酰化的GLP‑1衍生物的制作方法
本发明属于多肽技术领域,具体而言,本发明涉及glp-1(7-37)多肽类似物的脂肪酸修饰的缀合物。另外,本发明还涉及该肽缀合物的制备方法、含该肽缀合物的药物以及在制备药物中的用途和中间体等。
背景技术:
糖尿病是一种由遗传和环境等多种因素引起的糖代谢紊乱疾病,现已成为继肿瘤、心脑血管疾病之后威胁人类健康和生命安全的第三位重大疾病。糖尿病本身不一定造成危害,但长期血糖增高,大血管、微血管受损并危及心、脑、肾、周围神经、眼睛、足等,据世界卫生组织统计,糖尿病并发症高达100多种,是目前已知并发症最多的一种疾病。因糖尿病死亡者有一半以上是心脑血管所致,10%是肾病变所致。因糖尿病截肢是非糖尿病的10~20倍。为此治疗糖尿病进而预防其并发症是至关重要的社会问题。
糖尿病由于患病机理不同可分为几种类型。其中绝大部分属于二型糖尿病(约90%),主要是因体重过重和缺乏身体活动所致。ii型糖尿病患者多存在胰岛素抵抗和胰岛素分泌不足两方面异常,在发病的中晚期往往出现胰岛β细胞凋亡。目前,临床使用的口服降糖药的作用机理多为增强胰岛素敏感性,或促进胰岛素分泌以稳定血糖,均无法解决β细胞凋亡这一难题。而胰高血糖素样肽-1(glp-1)及其类似物药物由于具有减缓β细胞凋亡,增进其再生,促使胰岛β细胞分化并增殖的作用,使其成为治疗ii型糖尿病的研究重点。
glp-1是由回肠和结肠的l-细胞分泌的肠降血糖素。glp-1的作用是以葡萄糖依赖的方式增加胰岛素释放从而防止低血糖症发生。由于这种性质,其作用于2型糖尿病的潜在治疗而受到关注。然而,使用glp-1作为治疗药剂的主要障碍是其在血浆中小于4分钟的极其短的半衰期。
作为稳定肽并抑制它通过蛋白水解酶降解的方法,已经进行了一些实验来修饰对该蛋白水解酶敏感的特定氨基酸序列。由于glp-1(7-37或7-36酰胺)通过二肽基肽酶iv(dppiv)在第8位氨基酸(ala)和第9位氨基酸(asp)之间进行剪切而损失具有生物活性的glp-1的药物浓度,因此具有减少血液中葡萄糖浓度以治疗2型糖尿病作用的glp-1(7-37或7-36酰胺)具有4分钟或更短的生理活性半衰期(kreymann等,1987)。因此,已经进行了各种关于具有抗dppiv的glp-1类似物的研究,并且已经进行了用gly取代ala(deacon等,1998;burcelin等,1999)或用leu或d-ala取代ala(xiao等,2001)的实验,从而保持它的活性的同时增加了对dppiv的抗性。glp-1的n末端氨基酸,his7对glp-1的活性是重要的,并且作为dppiv的目标。因此,美国专利第5545618号描述了用烷基或酰基修饰n末端,并且gallwitz等描述了第7位his进行n甲基化或a甲基化,或者用整个his用咪唑取代来增加对dpp–iv的抗性并保持生理活性。
除了这些修饰,从希拉毒蜥蜴的唾液腺纯化的glp-1类似物exendin-4(美国专利第5424686号)具有对dppiv的抗性并比glp-1具有更高的生理活性。因此,它具有比glp-1的半衰期更长的2至4小时的体内半衰期。然而,仅适用增加dppiv抗性的方法,生理活性不能被充分地保持,并且在使用商业可获得的exendin-4(exenatide)的情况下,它需要一天两次被注射给病人,这对病人仍然是很痛苦的。
这些促胰岛素肽具有一个问题,通常肽的大小很小,从而,它们不能在肾中被重新获得,并且它们随后被排出体外。因此,已经使用以化学方法在肽的表面添加例如聚乙二醇的具有高溶解度的聚合物来抑制在肾中的损失的方法。美国专利第692464号描述了peg结合到exendin-4的赖氨酸残基来增加它的体内停留时间,然而,这种方法增加了peg的分子量,从而增加了肽类药物的体内停留时间,同时随着分子量的增加,该肽类药物的浓度显著地减少,并且肽的反应性也降低了。因此,产量被不希望的降低了。
此外,一系列其它不同方法也已经用于修饰胰高血糖素样肽-1化合物的结构以提供体内更长的作用持续时间。
wo96/29342公开了其中亲本肽激素已经通过在c-端氨基酸残基或在n-端氨基酸残基引入亲脂性取代物而修饰肽激素衍生物。
wo98/08871公开了其中亲本肽的至少一个氨基酸残基连接有亲脂性取代物的glp-1衍生物(liraglutide)。
wo99/43708公开了具有连接至c-端氨基酸残基的亲脂性取代物glp-1(7-35)和glp-1(7-36)衍生物。
wo00/34331公开了双酰化的glp-1类似物。
wo00/69911公开了用于注射进患者中的活化的促胰岛素肽,据认为在患者中它们与血液成分反应形成缀合物,从而据说提供体内更长的作用持续时间。
wo2006/097537公开了另一种酰化的glp-1类似物(semaglutide),通过将第8位氨基酸突变为非天然氨基酸,与wo98/08871的酰化的glp-1(liraglutide)相比,具有更长的半衰期。
国际专利公开第wo02/046227号申请描述了使用基因重组技术通过将glp-1,exendin-4或其类似物与人血清白蛋白或免疫球蛋白区(fc)结合来制备融合蛋白,这可以解决如聚乙二醇化产量低和非特异性的问题,但是它们仍然具有这样一个问题,即增加血液中半衰期的效果不像期待中的那么显著,并且有时候浓度也低。为了使增加血液中半衰期的效果最大化,使用了各种类型的肽连接器,但是可能引起免疫反应,在这其中就包括val8glu22-glp-1(7-37)-fc。
目前市场上获批的glp-1药物主要有从蜥蜴唾液中分离出的exenatide-4,以及采用脂肪酸,抗体fc段或血清白蛋白修饰的人源glp-1类似物。exenatide-4半衰期太短,仅2-4小时,一天需要至少两次注射。诺和诺德公司的脂肪酸修饰的利拉鲁肽在降血红蛋白糖基化及降体重方面最有效且副作用较少,但其不足方面是体内半衰期只有13小时,需要每天给药。为了进一步延长体内半衰期,减少给药频率,近年来进一步采用氨基酸序列突变和fc、脂肪酸或白蛋白等修饰的长效glp-1类似物已经被陆续开发。如礼来公司的杜拉鲁肽和诺和诺德公司即将上市的索玛鲁肽。这些长效化的glp-1类似物在人体内的半衰期可被进一步延长,可实现每周给药一次的给药频率。由于glp-1类似物需要长期注射给药,所以更长效期的药物的开发将具有更好的患者依从性及更大的市场竞争力。另外现有技术的药物生产成本较高,售价高昂。
现有技术中,通过fc或脂肪酸的方式修饰而开发的长效glp-1类似物,给药周期均被限定在1周或1周以内,本发明人独立地经过长期研究,令人意外地开发出了一种新的glp肽类似物的缀合物,在相同实验条件下,与现有公认的最好的技术产品代表杜拉鲁肽/索玛鲁肽相比,在普通老鼠及糖尿病老鼠模型上,其体内持续降糖活性时间可提高1倍左右,意味着在人体内可实现至少每周间隔给药、甚至每两周间隔或更长时间间隔的给药频率,并且能同时保证治疗特性,相当于、甚至优于进口昂贵的现有技术。另外,由于分子构建的特点,这种新的glp-1肽类似物生产体系更为便宜,极大的降低了类似物的生产成本,具有良好的市场开发前景。
技术实现要素:
本发明的目的在于提供一种新的glp-1肽缀合物。另外,本发明还提供了该肽缀合物的制备方法、含该肽缀合物的药物以及在制备药物中的用途和中间体等。
具体而言,在第一方面,本发明提供了如下结构式所示的肽缀合物或其药学上可接受的盐,
其中,
b为
其中m是0、1、2或3;n是1、2或3;p是1-8的任意整数;
a是含hooc(ch2)qco-的酰基,其中q是4-38的整数。
优选在本发明第一方面的肽缀合物或其药学上可接受的盐中,
所述b的结构为
a选自hooc(ch2)14co-、hooc(ch2)15co-、hooc(ch2)16co-、hooc(ch2)17co-、
hooc(ch2)18co-、hooc(ch2)19co-、hooc(ch2)20co-、hooc(ch2)21co-和
hooc(ch2)22co-,优选为hooc(ch2)16co-。
在本文中,如无矛盾或特别说明,(glp-1或glp-1(7-37)的)类似物可以与(glp-1或glp-1(7-37)的)衍生物互换使用,其与酰化的基团共同组合成肽缀合物。所述glp-1(7-37)类似物可以与氨基酸序列如seqidno:1所示的glp-1(7-37)相同,也可以具有一个氨基酸序列的不同(即一个氨基酸残基发生了取代、添加或缺失),或为具有两个氨基酸序列的不同,甚至为具有三个氨基酸序列的不同。具体地,所述glp-1(7-37)类似物是将氨基酸序列如seqidno:1所示的glp-1(7-37)中第8位氨基酸ala突变为val,第22位氨基酸gly突变为glu,第34位氨基酸lys突变为arg。
本发明人惊奇地发现,通过这种突变方式获得的酰化的glp-1类似物,与同样序列的其中就包括val8glu22-glp-1(7-37)-fc相比,体内降糖活性剂活性持续时间都要更强;而且,与同样为酰化的glp-1类产品,如利那鲁肽或索玛鲁肽(semaglutide)相比,小鼠体内也可以获得相对更长的活性持续时间,以及在相同时间点上更强的降糖活性。
可以通过这样一种方法制备本发明glp-1(7-37)类似物,该方法包括在允许肽表达的条件下,在适宜营养的培养基中,培养含有编码该多肽的dna序列并能表达该肽的宿主细胞,然后从培养物中回收产生的肽。
用来培养细胞的培养基可以是用于培养该宿主细胞的任何常规培养基,如基本培养基或含有适宜添加物的符合培养基。可以通过市售得到适宜的培养基,或根据已公开的制法制备适宜的培养基。然后可以通过常规方法从培养基中回收由该细胞产生的多肽,这些方法包括通过离心或过滤而从培养基中分离宿主细胞,用盐如硫酸铵沉淀上清液或滤液中的蛋白质成分,根据目的肽的种类而选用各种层析方法如例子交换层析、凝胶过滤层析、亲和层析等进行纯化。
编码母体肽的dna序列可以来源于基因组或cdna,如通过制备基因组或cdna文库,并根据标准技术(如,见sambrook,j,fritsch,ef和maniatis,t,分子克隆:实验操作指南,coldspringharborlaboratorypress,纽约,1989)使用合成的寡核苷酸探针,进行杂交而筛选出编码该肽的全部或部分dna序列。也可以通过已建立的标准方法,如beaucage和caruthers所述的磷酰胺(phosphoamidite)方法,或matthes等所述的方法(欧洲分子生物学组织杂志,1984)合成该编码肽的dna序列。也可以使用特定的引物,通过聚合酶链反应制备dna序列。
可以将dna序列插入任何便于进行dna充足过程的载体中,病且载体的选择常常取决于该载体将要被引入的宿主细胞,因此,载体可以是一种自主复制型载体,即作为一个染色体外实体存在的载体,其复制不依赖于染色体复制,如质粒。或者,载体可以是这样一种类型,当将其引入宿主细胞时,它将整合到宿主细胞基因组中,并与它所整合入的染色体一起复制。
载体优选是一种表达载体,其内编码所述肽的dna序列与该dna转录所需的其它区段(如启动子)有效相连。驱动自可以是在选用的宿主细胞中有转录活性的任何dna序列,它可以来源于编码宿主细胞同源性或异源性蛋白质的基因。本领域熟知适合于在多种宿主细胞中知道编码本发明肽的dna进行转录的启动子例子,参见如sambrook等(出处同前)。
载体还可以含有一个选择标记,如一个基因,其基因产物将弥补宿主细胞内的一个缺陷,或者能赋予对药物如氨苄青霉素、阿霉素、四环素、氯霉素、新霉素、链霉素或氨甲喋呤等的抗性。
为将本发明的母体肽引入宿主细胞的分泌途径,可以在重组载体中提供一个分泌信号序列(也称之为前导序列)。分泌信号序列以正确读框与编码该肽的dna序列连接。分泌信号序列通常位于编码该肽的dna序列的5’侧。分泌信号序列可以是正常地与该肽连接的分泌信号序列,或可以源于编码另一种分泌蛋白质的基因。
用于分别连接编码本发明肽的dna序列,启动子和可选择的终止子和/或分泌信号肽序列,并将其插入到适宜的含有复制所必需的信息的载体中的方法,对本领域的技术人员是已知的。
将导入dna序列或重组载体的宿主细胞可以是能够产生本发明肽的任何细胞,包括细菌、酵母、真菌和高等真核生物细胞。本领域技术人员熟知并使用的适宜的宿主细胞的例子包括但不限于:大肠杆菌、酿酒酵母、或哺乳动物bhk或cho细胞系。
在第二方面,本发明提供了制备本发明第一方面的肽缀合物或其药学上可接受的盐的方法,其包括:
(1)提供val8-glu22-arg34-glp-1类似物溶液,调整ph;
(2)向步骤(1)获得的溶液中加入三乙胺;
(3)将如下结构的脂肪酸溶于乙腈中;
其中m=1~3,n=1-3,优选为m=1,n=1;
(4)将步骤(2)获得的glp-1类似物溶液与步骤(3)获得的脂肪酸溶液混合,静置;
(5)调节ph终止反应,酸沉,离心,得沉淀;
(6)向步骤(5)获得的沉淀中加水溶解,并加入氢氧化钠,震荡使沉淀溶解,脱保护,调节ph终止反应;
(7)分离纯化。
优选本发明第二方面的方法包括:
(1)提供浓度为4~6mg/ml的val8-glu22-arg34-glp-1类似物溶液,调整ph至9-12;
(2)向步骤(1)获得的溶液中加入0.1-0.5%(v/v)的三乙胺;
(3)称取不低于该glp-1类似物2倍量(摩尔比)的如下结构的脂肪酸,优选为不低于3倍量的glp-1类似物,溶于乙腈中;
其中m=1~3,n=1-3,优选为m=1,n=1;
(4)将步骤(2)获得的glp-1类似物溶液与步骤(3)获得的脂肪酸溶液混合,于4℃静置一小时;
(5)加水稀释,调节ph至4.8终止反应,放于4℃静置酸沉,酸沉后4℃离心,得沉淀;
(6)向步骤(5)获得的沉淀中加水溶解,并加入1m氢氧化钠至终浓度100mmnaoh,震荡使沉淀溶解,置于室温静置脱保护,反应液调节ph至8.0-8.5终止反应;
(7)分离纯化。
在第三方面,本发明提供了一种药物组合物,其包括本发明第一方面的肽缀合物或其药学上可接受的盐,以及药学上可接受的辅料。
在本文中,如无矛盾或特别说明,药物、药物组合物和药物制剂(药剂)可以互换使用。在本文中,药学上可接受的辅料指无毒的填充剂、稳定剂、稀释剂、载体、溶剂或其他制剂辅料。例如,稀释剂、赋形剂,如微晶纤维素、甘露醇等;填充剂,如淀粉、蔗糖等;粘合剂,如淀粉、纤维素衍生物、藻酸盐、明胶和/或聚乙烯吡咯烷酮;崩解剂,如碳酸钙和/或碳酸氢钠;吸收促进剂,如季铵化合物;表面活性剂,如十六烷醇;载体、溶剂,如水、生理盐水、高岭土、皂粘土等;润滑剂,如滑石粉、硬脂酸钙/镁、聚乙二醇等。另外,本发明的药物组合物优选为注射剂。
优选在本发明第三方面的药物组合物中,本发明第一方面的肽缀合物或其药学上可接受的盐以0.1mg/ml至25mg/ml的浓度存在,优选以0.1mg/ml至10.0mg/ml的浓度存在。
也优选本发明其中所述的本发明第三方面的药物组合物具有3.0至9.0的ph。其中,可进一步包含缓冲系统、防腐剂、表面张力剂、螯合剂、稳定剂和表面活性剂。在本发明的一个实施方案中,本发明第三方面的药物组合物是含水制剂。这种制剂通常是溶液或悬浮。本发明的具体实施方案中,该药物组合物是稳定的含水溶液。在本发明的另一个具体实施方案中中,该药物组合物是一种冻干制剂,在使用前医师或患者加入溶剂和/或稀释液至其中。
本发明第三方面的药物组合物也可以包括另一种或多种药理活性物质,与本发明第一方面的肽缀合物或其药学上可接受的盐组合使用。所述药理活性物质可以选自抗糖尿病药物、减肥药、食欲调节剂、抗高血压药、用于治疗和/或预防由糖尿病引发或与其相关的并发症的制剂以及用于治疗和/或预防由肥胖症引发或与其相关的并发症和障碍的药物化合物或组合物。这些药理活性物质的实例是:胰岛素、磺脲类、双胍、氯茴苯酸类、葡萄糖苷酶抑制剂、胰高血糖素拮抗剂、涉及刺激糖异生和/或糖原分解的肝脏酶的抑制剂、葡萄糖摄取调节剂、调节脂类代谢的化合物(如抗高血脂剂,如hgm、coa抑制剂)、胃抑制性多肽、降低食物摄取的化合物、rxr激动剂和作用于β细胞的atp依耐性钾通道制剂、β阻滞剂如吲哚心安、萘心安和甲养乙心安、ace抑制剂如贝那普利和卡托普利;cart激动剂、npy拮抗剂、pyy激动剂、pyy2激动剂、pyy4激动剂、tnf激动剂、促皮质素释放因子激动剂、5ht、蛙皮素激动剂、神经节肽拮抗剂、生长激素、促甲状腺激素释放激素激动剂、瘦素激动剂、trβ激动剂;组胺h3拮抗剂、脂肪酶/淀粉酶抑制剂、胃抑制性多肽激动剂或拮抗剂、胃泌素和胃泌素类似物等。
在第四方面,本发明提供了本发明第一方面的肽缀合物或其药学上可接受的盐在制备具有治疗或预防糖尿病、肥胖症、高血糖、血脂代谢障碍和/或非酒精性脂肪肝的药物中的应用。
优选在本发明第四方面的应用中,药物是本发明第三方面的药物组合物。
优选在本发明第四方面的应用中,糖尿病为2型糖尿病。
在第五方面,本发明提供了本发明第一方面的肽缀合物或其药学上可接受的盐在制备减少食物摄取、减少胰岛β-细胞凋亡、增加胰岛β-细胞功能和胰岛β-细胞数量和/或恢复胰岛β-细胞的葡萄糖敏感性的药物中的应用。
优选在本发明第五方面的应用中,药物是本发明第三方面的药物组合物。
另外,本发明还提供了本发明第一方面的肽缀合物的中间体及其应用等。
具体而言,在第六方面,本发明提供了如下结构式所示的化合物,
在第七方面,本发明提供了本发明第六方面的化合物在制备本发明第一方面的肽缀合物或其药学上可接受的盐中的应用。
在第八方面,本发明提供了glp-1(7-37)类似物,其是val8-glu22-arg34-glp-1(7-37),即将氨基酸序列如seqidno:1所示的glp-1(7-37)中第8位氨基酸ala突变为val,第22位氨基酸gly突变为glu,第34位氨基酸lys突变为arg。
在第九方面,本发明提供了本发明第八方面的val8-glu22-arg34-glp-1(7-37)在制备本发明第一方面的肽缀合物或其药学上可接受的盐中的应用。
在第十方面,本发明提供了val8-glu22-arg34-glp-1(7-37)-fc,即本发明第八方面的val8-glu22-arg34-glp-1(7-37)和fc的融合蛋白。
本发明的有益效果在于:能每周间隔、甚至每两周间隔地给药,并且能同时保证临床治疗特性,相当于、甚至优于进口、昂贵的现有技术。
本发明通过下面实施例来进一步阐述,然而,所述的实施例不应理解为限制本专利的保护范围,在前面描述和下列实施例中公开的特征(个别地和它们的任何组合),可以是用于以基本不同形式实现本发明的材料。另外,本发明引用了公开文献,这些文献是为了更清楚地描述本发明,它们的全文内容均纳入本文进行参考,就好像它们的全文已经在本文中重复叙述过一样。
说明书附图
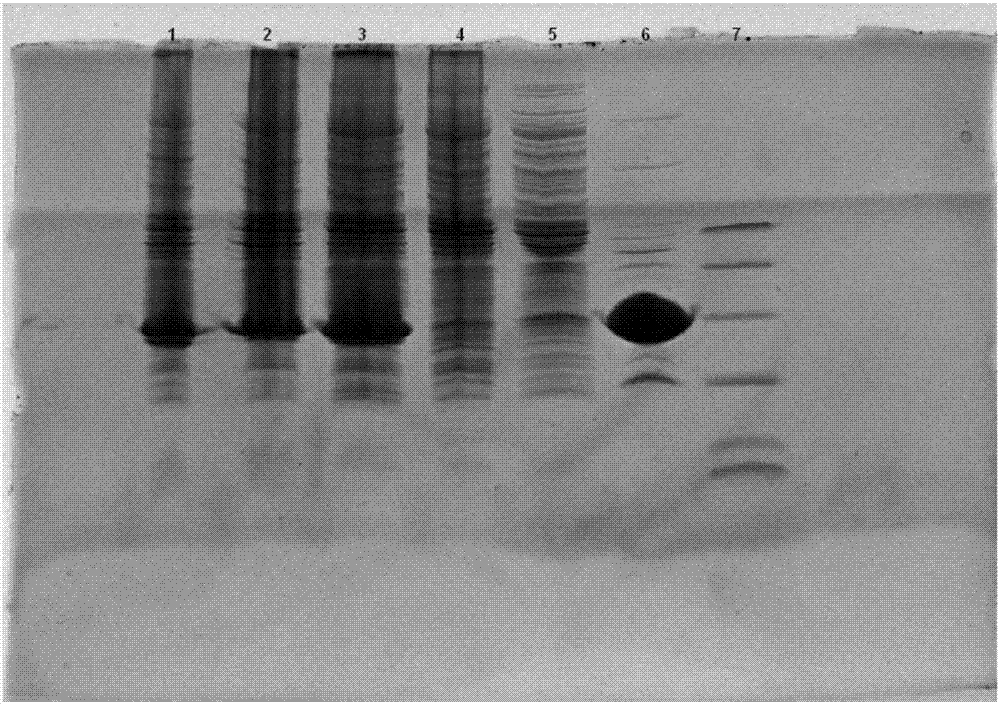
图1为glp-1/glp-1-fc发酵液电泳图,泳道1为marker,泳道2为glp-1-fc,泳道3为val8glu22arg34-glp-1。
图2为val8glu22arg34-glp-1纯化过程中的电泳图谱,泳道1为溶菌后全菌电泳,泳道2为破菌后全菌电泳,泳道3为离心后上清电泳,泳道4为ni柱层析上样穿透电泳,泳道5为ni柱层析淋洗穿透电泳,泳道6为ni柱层析洗脱电泳,泳道7位marker。
图3为val8glu22arg34-glp-1精纯电泳图谱,泳道1为酶切后的电泳,泳道2为butyl柱层析上样穿透电泳;泳道3为butyl柱层析洗脱电泳,泳道4为marker,泳道5为val8glu22arg34-glp-1酸沉后电泳。
图4为glp-1-fc纯化过程中电泳图谱,泳道1为glp-fc离心后上清;泳道2为glp-fc离心后沉淀;泳道3为glp-fcni柱层析酶切后目的蛋白;泳道4为glp-fcni柱层析上样穿透;泳道5为glp-fcni柱层析洗脱;泳道6为marker;泳道7为glp-fcq柱层析穿透;泳道8为glp-fcq柱层析洗脱1;泳道9为glp-fcq柱层析洗脱2;泳道10为glp-fcq柱层析碱洗
图5为正常小鼠给予不同治疗药物后的降糖效果图。
图6为糖尿病小鼠给予不同药物后的降糖效果图。
具体实施例
以下本文将通过具体的实施例来描述发明。如未特别指明之处,可根据本领域技术人员所熟悉的《分子克隆实验指南》、《细胞实验指南》等实验手册以及cfda的试验指引等所列方法来实施。其中,所用的试剂原料均为市售品,可以通过公开渠道购买获得。
实施例1val8glu22arg34-glp-1(7-37)及val8glu22-glp-1(7-37)-fc表达质粒的构建
实施例1a构建val8-glu22-arg34-glp-1的dna
将6-his标签,sumo标签和val8-glu22-arg34-glp-1编码基因序列(seqidno:3)依次串联融合,并使用化学合成的方式获得基因片段。通过bamhi和xhoi位点,将上述片段插入原核表达质粒pet-24(+)中并测序验证。得到的用于转化测定的表达质粒,称作pet-24(+)-his-sumo-val8-glu22-arg34-glp-1。
实施例1b构建val8-glu22-glp-1-fc的dna:
将6-his标签,sumo标签,val8-glu22-glp-1编码基因(seqidno:4),linker(ggggsggggsggggs)编码基因、和人igg4-fc片段编码基因(包括铰链区,ch2和ch3结构域)依次串联融合,并使用化学合成的方式获得基因片段。通过bamhi和xhoi位点,将上述片段插入原核表达质粒pet-24(+)中并测序验证。得到的用于转化测定的表达质粒,称作pet-24(+)-his-sumo-val8-glu22-glp-1-fc。
实施例2融合蛋白表达
使用实施例1中所述dna构建进行融合蛋白的表达,通过表达细胞bl21(thermofisherscientificinc.,catalog#c600003)获得目的蛋白。将bl21感受态细胞50μl置于冰浴上融化,加入目的dna,轻轻摇匀,并在冰浴中放置30分钟。继而42℃水浴热激30秒,然后快速将离心管转移到冰浴中放置2分钟,该过程不要摇动离心管。向离心管中加入250μl无菌的lb培养基(不含抗生素),混匀后置于37℃,225rpm培养1小时,使细菌复苏。吸取100μl已转化的感受态细胞加到含有卡那霉素抗性的lb琼脂培养基平板上,将细胞均匀涂开。将平板置于37℃至液体被吸收,倒置平板,37℃过夜培养。次日,使用接种环挑取转化平皿中的单克隆菌落,并接种于3ml的无菌lb培养基(含抗生素),37℃过夜培养。
实施例3重组glp-1/glp-1-fc的发酵:
向50ml的lb培养基中加入100μl菌液(表达glp-1菌液,或表达glp-1-fc菌液),同时加入50μl卡那霉素,混匀后放30℃恒温振荡器中,接种过夜。取过夜接种的菌液7ml加入800ml的lb培养基中,同时加入800μl卡那霉素。摇匀后放于30℃摇床内,180rpm,接种4h后向培养基中接入终浓度为0.1mol/l的iptg,摇匀后放于30℃摇床内,180rpm,过夜诱导表达。将过夜表达的菌液以13000g离心60min。菌体收率约为4g菌/l发酵液,sds-page测定目的蛋白表达量约为40%。
实施例4重组glp-1/glp-1-fc的纯化
称取100g细胞浆重悬于500ml的50mmtris-hcl、ph8.0,50mmnacl中,用超声波细胞粉碎机中超声30min,以使细胞破碎。所述匀浆在4℃下以13000g离心60min,离心完成后收集上清即为ni柱层析样品。
将所得上清液经事先用50mmtris-hcl、ph8.0,50mmnacl,10mm咪唑(平衡液1)平衡过的chelatingsepharoseff浓缩。平衡液淋洗后,再用50mmtris-hcl、ph8.0,0.5mnacl,0.3m咪唑(洗脱液)洗脱。经sds-page分析,由上述纯化过程生成glp-1中间产物纯度高于70%。
使用ulp酶将sumo标签序列切除:中间产物中加入20mmpb、ph7.4缓冲液使其稀释三倍,4℃条件下按照ulp酶:中间产物为1:150加入ulp混匀后酶切过夜。经sds-page分析酶切率近100%。
glp-1精纯:将酶切后所得产物经事先用20mmna2hpo4,0.7mnacl(平衡液2)平衡过的东曹butyl550c浓缩。平衡液淋洗后,再用20%乙醇洗脱,经sds-page纯度约为90%。
洗脱样品中加入0.2mna2hpo4使其终浓度为20mmna2hpo4,用1m柠檬酸调节ph至4.8-5.0,4℃酸沉过夜。sds-page检测收率90%以上。4℃下以13000g离心60min,收取沉淀放于-20℃保存。
glp-1-fc精纯:将酶切后所得产物经事先用20mmtris-hcl、ph8.0,0.05mnacl(平衡液4)平衡过的geq-xl色谱柱。平衡液淋洗后,再用20mmtris-hcl、ph8.0,1mnacl梯度洗脱,收集洗脱液经sds-page纯度约为90%。
收集的洗脱液用淋洗液稀释至低于1mg/ml,加入等体积的2m盐酸胍(盐酸胍调节ph至9.0左右,缓慢滴入搅拌的蛋白中,以免沉淀),再加入1m硫酸铜(用氢氧化钠调节ph至9.0左右)至其终浓度为10mm硫酸铜,混匀后放于室温静置6h。
将蛋白注入透析卡内,透析于100倍体积的4℃预冷的复性液50mmtris-hcl、ph9.0,3%甘油,0.3m精氨酸中,外液搅拌过夜。过夜后将蛋白抽出。
将复性后所得产物再次过先用10mmpb、ph8.0(平衡液4)平衡过的geq-xl色谱柱。平衡液淋洗后,再用10mmpb、ph8.0,0.5mnacl洗脱。
实施例5n-ε26-[2-(2-[2-(2-[4-(17-羧基十七烷酰氨基)-4(s)-羧基丁酰基氨基]乙氧基)乙氧基]乙酰氨基)乙氧基]乙氧基]乙酰基](val8glu22arg34-glp-1(7-37))肽的制备(val8glu22arg34-glp-1(7-37)-脂肪酸)
脂肪酸修饰:收集的val8glu22arg34-glp-1(7-37)沉淀中加水配为4~6mg/ml溶解液,加入1m氢氧化钠调整ph至11.0-11.5,摇匀使蛋白完全溶解,hplc定量多肽浓度。按多肽与脂肪酸(结构如下)摩尔比1:3称取脂肪酸粉末溶于乙腈中。向多肽溶液中加入体积为千分之二的三乙胺,并与脂肪酸溶液混合,将混合液于4℃静置一小时。
样品加水稀释5倍,用1m柠檬酸(或10%乙酸)调ph至4.8终止反应,放于4度静置酸沉10min,酸沉后离心13000g,4℃离心30min,将沉淀放于-80℃保存
脂肪酸脱保护与纯化:向酸沉样品中加水溶解(终体积与修饰体积相同),加入1m氢氧化钠至终浓度100mmnaoh,震荡使沉淀溶解,置于室温静置脱保护30min,向反应液中滴入10%乙酸(或1m柠檬酸)调节ph至8.0-8.5终止反应。
用制备液相仪(岛津lc-8a)将终止后反应液按4ml/min流速,泵入事先用10mm乙酸铵,20%乙醇(平衡液3)平衡过的unisil10-120c18(购自纳微)进行浓缩。平衡液3淋洗后,再按0-100%洗脱液(10mm乙酸铵,80%乙醇)梯度洗脱,收集洗脱峰经rp-hplc检测纯度约为90%。
洗脱峰用水稀释5倍,酸沉调整ph至4.80,4℃酸沉30min。离心后沉淀中加入dpbs缓冲液(ph7.0)复溶后-80℃冻存。
实施例6使用正常小鼠的药效研究
选用28只周龄4~6周的健康cd-1雌性小鼠,分为四组,分别皮下注射v+e脂肪酸(通过实施例5制备的val8glu22arg34-glp-1(7-37)-脂肪酸)、v+e-fc(通过实施例4制备的glp-1-fc)、杜拉鲁肽和索马鲁肽剂量为0.3mg/kg体重。按照4h、1天、2天、3天、4天、6天的时间间隔灌胃20%葡萄糖剂量为2g/kg体重,给糖前禁食过夜,并在给糖后0、0.5、1、2h分别尾尖取血并使用罗氏血糖试纸实时检测血糖值,并计算出0~120分钟内的血糖auc(血糖~时间曲线下面积),算出血糖抑制率(表1、图5)。
表1:不同glp-1类似物肽对正常小鼠体内降糖效果
p值:与给药前血糖比较
从表中可以看出,对于正常小鼠的血糖影响,v+e-脂肪酸偶联物在各个时间点的活性均要强于v+e-fc,。索玛鲁肽和杜拉鲁肽在正常小鼠体内的降糖活性可以持续大约3d,第4天后基本不再具有降糖活性,而v+e脂肪酸在正常小鼠体内的降糖活性在第6天仍然表现出一定的活性,体内维持持续降糖活性的时间要明显较索玛鲁肽或杜拉鲁肽延长,并且在给药第2d后的各个时间点上,v+e-脂肪酸的降糖效果也都要明显强于索玛鲁肽或杜拉鲁肽。
实施例7使用db/db小鼠的药效研究
糖尿病小鼠ogtt试验:选用15只周龄4~6周的db/db转基因糖尿病小鼠,分为三组,分别皮下注射溶酶(pbst溶液,10ml/kg)、v+e脂肪酸和杜拉鲁肽剂量为0.3mg/kg体重。按照4h、1天、2天、3天、4天、5天的时间间隔灌胃20%葡萄糖剂量为1g/kg体重,给糖前禁食过夜,并在给糖后0、0.5、1、2h分别尾尖取血并使用罗氏血糖试纸实时检测血糖值。测定结束后给食8h以上,尾尖取血并使用罗氏血糖试纸实时检测血糖值,并计算出0~120分钟内的血糖auc(血糖~时间曲线下面积),算出血糖抑制率(表2、图6)。
表1:不同glp-1类似物肽对糖尿病小鼠体内降糖效果
p值:与阴性对照小鼠比较
从上表结果可以看出,对于糖尿病模型小鼠,无论是禁食血糖还是非禁食血糖,给药后各个时间点上,v+e-脂肪酸的降糖活性均要强于杜拉鲁肽,对糖尿病小鼠的降糖活性明显更强;杜拉鲁肽在第二天后,对糖尿病小鼠就活性明显下降,虽表现出一定降糖活性,但与正常组间差异已不显著,到第三天几乎没有降糖活性,而v+e-脂肪酸给药后第二天活性依然很强,第三天也有一定的降糖活性,v+e-脂肪酸在糖尿病小鼠体内维持持续降糖活性的时间更长。
sequencelisting
<110>北京凯因科技股份有限公司
<120>酰化的glp-1衍生物
<130>参考文献在背景技术中
<160>7
<170>patentinversion3.5
<210>1
<211>31
<212>prt
<213>人工序列
<220>
<223>glp-1(7-37)
<400>1
hisalagluglythrphethrseraspvalsersertyrleuglugly
151015
glnalaalalysglupheilealatrpleuvallysglyarggly
202530
<210>2
<211>31
<212>prt
<213>人工序列
<220>
<223>val8glu22arg34-glp-1类似物
<400>2
hisvalgluglythrphethrseraspvalsersertyrleugluglu
151015
glnalaalalysglupheilealatrpleuvalargglyarggly
202530
<210>3
<211>93
<212>dna
<213>人工序列
<220>
<223>val8glu22arg34-glp-1类似物编码基因
<400>3
cacgttgaaggtaccttcacctctgacgtttcttcttacctggaagaacaggctgctaaa60
gaattcatcgcttggctggttcgtggtcgtggt93
<210>4
<211>31
<212>prt
<213>人工序列
<220>
<223>val8glu22-glp-1类似物
<400>4
hisvalgluglythrphethrseraspvalsersertyrleugluglu
151015
glnalaalalysglupheilealatrpleuvallysglyarggly
202530
<210>5
<211>93
<212>dna
<213>人工序列
<220>
<223>val8glu22-glp-1类似物编码基因
<400>5
cacgttgaaggtaccttcacctctgacgtttcttcttacctggaagaacaggctgctaaa60
gaattcatcgcttggctggttaaaggtcgtggt93
<210>6
<211>421
<212>dna
<213>人工序列
<220>
<223>带sumo标签的val8glu22arg34-glp-1类似物编码基因
<400>6
attttgtttaactttaataaggagatataccatgcatcaccatcatcaccacgctaaacc60
ggaagttaaaccggaagttaaaccggaaacccacatcaacctgaaagtttctgacggttc120
ttctgaaatcttcttcaaaatcaaaaaaaccaccccgctgcgtcgtctgatggaagcttt180
cgctaaacgtcagggtaaagaaatggactctctgcgtttcctgtacgacggtatccgtat240
ccaggctgaccagaccccggaagacctggacatggaagacaacgacatcatcgaagctca300
ccgtgaacagatcggtggtcacgttgaaggtaccttcacctctgacgtttcttcttacct360
ggaagaacaggctgctaaagaattcatcgcttggctggttcgtggtcgtggttaataata420
a421
<210>7
<211>1153
<212>dna
<213>人工序列
<220>
<223>带sumo标签的val8glu22-glp-1-fc编码基因
<400>7
attttgtttaactttaataaggagatataccatgcatcaccatcatcaccacgctaaacc60
ggaagttaaaccggaagttaaaccggaaacccacatcaacctgaaagtttctgacggttc120
ttctgaaatcttcttcaaaatcaaaaaaaccaccccgctgcgtcgtctgatggaagcttt180
cgctaaacgtcagggtaaagaaatggactctctgcgtttcctgtacgacggtatccgtat240
ccaggctgaccagaccccggaagacctggacatggaagacaacgacatcatcgaagctca300
ccgtgaacagatcggtggtcacgttgaaggtaccttcacctctgacgtttcttcttacct360
ggaagaacaggctgctaaagaattcatcgcttggctggttaaaggtcgtggtggtggtgg420
tggttctggtggtggtggttctggtggtggtggttctgctgaatctaaatacggtccgcc480
gtgcccgccgtgcccggctccggaagctgctggtggtccgtctgttttcctgttcccgcc540
gaaaccgaaagacaccctgatgatctctcgtaccccggaagttacctgcgttgttgttga600
cgtttctcaggaagacccggaagttcagttcaactggtacgttgacggtgttgaagttca660
caacgctaaaaccaaaccgcgtgaagaacagttcaactctacctaccgtgttgtttctgt720
tctgaccgttctgcaccaggactggctgaacggtaaagaatacaaatgcaaagtttctaa780
caaaggtctgccgtcttctatcgaaaaaaccatctctaaagctaaaggtcagccgcgtga840
accgcaggtttacaccctgccgccgtctcaggaagaaatgaccaaaaaccaggtttctct900
gacctgcctggttaaaggtttctacccgtctgacatcgctgttgaatgggaatctaacgg960
tcagccggaaaacaactacaaaaccaccccgccggttctggactctgacggttctttctt1020
cctgtactctcgtctgaccgttgacaaatctcgttggcaggaaggtaacgttttctcttg1080
ctctgttatgcacgaagctctgcacaaccactacacccagaaatctctgtctctgtctct1140
gggttaataataa1153
- 还没有人留言评论。精彩留言会获得点赞!
- 一类苯內酰烯酯酰胺类衍生物在制备抗骨质疏松药物中的应用的制作方法与工艺
- 一种丹参酮类化合物17位酯化衍生物及其制备工艺和应用的制作方法与工艺
- 用于治疗癌症的异喹啉酮衍生物的制作方法与工艺
- 二氢碟啶酮衍生物及其用途的制作方法与工艺
- 一种酞嗪酮衍生物、该衍生物的制备方法及其制备用催化剂与流程
- 环烃基取代的甲磺酰基苯甲酰胺衍生物、其制法与医药上的用途的制作方法与工艺
- 一种以喹唑啉酮衍生物为核心的化合物及其在OLED上的应用的制作方法与工艺
- 一种母核为喹唑啉酮衍生物的化合物及其在OLED上的应用的制作方法与工艺
- 一种母核为喹唑啉酮衍生物的化合物及其应用的制作方法与工艺
- 蝶啶酮衍生物作为EGFR抑制剂的应用的制作方法与工艺