(Lys-Leu-Pro-Phe-Phe-Asp-Lys)。
优选地,R2为
中的任意一种,其中表示基团连接位点。
即在本发明中,R2的供体的结构为:(即GGDGR(Gly-Gly-Asp-Gly-Arg))、(即YIGSR(Tyr-Ile-Gly-Ser-Arg))、(即DGEA(Asp-Gly-Glu-Ala))、(即KQAGDV(Lys-Gln-Ala-Gly-Asp-Val))或(即GGNGR(Gly-Gly-Asn-Gly-Arg))中的任意一种。
优选地,R3为
中的任意一种,其中表示基团连接位点。
即在本发明中,所述R3的供体结构为:(即GGDGR(Gly-Gly-Asp-Gly-Arg))、(即YIGSR(Tyr-Ile-Gly-Ser-Arg))、(即DGEA(Asp-Gly-Glu-Ala))、(即KQAGDV(Lys-Gln-Ala-Gly-Asp-Val))或(即GGNGR(Gly-Gly-Asn-Gly-Arg))中的任意一种。
优选地,所述多肽纳米材料为具有以下式a-c所示结构的化合物中的任意一种或至少两种的组合:
另一方面,本发明提供了如上所述含有双芘基团的多肽纳米材料的制备方法,所述方法为以树脂为载体,以式II所示双芘化合物和氨基酸为原料,利用固相合成方法制备得到所述含有双芘基团的多肽纳米材料。
优选地,所述树脂为王氏树脂。
在本发明中,所述固相合成法是本领域技术人员所熟知的技术,本发明采用王氏树脂作为载体,在无水DMF中溶胀6个小时后,按照多肽合成顺序,室温下,在多肽合成管中加入氨基酸(10倍当量,所述氨基酸为将一些活泼基团进行了特定保护的氨基酸)和HBTU(苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐,10倍当量),通过偶联剂(氮甲基吗啉:DMF=5:95,体积比)进行偶联45min,依照多肽顺序将氨基酸依次偶联,双芘化合物也同样按照氨基酸的偶联方法进行反应,偶联双芘化合物时将预发生反应的氨基酸上由保护基保护的氨基进行脱保护(例如进行Fmoc脱保护,Fmoc脱保护剂为DMF:六氢吡啶=4:1),偶联成功后对氨基酸上其他由保护基保护的基团进行脱保护(Dde脱保护剂为水合肼:DMF=2:98,Boc、tBu、Pbf三种保护基均在后续的裂解过程中在三氟乙酸的作用下脱除)15min左右,最后经三氟乙酸裂解、旋蒸、乙醚纯化而得。
另一方面,本发明提供了如上所述含有双芘基团的多肽纳米材料的形变自组装方法,所述方法包括以下步骤:
(1)将如上所述含有双芘基团的多肽纳米材料溶于有机溶剂中,得到所述多肽纳米材料溶液;
(2)向步骤(1)得到的多肽纳米材料溶液中加入水,自组装得到纳米球颗粒体系;
(3)向步骤(2)得到的纳米球颗粒体系中加入钙盐,诱导纳米球形颗粒自组装成纳米纤维网状结构。
本发明中,所述形变自组装是指本发明所述的含有双芘基团的多肽纳米材料在不同的自组装条件下可以由一种自组装形态转变为另一种自组装形态的行为。
优选地,步骤(1)所述有机溶剂为二甲基亚砜(DMSO)和或六氟异丙醇(HFIP);
优选地,步骤(1)所述多肽纳米材料溶液的浓度为(1-2)×10-3M,例如1×10-3M、1.1×10-3M、1.2×10-3M、1.3×10-3M、1.4×10-3M、1.5×10-3M、1.6×10-3M、1.7×10-3M、1.8×10-3M、1.9×10-3M或2×10-3M,优选1.5×10-3M。
优选地,步骤(2)所述水与步骤(1)所述有机溶剂的体积比为(40-80):1,例如40:1、43:1、45:1、48:1、50:1、52:1、55:1、58:1、60:1、65:1、68:1、70:1、73:1、75:1、78:1或80:1。
在本发明的步骤(2)自组装形成纳米球颗粒体系时,此纳米球颗粒呈现β折叠结构,在这种聚集的状态下,所述多肽纳米材料中包含的双芘结构可自发产生绿色荧光。该荧光可检测纳米材料在细胞表面的聚集以及在活体内的分布与滞留。
优选地,步骤(3)所述钙盐为氯化钙。
优选地,步骤(3)中加入钙盐使得体系中钙离子的浓度为(2-8)×10-4M,例如2×10-4M、2.5×10-4M、3×10-4M、3.5×10-4M、4×10-4M、4.5×10-4M、5×10-4M、5.5×10-4M、6×10-4M、6.5×10-4M、7×10-4M、7.5×10-4M或8×10-4M,优选6×10-4M。
在本发明中,步骤(3)所述纳米纤维网状结构为β折叠纳米纤维网状结构。
在本发明中,于所述多肽纳米材料形成的纳米球颗粒体系中加入合适浓度的钙盐达到合适浓度范围的钙离子浓度后可以使得其纳米球形态转变为纳米纤维网状形态。该钙离子浓度是模拟肿瘤细胞区域钙离子浓度,说明本发明的材料在肿瘤细胞区域有转变为纳米纤维网状结构的性能,在正常血浆和组织液中的钙离子含量为(2.4-2.5)×10-3M,在该浓度下虽然可以发生形变自组装,但是其需要的时间较长,如果在体内,则已经被体内循环机制清除体外了,因此在该材料输入体内时,优先在肿瘤组织区域发生形变,选择性粘附在肿瘤区域。
另一方面,本发明提供一种人造细胞外基质,所述人造细胞外基质为如上所述含有双芘基团的多肽纳米材料形成的纳米纤维网状结构。
将本发明的含有双芘基团的多肽纳米材料形成的纳米球颗粒可以在肿瘤细胞或肿瘤细胞团与膜表面受体结合,发生形变自组装成β折叠纳米纤维网状结构。
另一方面,本发明提供了所述含有双芘基团的多肽纳米材料在制备抑制肿瘤侵袭和转移的材料中的应用。
将本发明的含有双芘基团的多肽纳米材料形成的纳米球颗粒输入活体(例如动物或人),该多肽纳米材料具有良好的生物兼容性,其能够大大提高了药物的利用度以及材料在肿瘤区域的滞留时间,并且其可以在肿瘤细胞表面与受体结合,发生形变自组装成纳米纤维网状结构,在活体内自组装成的纳米纤维网状结构原位构筑了人造细胞外基质,该人造细胞外基质具有选择性粘附在肿瘤区域形成迁移屏障的优点,可以高效抑制肿瘤的侵袭和转移。
相对于现有技术,本发明具有以下有益效果:
本发明的含有双芘基团的多肽纳米材料可发生形变自组装,可以在肿瘤区域自组装成纳米纤维网状结构,原位构筑了人造细胞外基质,该人造细胞外基质具有选择性粘附在肿瘤区域形成迁移屏障,可以高效抑制肿瘤的侵袭和转移,在医学上具有广泛的应用前景。
附图说明
图1为本发明实施例1制备得到的多肽纳米材料的质谱图。
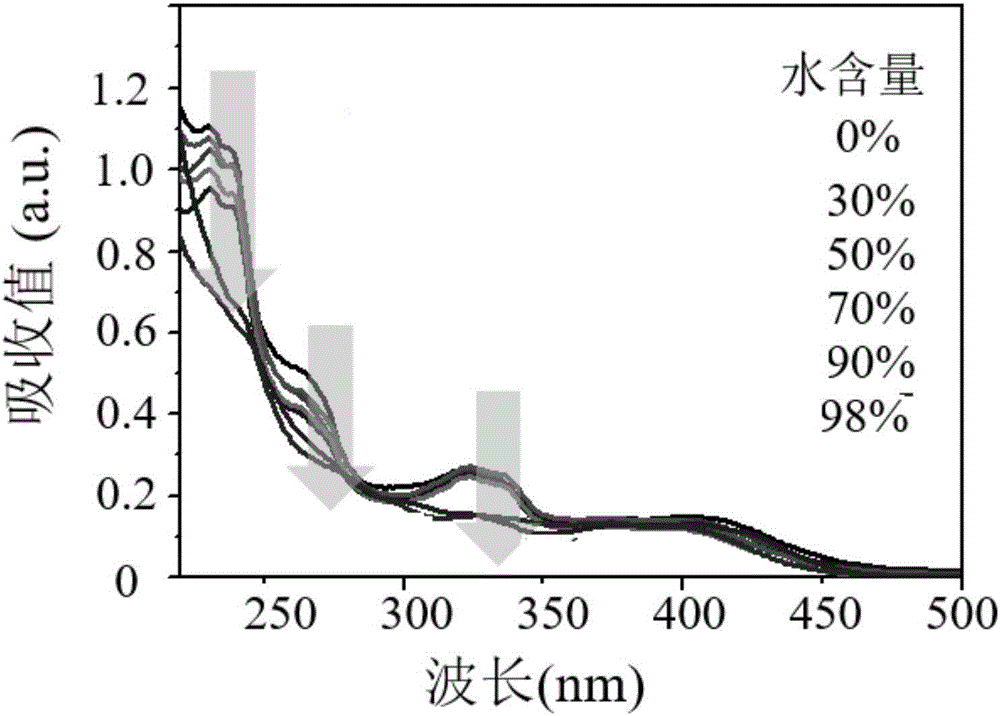
图2为本发明实施例1制备得到的多肽纳米材料在不同水含量的自组装体系中形成的纳米颗粒体系的紫外光谱图,图中沿着箭头标示方向水含量由0%逐渐升高至98%。
图3为本发明实施例1制备得到的多肽纳米材料在不同水含量的自组装体系中形成的纳米颗粒体系的荧光光谱图,图中沿着箭头标示方向水含量由0%逐渐升高至98%。
图4A为本发明实施例4制备得到的纳米颗粒体系的透射电镜图。
图4B为本发明实施例4制备得到的纳米纤维分散液的透射电镜图。
图5为本发明实施例4制备得到的纳米纤维分散液的圆二色谱表征结果图。
图6为利用本发明的多肽纳米材料培养人源乳腺癌细胞MDA-MB-231后的单光子激光共聚焦成像图;
图7为利用本发明的多肽纳米材料培养人源乳腺癌细胞MDA-MB-231后对细胞表面的扫描电镜结果图;其中A图为空白对照组结果图,标尺为3μm,B图为实验组结果图,标尺为3μm。
图8为本发明实施例8对本发明的多肽纳米材料对肿瘤细胞的伤口愈合的抑制情况的考察结果图,其中A图是空白对照组细胞的伤口愈合情况,标尺为500μm,B图为实验组细胞的伤口愈合情况,标尺为500μm。
图9为本发明实施例9对本发明的多肽纳米材料对肿瘤细胞的细胞迁移的抑制情况的考察结果图,其中A图是空白对照组细胞的侵袭情况,标尺为500μm,B图为实验组细胞的侵袭情况,标尺为500μm。
图10为本发明实施例10对本发明的多肽纳米材料对肿瘤细胞的细胞侵袭的抑制情况的考察结果图,其中A图是空白对照组细胞的迁移情况,标尺为500μm,B图为实验组细胞的迁移情况,标尺为500μm。
图11为利用本发明的多肽纳米材料治疗荷瘤小鼠后,多肽纳米材料在在小鼠体内分布的荧光成像图。
图12为利用本发明的多肽纳米材料治疗荷瘤小鼠后其肿瘤大小的变化图,其中A图为空白对照组肿瘤大小,B图为治疗后肿瘤大小。
图13为利用本发明的多肽纳米材料治疗荷瘤小鼠后,小鼠肿瘤组织切片的微血管密度变化图,其中A图为空白对照组微血管密度图,B图为治疗后肿瘤组织切片的微血管密度图。
图14为利用本发明的多肽纳米材料治疗荷瘤小鼠后,小鼠肺部情况变化图,其中A图为空白对照小鼠肺部情况图,B图为治疗后小鼠肺部情况图。
图15为利用本发明的多肽纳米材料治疗荷瘤小鼠后,小鼠肺部结节情况变化图,其中A图为空白对照组小鼠肺部结节情况图,B图为治疗后小鼠肺部结节情况图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
在本实施例中,含有双芘基团的多肽纳米材料的结构如下所示:
即在式I所示的结构中,R1的供体为KLVFFK,R2的供体为YIGSR,R3的供体为GGDGR。
所述含有双芘基团的多肽纳米材料以多肽的固相合成(SPPS)方法来合成,多肽固相合成法实验步骤如下:
采用王氏树脂作为载体,在无水DMF中溶胀6个小时后,按照多肽合成顺序,室温下,在多肽合成管中加入氨基酸(10倍当量,所述氨基酸为将一些活泼基团进行了特定保护的氨基酸)和HBTU(苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐,10倍当量),通过偶联剂(氮甲基吗啉:DMF=5:95,体积比)进行偶联45min,依照多肽顺序将氨基酸依次偶联,双芘化合物也同样按照氨基酸的偶联方法进行反应,偶联双芘化合物时将预发生反应的氨基酸上由保护基保护的氨基进行脱保护(例如进行Fmoc脱保护,Fmoc脱保护剂为DMF:六氢吡啶=4:1),偶联成功后对氨基酸上其他由保护基保护的基团进行脱保护(Dde脱保护剂为水合肼:DMF=2:98,Boc、tBu、Pbf三种保护基均在后续的裂解过程中在三氟乙酸的作用下脱除)15min左右,最后经三氟乙酸裂解、旋蒸、乙醚纯化而得,其合成流程可表示如下(其中黑色的球代表王氏树脂载体):
对本实施例合成的含有双芘基团的多肽纳米材料进行质谱(Microflex LRF MALDI-TOF)表征,结果如图1所示,由该图可知所得质谱峰值与所得式I分子量[M+H+]相吻合。
实施例2
在本实施例中,含有双芘基团的多肽纳米材料的结构如下所示:
即在式I所示的结构中,R1的供体为KLPFFDK,R2的供体为YIGSR,R3的供体为GGDGR。
所述含有双芘基团的多肽纳米材料以多肽的固相合成(SPPS)方法来合成,选用王氏树脂为载体,按照结构中的多肽顺序,选择相对应的氨基酸进行固相合成,双芘化合物也同样按照氨基酸的偶联方法进行反应,其合成过程与实施例1相似,合成得到本实施例的含有双芘基团的多肽纳米材料。
实施例3
在本实施例中,含有双芘基团的多肽纳米材料的结构如下所示:
即在式I所示的结构中,R1的供体为KLVFFK,R2的供体为DGEA,R3的供体为KQAGDV。
所述含有双芘基团的多肽纳米材料以多肽的固相合成(SPPS)方法来合成,选用王氏树脂为载体,按照结构中的多肽顺序,选择相对应的氨基酸进行固相合成,双芘化合物也同样按照氨基酸的偶联方法进行反应,其合成过程与实施例1相似,合成得到本实施例的含有双芘基团的多肽纳米材料。
实施例4
在本实施例中对实施例1制备的得到的多肽纳米材料进行形变自组装,方法如下:
(1)将实施例1制备得到的多肽纳米材料溶于DMSO溶剂中,得到浓度为1.5×10-3M的多肽纳米材料溶液,取20μL上述溶液置于离心管中,将980μL超纯水缓慢加入离心管中,所得混合溶液中多肽纳米材料的浓度为3.0×10-5M,超声5分钟,即可得到显示绿色荧光的纳米颗粒体系;
(2)模拟体内肿瘤部位钙离子环境,以在体外溶液中诱导材料自组装形变,具体操作为:在上述纳米颗粒体系中加入无水CaCl2,完全溶解后使得Ca2+浓度为6.0×10-4M,两天后即可得到纳米纤维分散液。
利用紫外光谱仪(Lambda 950)对依照本实施例的方法在不同水含量(0%、10%、30%、50%、70%、90%和98%)的自组装体系中形成的纳米颗粒体系进行表征,结果如图2所示,由该图可知在吸收谱峰232nm(多肽),265nm(芳香族残基)和350nm(双芘材料)处峰值降低并谱带变宽,表征所述纳米材料发生聚集,特别地,265nm处峰值降低表明多肽的芳香族残基通过π-π相互作用聚集。
利用荧光光谱仪(FS5Spectrofluorometer)对依照本实施例的方法在不同水含量(0%、10%、30%、50%、70%、90%和98%)的自组装体系中形成的纳米颗粒体系进行表征,结果如图3所示,由该图可知,伴随水含量的增加,荧光图谱中535nm处荧光强度增大并呈现最大值,表明所述纳米材料形成双芘(BP)分子的J型聚集体。
对本实施例中步骤(1)形成的纳米颗粒体系以及步骤(2)得到的纳米纤维分散液进行透射电镜(Tecnai G2 20S-TWIN)表征,结果分别如图4A和图4B所示,图4A表明将本发明所述多肽纳米材料在水环境中自组装可以形成纳米球状颗粒,其粒径小于100nm,粒径比较均一;图4B表明在加入适当浓度的钙离子后,纳米球状颗粒自组装成纳米纤维网状结构,发生形貌变化。
对本实施例中得到的纳米纤维分散液进行了圆二色谱(J-1500)表征,结果如图5所示,由结果可知,其形成的为β折叠结构。
实施例5
在本实施例中对实施例2制备的得到的多肽纳米材料进行形变自组装,方法如下:
(1)将实施例2制备得到的多肽纳米材料溶于DMSO溶剂中,得到浓度为1×10-3M的多肽纳米材料溶液,取20μL上述溶液置于离心管中,将1200μL超纯水缓慢加入离心管中,所得混合溶液中多肽纳米材料的浓度为1.64×10-5M,超声5分钟,即可得到显示绿色荧光的纳米颗粒体系;
(2)模拟体内肿瘤部位钙离子环境,以在体外溶液中诱导材料自组装形变,具体操作为:在上述纳米颗粒体系中加入无水CaCl2,完全溶解后使得Ca2+浓度为2.0×10-4M,两天后即可得到纳米纤维分散液。
实施例6
在本实施例中对实施例3制备的得到的多肽纳米材料进行形变自组装,方法如下:
(1)将实施例3制备得到的多肽纳米材料溶于DMSO溶剂中,得到浓度为2×10-3M的多肽纳米材料溶液,取20μL上述溶液置于离心管中,将800μL超纯水缓慢加入离心管中,所得混合溶液中多肽纳米材料的浓度为4.9×10-5M,超声5分钟,即可得到显示绿色荧光的纳米颗粒体系;
(2)模拟体内肿瘤部位钙离子环境,以在体外溶液中诱导材料自组装形变,具体操作为:在上述纳米颗粒体系中加入无水CaCl2,完全溶解后使得Ca2+浓度为8.0×10-4M,两天后即可得到纳米纤维分散液。
实施例7
在本实施例中,考察实施例1制备得到的多肽纳米材料在人源乳腺癌细胞表面的形貌转化情况,考察方法如下:
本实验中采用MDA-MB-231人源乳腺癌细胞,在含10.0%的胎牛血清和100.0U/mL青霉素以及100.0μg/mL链霉素的DMEM培养基中培养,培养温度为37.0℃,湿润空气中CO2的浓度为5.0%。培养细胞至对数生长期,用胰酶消化分散细胞。将合适比例的细胞悬浮液(104个细胞/mL)加至共聚焦(confocal)皿中,在上述条件的细胞培养箱中培养24小时,然后将含有实施例4制备得到的纳米颗粒溶液(3.0×10-5M)的培养基替换原培养液再培养2h,用PBS清洗三遍,加入Hoechst 33342染色20分钟,再用PBS洗三遍,进行单光子激光共聚焦成像实验,结果如图6所示,由该图可知,所述纳米材料在体外细胞实验中,可以高效抑制MDA-MB-231细胞的伤口愈合能力以及迁移和侵袭能力。
在细胞培养过程中在培养皿的底部放入硅片,使细胞在硅片上生长24h,20%的戊二醛溶液(戊二醛:PBS缓冲液=1:4)固化2-3h,再依次用PBS稀释的30%、50%、70%、90%、100%的乙醇溶液脱水处理,每个浓度脱水三次,每次10分钟。然后用叔丁醇溶液置换出水和乙醇,置换3次,每次10分钟。干燥,利用扫描电镜进行扫描观察,结果如图7所示,A图为未加入实施例4制备得到的纳米颗粒溶液培养的细胞(即空白对照)的扫描结果,B图为加入实施例4制备得到的纳米颗粒溶液培养的细胞(即实验组)的扫描结果,由结果可知,本发明的多肽纳米材料在MDA-MB-231细胞表面由纳米颗粒形态转变为纳米纤维网状结构。
实施例5和6制备得到的纳米颗粒溶液在MDA-MB-231细胞表面同样能由纳米颗粒形态转变为纳米纤维网状结构
实施例8
在本实施例中,考察本发明的多肽纳米材料对肿瘤细胞的伤口愈合的抑制作用,考察方法如下:
将细胞接种到24孔细胞培养板中(104个细胞/mL),生长24h后单层细胞的融合度达70-80%,不更换培养基,用新的20μL枪头轻轻在单层细胞间划痕,划痕横穿过孔,枪头垂直于孔板底部。划痕后用培养基轻轻的清洗孔板2次,以除去脱落的细胞,然后每孔加入新鲜的培养基,其中实验组所加新鲜培养基中添加实施例4制备得到的纳米颗粒溶液(3.0×10-4M),以不加所述纳米颗粒溶液的细胞组作为空白对照组,细胞生长24h,PBS清洗两次,然后用无水甲醇固定30min,0.1%的结晶紫(2%乙醇溶解)染色30min,PBS清洗三遍,染色的单层细胞选取不同的视野,显微镜(ZEISS Axio observer Z1)拍照,结果如图8所示,其中A图是空白对照组细胞的伤口愈合情况,B图为实验组细胞的伤口愈合情况,由结果可以看出,本发明的多肽纳米材料可明显抑制癌细胞MDA-MB-231的伤口愈合。
实施例9
在本实施例中,考察本发明的多肽纳米材料对肿瘤细胞的细胞侵袭的抑制作用,考察方法如下:
将铺有人工基质胶的Transwell小室放入24孔板中,在上室加入300μL预温的无血清培养基室温下静置30min,使基质胶再水化,再吸去剩余培养液。制备细胞悬浮液前,细胞撤血清饥饿处理12h,进一步除去血清的影响。胰酶消化细胞制备细胞悬浮液,用培养基将细胞悬浮液调至5×105个细胞/mL,取200μL加入Transwell小室内,并在上室加入所述纳米材料(3.0×10-5M),在小室下的24孔板中加入500μL含有10%的胎牛血清的培养基,培养细胞24h,将小室取出,用棉签轻轻刮去小室上层的细胞,将小室下表面的细胞用无水甲醇固定30min,0.1%的结晶紫(2%乙醇溶解)染色30min,PBS清洗三遍,染色的单层细胞选取不同的视野,显微镜拍照,结果如图9所示,其中A图是空白对照组细胞的侵袭情况,B图为实验组细胞的侵袭情况,由结果可以看出,本发明的多肽纳米材料可明显抑制癌细胞MDA-MB-231的侵袭。
实施例10
在本实施例中,考察本发明的多肽纳米材料对肿瘤细胞的细胞迁移的抑制作用,考察方法与实施例9所述细胞侵袭的考察方法不同之处仅在于,将实施例9所述细胞侵袭中Transwell小室中的人工基质胶刮去,清洗后紫外灭菌12h后使用,按照实施例9所述细胞侵袭实验的方法进行细胞处理,而后对细胞进行显微镜拍照考察多肽纳米材料对肿瘤细胞的细胞迁移的抑制作用,结果如图10所示,其中A图是空白对照组细胞的迁移情况,B图为实验组细胞的迁移情况,由结果可以看出,本发明的多肽纳米材料可明显抑制癌细胞MDA-MB-231的迁移。
实施例11
在本实施例中,考察本发明的多肽纳米材料对活体肿瘤细胞的治疗以及肿瘤迁移抑制作用,考察方法如下:
小鼠右胸处皮下注射约106个MDA-MA-231细胞,用于肿瘤生长建立老鼠模型。当小鼠肿瘤体积约100mm3左右时,开始给药治疗。每隔72小时静脉给药(实施例4制备的多肽纳米材料的纳米颗粒)200μL,药物浓度100μM,给药后分别在4、10、24、48、72小时进行小动物成像实验,收集波段550-600nm。所述多肽纳米材料在在小鼠体内分布的荧光成像图如图11所示,给药后所述多肽纳米材料在小鼠肿瘤组织部位聚集,实验表明所述纳米材料可以在肿瘤部位滞留72h,与同类纳米球形药物对比具有长效滞留的特点。
治疗结束后测定小鼠肿瘤大小以及肿瘤组织切片内的微血管密度,图12为治疗前后小鼠肿瘤大小,由A图的治疗前和B图的治疗后结果可知,用本发明的多肽纳米材料治疗小鼠后,小鼠的肿瘤体积变小。图13为治疗前后小鼠肿瘤组织切片的微血管密度,由A图的治疗前和B图的治疗后结果可知,用本发明的多肽纳米材料治疗小鼠后,小鼠肿瘤组织切片的微血管密度降低,说明对肿瘤的转移具有抑制作用。
图14为治疗前后小鼠肺部情况图,由A图的治疗前和B图的治疗后结果可知,所述纳米材料可以在肿瘤区域长效滞留,并且在肺部节结数明显较少,表征所述材料可以高效抑制肿瘤的浸润和转移。
图15为治疗前后小鼠肺部切片显示的肺部结节情况图,从A图的治疗前和B图的治疗后结果可知利用本发明的多肽纳米材料治疗小鼠后,其肺部结节数量明显减少,说明本发明的多肽纳米材料对肿瘤转移具有明显的抑制作用。
申请人声明,本发明通过上述实施例来说明本发明的含有双芘基团的多肽纳米材料,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!