一种提高极低拷贝数目标DNA检测灵敏度的方法与流程
本发明属于核酸检测技术领域,具体涉及一种提高现有核酸检测技术对极低拷贝数目标DNA检测灵敏度的方法。
背景技术:
基于体外DNA扩增技术的核酸检测方法具有高灵敏度与高特异性的优点,比较具有代表性的是PCR(变温反应)以及环介导等温扩增(LAMP),在生物学、医学、环境科学、法医学、食品科学等领域发挥着极其重要的作用。在食品安全检测领域,传统的微生物培养鉴定法需要数天检测周期,而核酸检测方法在几小时内便可完成,且灵敏度与特异性均高于传统方法;在医学领域核酸检测方法被广泛应用于多种疾病的快速准确检测以及药物研发等多种项目中;在法医学中DNA检测是侦办各类刑事案件的重要手段。
但是核酸检测法高灵敏度结果的获得往往需要在使用较为理想的样品的条件下获得,相对于一些目标DNA数量极低(或极微量目标DNA混杂于大量背景核酸中)的样品,核酸检测方法的灵敏度与特异性可能降低从而导致假阴性或假阳性的结果,如医学检测中“分析”灵敏度与“临床”灵敏度不一致的情况。为此,针对这一类样品进行纯化以消除大量背景核酸的影响成为提高这类样品检测效率的方法之一,如美国专利US 13/933,919(Richmond et al,2013)公开了一种利用阴离子交换技术从大量背景核酸中选择性富集特定片段长度DNA的方法,但该方法适用于较大量样品的纯化且需要制备特定的阴离子交换微珠,极低数量的目标DNA容易在吸附和洗脱过程中丢失;Zhou等报道了一种特异性去除人基因组DNA从而提高血液样品中沙门氏菌检测灵敏度的方法,其先使用牛胆汁和微球菌核酸酶将人基因组DNA降解后再单独提取沙门氏菌DNA进行检测,但此方法只适合于全血样品的处理且操作过程相对较为繁琐(Zhou L,Pollard A J.A novel method of selective removal of human DNA improves PCR sensitivity for detection of Salmonella Typhi in blood samples[J].BMC infectious diseases,2012,12(1):164.)。针对不含背景核酸的纯DNA样品,虽然PCR法可以达到理论上1拷贝的检测灵敏度,但由于需要极其严格的实验环境以及最优的实验条件(引物、试剂、参数等),因此实际操作中因很难达到这一目标,很多文献报道中PCR的检测阈值约为100拷贝甚至更多(王怡倩,叶长芸.建立检测产气肠杆菌的TaqMan荧光定量PCR和普通PCR方法[J].疾病监测,2016,31(2):153-158;Sritunyalucksana K,Srisala J,McColl K,et al.Comparison of PCR testing methods for white spot syndrome virus(WSSV)infections in penaeid shrimp[J].Aquaculture,2006,255(1):95-104.),增加扩增循环数虽可一定程度上提高检测灵敏度但同时增加了假阳性风险。
显然,如果研发一种成本低廉、操作简便、对实验设备等要求低的方法可以提高极低丰度(低于百万分之一)DNA的检测灵敏度,同时保证检测的高特异性,将有效提高核酸检测方法在各种实际复杂情况下检测的效率,应用前景广阔。
技术实现要素:
针对上述问题,本发明旨在提供一种高效、简便的可以提高现有核酸检测灵敏度的方法,使极低数量的目标DNA可被高灵敏度、高特异性检出。
一种提高极低拷贝数目标DNA检测灵敏度的方法,主要步骤如下:向待检测样品DNA中加入外源背景基因组DNA;使用特定的可以对目标DNA切割产生随机碱基粘性末端的限制性内切酶对混合后的样品DNA进行酶切;根据目标DNA切割后所产生的粘性末端设计与之互补的接头并与之进行连接;使用接头中含有的通用引物对连接产物进行PCR反应;以PCR产物为模板,使用针对目标DNA设计的特异性引物进行特异性核酸扩增检测。
更具体的步骤为:
a)向待检测样品DNA中加入终浓度为10-500ng/μL的外源背景基因组DNA;
b)使用可以对目标DNA切割产生随机碱基粘性末端的限制性内切酶对步骤a)得到的样品进行酶切,酶切反应参数由所选内切酶确定;
c)向酶切产物中加入针对目的基因酶切片段设计的双链DNA接头,并在DNA连接酶的作用下进行连接反应,使酶切片段的两端均连接上接头,所述接头上均带有通用引物序列,接头最终浓度为50-400nM,连接反应参数根据所选连接酶确定;
d)以连接产物为模板,使用通用引物进行PCR反应,反应参数根据最终目标片段长度确定;
e)以上一步PCR反应产物为模板,使用目标DNA的特异性引物进行PCR或其他核酸扩增方法实现对目标基因的最终检测,反应条件及参数根据所选特异性检测方法确定。
所述极低拷贝数一般指拷贝数低于50,当高于该拷贝数时,常规PCR在优化的实验条件下可以检出目标DNA,低于该拷贝数时则很难检出,且易产生假阳性。
其中,步骤a)所述外源背景基因组DNA包括所有真核生物的基因组DNA,优选鲑鱼精DNA等工业化生产较为成熟、可大量制备、纯度高且价格较为低廉的基因组DNA。
步骤b)中所述限制性内切酶是一类可以对目标DNA切割产生随机碱基粘性末端的内切酶,即酶切所产生的片段的粘性末端由随机碱基组成。选择识别位点为4-6碱基、产生的粘性末端长度为4-9碱基内切酶进行酶切反应,且所选内切酶应至少在目标基因中含有两个酶切位点或使用两种在目标基因中各含有一个酶切位点的内切酶,以便产生两端均具有随机碱基粘性末端的酶切片段,所述目标基因上两个酶切位点的距离为70-2000bp。所述内切酶包括但不限于Alw26I,BbvI,FokI,HgaI,TspRI等。
目标DNA(序列已知)和背景基因组DNA经过内切酶充分酶切后,产生了大量具有随机碱基粘性末端的片段,其中目标基因酶切片段的粘性末端的序列及内部序列作为接头及特异性引物设计的根据;背景基因组DNA被同一种酶酶切后的片段数量中具有与目标基因酶切片段完全相同粘性末端的片段比例大约为1/42n(n为一个粘性末端中随机序列的碱基数),与目标基因酶切片段两个粘性末端中任意一端碱基相同的比例为1/4n。由于所加入的背景基因组DNA经酶切后产生了数以亿计的片段数量,因此其中含有与目标酶切片段相同的粘性末端的片断数高达数千以上。
步骤c)中所述“针对目的基因酶切片段设计的双链DNA接头”由两条长度不同的单链DNA组成,其中长链部分5’端含有一段通用引物序列,短链部分5’端含有可与目标基因酶切片段粘性末端完全互补配对的“衔接”序列。长链与短链之间则含有≥8碱基的互补配对部分,因此可形成稳定的部分双链结构。所有接头长链均相同,而短链则具有不同的衔接序列(根据目标酶切片段粘性末端的不同而异)。
所述DNA连接酶主要包括T3、T4、T7DNA连接酶以及E.coli DNA连接酶和Taq DNA连接酶,优选连接效率高、反应条件温和的T3、T4、T7DNA连接酶进行连接反应。连接时间0.5-2.5h,过长的连接时间将导致接头与酶切片段间错配的几率增大,而过短的连接时间可能导致目标片段未能完全与接头进行连接。
在加入针对目的基因酶切片段设计的双链DNA接头以及DNA连接酶后,接头与目的基因片段发生连接反应,使目标酶切片段的两端均含有一个接头结构;同时,背景基因组DNA中含有与目标酶切片段相同粘性末端的酶切片段也同时与接头连接,即:经过连接反应后,酶切片段中与接头连接的片段包括目标基因以及一部分背景基因组DNA片段,在接下来的PCR反应过程中,背景基因组DNA中的这部分片段会同目标DNA同时被通用引物扩增,提高了PCR反应的起始模板数量,达到数十万拷贝之上,满足了PCR对扩增起始模板数的最低要求,因此可使反应的效率及灵敏度提高。
步骤d)中所述通用引物指的是一段自行设计的寡核苷酸序列,同时也是所有双链DNA接头的长链部分,长度为18-26个碱基,熔点为60℃±5℃,可根据与目标DNA间是否形成不利于扩增的二级结构而更改。通用引物在PCR体系中的浓度为0.1-1μM。本发明实施例中所用的通用引物的序列为:TCGTCACTGCACGGTGACTCAGCA。
步骤e)中所述特异性引物为针对目标酶切片段所设计,扩增产物为目标片段内部的一段DNA序列,特异性检测方法包括但不限于PCR、LAMP、RCA等常用核酸检测手段,最终使用凝胶电泳或测序等手段对反应产物进行检测鉴定。
相比于现有技术的不足,本发明具有以下有益效果:
(1)可提高现有核酸检测方法的检测灵敏度。通常情况下核酸检测方法对起始模板数量有一定要求(拷贝数高于50),低于该阈值则无法有效扩增,本发明通过加入与目标基因不相关的外源背景DNA并通过酶切连接等步骤使一部分背景DNA充当了起始扩增模板,使最终的扩增起始模板数超过了扩增阈值,因此可以有效启动扩增反应,最终结合其他特异性检测方法可实现对低于阈值目标基因的高灵敏度、高特异性检测。
(2)相对于常规核酸检测方法,提高了检测的特异性。由于酶切及接头连接过程均对特异性有一定要求,起到双重控制的作用,因此最终检测的特异性得到了一定的提高。
(3)方法操作简便,不需要特殊、较为昂贵的仪器及试剂。本发明所需仪器主要为常规热循环仪,所需试剂为分子生物学中常见的工具酶,最终检测手段为常规电泳或测序,因此对实验条件要求较低,可满足大多数实验室的要求。
(4)方法适用性强,最终特异性检测除可使用PCR方法外,还可选择LAMP以及RCA等恒温扩增方法。
附图说明
图1.本发明实施例1技术流程图;
图2.实施例1对八宝粥中黄曲霉菌的检测结果(A)和常规PCR法的检测结果(B)。M:DNA ladder;N:阴性对照。泳道1-5分别表示不同稀释度的黄曲霉菌,泳道1:稀释度为104,泳道2:稀释度为105,泳道3:稀释度为106,泳道4:稀释度为107,泳道5:稀释度为108;
图3.实施例2对饮用水中霍乱弧菌的检测结果(A)和常规LAMP法的检测结果(B)。泳道1-5分别表示不同稀释度的霍乱弧菌,泳道1:稀释度为104,泳道2:稀释度为105,泳道3:稀释度为106,泳道4:稀释度为107,泳道5:稀释度为108;
图4.实施例3对番茄酱中沙门氏菌的检测结果(A)和常规LAMP法的检测结果(B)。泳道1-5分别表示不同稀释度的沙门氏菌,泳道1:稀释度为104,泳道2:稀释度为105,泳道3:稀释度为106,泳道4:稀释度为107,泳道5:稀释度为108;
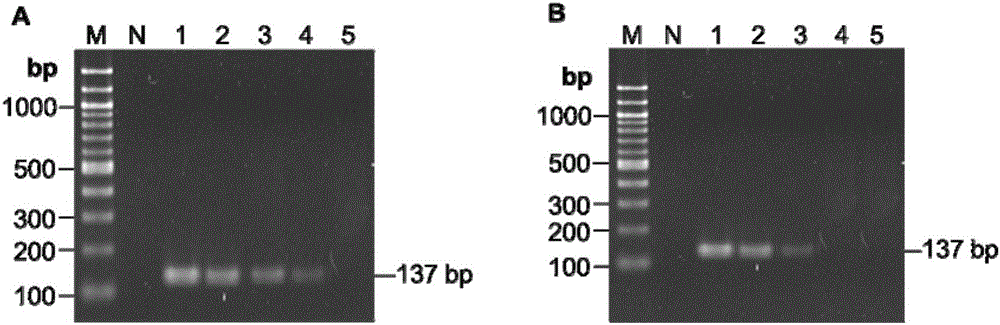
图5.使用本发明对克雷伯噬菌体p13的检测结果(A)和常规PCR法的检测结果(B)。M:DNA ladder;N:阴性对照。泳道1-5分别表示不同拷贝数的目标DNA,泳道1:5000拷贝,泳道2:500拷贝,泳道3:50拷贝,泳道4:5拷贝,泳道5:0.5拷贝;
图6.使用本发明对实际牡蛎样品中霍乱弧菌的检测结果(A)和常规PCR法的检测结果(B)。样品编号标注于泳道顶部。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图和实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
实施例中所用的部分试剂如下,未特殊说明的均为常规市售商品:
基因组提取试剂盒:天根,#DP350;
细菌基因组提取试剂盒:天根,#DP302;
海洋动物组织基因组提取试剂盒:天根,#DP324;
鲑鱼精DNA:Sigma-Aldrich,#D1626;
FastDigest BbvI内切酶:Thermo Scientific,#FD2074;
FastDigest HgaI内切酶:Thermo Scientific,#FD1904;
T3 DNA连接酶:New England Biolabs,#M0317S;
T4 DNA连接酶:New England Biolabs,#M0202S;
T7 DNA连接酶:New England Biolabs,#M0318S;
pfu DNA聚合酶:Thermo Scientific,#EP0502。
实施例1.应用本发明对八宝粥中黄曲霉菌进行检测
1、无菌条件下向5g八宝粥样品(娃哈哈)中加入100μL经过系列稀释的黄曲霉菌液(稀释度分别为104、105、106、107、108),对混合后的样品进行离心(4C,1792×g)后收集上清,对上清液进行离心(4℃,16128×g)收集菌渣,使用基因组提取试剂盒对收集到的菌渣进行DNA提取。同时对各稀释度的黄曲霉菌进行菌落计数以计算其大体数量。
2、向步骤1中提取到的各DNA样品中加入鲑鱼精DNA,使最终鲑鱼精DNA在各样品中的浓度为20ng/μL。
3、目标DNA片段的选择:对黄曲霉基因组(GenBank:EQ963478.1)使用BbvI进行酶切分析,选择其中一段441bp的酶切片段作为待检测目标,该酶切片段序列信息如序列表中SEQ ID No.1所示:
4、样品酶切(图1中步骤1):使用BbvI内切酶对黄曲霉DNA进行酶切反应,10μL酶切体系中含有1μL 10×FastDigest buffer,1μL步骤2中样品,0.5μL FastDigest BbvI内切酶,用ddH2O补足至10μL后于37℃保温5min后65℃灭酶5min。
5、接头连接反应(图1中步骤2):向酶切体系中依次加入0.5μL针对BbvI酶切片段设计的双链DNA接头(终浓度50nM,表1)、0.5μL ATP(10mM)和1μL T4DNA连接酶,于25℃保温2h。
表1.实施例1中使用的接头序列
注:长链接头同时作为本发明中所有实施例PCR反应中的通用引物;下划线部分是与目标片段粘性末端互补的“衔接”序列,下同。
6、使用通用引物的PCR反应(图1中步骤3):向连接产物中直接加入5μL 10×pfu DNA聚合酶buffer、1μL 10mM dNTPs、1.5μL通用引物(10μM,表1)、0.5μL pfu DNA聚合酶(2.5U/μL),用ddH2O将总体系补足至50μL,反应参数为:72℃5min,94℃3min,25个循环扩增反应(94℃30s、63℃30s、72℃75s)后72℃保温5min。
7、特异性PCR检测(图1中步骤4):针对黄曲霉菌DNA目标酶切片段设计特异性引物(表2)进行PCR反应,20μL体系中含有1μL步骤6中产物、2μL pfu buffer(10×)、0.4μL10mM dNTPs、各0.4μL特异性上下游引物(表2)。反应参数为:94℃3min,25个循环扩增反应(94℃30s、63℃30s、72℃45s)后72℃保温5min。
表2.实施例1中使用的特异性PCR引物
同时设置常规PCR对照实验,直接以步骤1获得的DNA作为模板进行步骤7的特异性PCR检测(除循环数为50循环外其他条件相同),最终产物使用2%琼脂糖凝胶电泳检测。
图2结果显示,使用本发明的方法对黄曲霉菌DNA检测灵敏度达到107稀释度(图2A),通过菌落计数结果可知此时的黄曲霉菌数量为20拷贝;常规PCR法可检测至106稀释度(图2B),相当于大约300拷贝,对比结果表明本发明可使PCR法对黄曲霉菌检测灵敏度提高约15倍。由于加入黄曲霉菌再进行提取会导致一定量的损失,因此推测本发明的实际检测灵敏度应高于本实施例结果,但通过使用相同样品的对比结果显示本发明可以提高PCR法的检测灵敏度。
实施例2.应用本发明对饮用水中霍乱弧菌进行检测
1、无菌条件下向1mL饮用纯净水(冰露)中加入100μL经过系列稀释的培养对数期的霍乱弧菌(稀释度分别为104、105、106、107、108),离心(4℃,16128×g)后弃上清,使用细菌基因组提取试剂盒对收集到的菌渣进行DNA提取。同时对各稀释度的霍乱弧菌进行菌落计数以计算其大体数量。
2、向步骤1中提取到的各DNA样品中加入鲑鱼精DNA(Sigma-Aldrich),使最终鲑鱼精DNA在各样品中的浓度为50ng/μL。
3、目标DNA片段的选择:对霍乱弧菌16S rRNA基因(GenBank:U10955.1)使用HgaI进行酶切分析,选择其中一段444bp的酶切片段作为待检测目标,该酶切片段序列信息如序列表中SEQ ID No.7所示:
4、使用HgaI内切酶对步骤2中制备的样品进行酶切反应,10μL酶切体系中含有1μL10×FastDigest buffer,1μL步骤2中样品,0.5μL FastDigest HgaI内切酶,用ddH2O补足至10μL后于37℃保温5min后65℃灭酶5min。
5、使用针对HgaI酶切片段设计的双链DNA接头以及T3DNA连接酶进行连接反应(表3),体系中接头终浓度为100nM,其他反应条件与参数与实施例1中步骤5相同。
表3.实施例2中使用的接头序列
6、以步骤5中的连接产物为模板,使用通用引物的PCR反应,实验条件及反应参数与实施例1中步骤6相同。
7、特异性LAMP检测:向薄壁管中依次加入8μL ddH2O,2.5μL 10×isothermal amplification buffer,3.5μL dNTPs(10mM),2μL MgSO4(25mM),5μL betaine(5M),1μL模板(步骤6中产物),2μL混合引物(各引物在混合引物中浓度分别为:内引物FIP和BIP均为10μM,外引物F3和B3均为2.5μM,见表4),混匀后置于热盖温度为95℃的热循环仪中保持2min后取出并立即插入冰中5min,然后冰上操作向管中加入1μL Bst 2.0DNA聚合酶(8U/μL,NEB),并将反应管立即放入已预热好的热循环仪中,65℃反应45min后95℃灭酶2min以终止反应。最终产物使用0.8%琼脂糖凝胶电泳检测。
表4.实施例2中使用的LAMP引物
同时设置以步骤1中获得的DNA为模板直接进行LAMP检测的对照组,除使用模板不同之外,其他反应条件与参数与步骤7中完全一致。
图3结果显示,使用本发明的方法对饮用水中霍乱弧菌的检测灵敏度达到107稀释度(图3A),通过菌落计数结果可知此时的霍乱弧菌数量为16拷贝;常规LAMP法可检测至106稀释度(图3B),相当于大约280拷贝,对比结果表明本发明可使LAMP法对霍乱弧菌检测灵敏度提高约十数倍。相对于实施例1所使用的BbvI内切酶,本实施例使用了HgaI内切酶,表明本发明在内切酶选择上具有一定的灵活性。
实施例3.应用本发明对番茄酱中沙门氏菌进行检测
1、无菌条件下向1g番茄酱(亨氏)中加入100μL经过系列稀释的培养对数期的沙门氏菌(稀释度分别为104、105、106、107、108),对混合后的样品进行离心(4℃,4032×g)后收集上清,对上清液进行离心(4℃,16128×g)收集菌渣,使用细菌基因组提取试剂盒对收集到的菌渣进行DNA提取。同时对各稀释度的沙门氏菌进行菌落计数以计算其大体数量。
2、向步骤1中提取到的各DNA样品中加入鲑鱼精DNA(Sigma-Aldrich),使最终鲑鱼精DNA在各样品中的浓度为150ng/μL。
3、目标DNA片段的选择:对沙门氏菌SodC2基因(GenBank:DQ023313.1)使用HgaI进行酶切分析,选择其中一段346bp的酶切片段作为待检测目标,该酶切片段序列如序列表中SEQ ID No.14所示:
4、使用HgaI内切酶对步骤2中的样品进行酶切反应,10μL酶切体系中含有1μL10×FastDigest buffer,1μL步骤2中样品,0.5μL FastDigest HgaI内切酶,用ddH2O补足至10μL后于37℃保温5min后65℃灭酶5min。
5、使用针对沙门氏菌DNA设计的双链DNA接头以及T7DNA连接酶进行连接反应(表5),体系中接头终浓度为200nM,其他反应条件与参数与实施例1中步骤5相同。
表5.实施例3中使用的接头序列
6、以步骤5中连接产物作为模板,使用通用引物进行PCR反应,其他实验条件及反应参数与实施例1中步骤6相同。
7、特异性LAMP检测:以步骤6中产物作为LAMP反应模板进行检测(表6),使用针对沙门氏菌DNA设计的LAMP引物进行扩增反应,其他反应条件及参数同实施例2中步骤7完全一致。
表6.实施例3中使用的LAMP引物
同时设置以步骤1中获得的DNA为模板直接进行LAMP检测的对照组,除使用模板不同之外,其他反应条件与参数与步骤7中完全一致。
图4结果显示,使用本发明的方法对番茄酱中沙门氏菌的检测灵敏度达到107稀释度(图4A),通过菌落计数结果可知此时的沙门氏菌数量为12拷贝;常规LAMP法可检测至106稀释度(图4B),相当于大约180拷贝,对比结果表明本发明可使LAMP法对沙门氏菌检测灵敏度提高十数倍。
实施例4.应用本发明对克雷伯噬菌体p13进行检测
为了更加直观地对本发明的检测灵敏度进行量化评价,使用已知精确数量的纯化后的噬菌体DNA作为待检测目标,应用本发明方法以及常规PCR法进行检测并进行结果比较评价。
1、梯度稀释已知浓度的噬菌体DNA,使其浓度依次为5000、500、50、5、0.5拷贝/μL,然后向各样品中加入鲑鱼精DNA(Sigma-Aldrich),使最终鲑鱼精DNA在各样品中的浓度均为400ng/μL。
2、目标DNA片段的选择:对噬菌体基因组(GenBank:SRX270599)使用BbvI进行酶切分析,选择其中一段511bp的酶切片段作为待检测目标,该酶切片段序列如序列表中SEQ ID No.21所示:
3、使用BbvI内切酶进行酶切反应,10μL酶切体系中含有1μL 10×FastDigest buffer,1μL样品,0.5μL FastDigest BbvI内切酶,用ddH2O补足至10μL后于37℃保温5min后65℃灭酶5min。
4、向酶切体系中依次加入0.5μL针对噬菌体DNA酶切片段设计的双链DNA接头(终浓度300nM,表7)、0.5μL ATP(10mM)和1μL T4DNA连接酶,于25℃保温0.5h。
表7.实施例4中使用的接头序列
5、以步骤4中连接产物作为模板,使用通用引物进行PCR反应,其他实验条件及反应参数与实施例1中步骤6相同。
6、使用针对目标酶切片段设计特异性引物进行PCR反应,20μL体系中含有1μL步骤5中产物、2μL pfu buffer(10×)、0.4μL 10mM dNTPs、各0.4μL特异性上下游引物(表8)。反应参数为:94℃3min,25个循环扩增反应(94℃30s、63℃30s、72℃45s)后72℃保温5min。
同时设置常规PCR对照实验,直接以未加鲑鱼精DNA的噬菌体p13DNA作为模板进行步骤6的特异性PCR检测(除循环数为50循环外其他条件相同),最终产物使用2%琼脂糖凝胶电泳检测。
表8.实施例4中使用的特异性PCR引物
图5结果显示,使用本发明的方法可使检测灵敏度达到5拷贝(图5A),常规PCR法检测灵敏度为50拷贝(图5B),本发明使检测灵敏度提高了10倍。
实施例5.应用本发明对实际牡蛎样品中霍乱弧菌进行检测
1、采集从6月至8月间市场上购买的23份牡蛎样品,使用海洋动物组织基因组提取试剂盒对牡蛎组织的全DNA进行提取。
2、目标DNA片段的选择:同实施例2。
3、使用BbvI内切酶进行酶切反应,10μL酶切体系中含有1μL 10×FastDigest buffer,1μL步骤1中提取的牡蛎DNA样品,0.5μL FastDigest BbvI内切酶,用ddH2O补足至10μL后于37℃保温5min后65℃灭酶5min。
4、向酶切体系中依次加入0.5μL针对酶切片段设计的双链DNA接头(终浓度400nM,表3)、0.5μL ATP(10mM)和1μL T4DNA连接酶,于25℃保温1h。
5、以步骤4中的连接产物为模板,使用通用引物的PCR反应,其他反应条件与实施例1中步骤6相同。
6、以步骤5中的产物作为模板,使用针对霍乱弧菌酶切片段设计特异性引物进行PCR反应(表9),除模板与引物不同之外,其他反应条件与参数和实施例1中步骤7相同。
表9.实施例5中使用的特异性PCR引物
同时设置常规PCR对照实验,直接以步骤1中提取到的牡蛎DNA作为模板进行步骤6的特异性PCR检测(除循环数为50循环外其他条件相同),最终产物使用2%琼脂糖凝胶电泳检测。
图6结果显示,使用本发明的方法最终共检测出3例霍乱弧菌阳性(图6A),常规高循环数PCR法共检测出2例阳性样品(图6B),表明本发明可以应用于实际样品的检测中,并提高检测灵敏度。由于实际样品检测中不可避免的会引入大量的背景基因组DNA,所以在酶切过程中不需要额外加入背景基因组DNA,因此本发明尤其适合于实际样品中极微量目标DNA的检测,具有显著的实际应用意义。
上述的实施例的结果表明本发明的方法可有效提高现有常用核酸检测方法对极低数量目标DNA的检测灵敏度,通过多个实施例分别验证了本发明方法对不同类型微生物(包括病毒、真菌、细菌)的检测效果;验证了多种不同内切酶均适用,表明本发明方法的选择灵活性;通过对实际样品的检测结果表明本方法具有十分明显的实际应用价值,且整体上对仪器和试剂的要求较低,因此具有广阔的实际应用前景。
应当理解的是,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,而所有这些改进和变换都应属于本发明所附权利要求的保护范围。
SEQUENCE LISTING
<110> 青岛千卓分子生物科技有限公司
<120> 一种提高极低拷贝数目标DNA检测灵敏度的方法
<130> 2017
<160> 27
<170> PatentIn version 3.5
<210> 1
<211> 441
<212> DNA
<213> Aspergillus flavus
<400> 1
atcaattacc tcgctgccaa gaggaaagaa ctcggtctcg atagctcgac ggtcgtcctt 60
caacaaagct cactgggatt cgacatggga cttgcacaga ctctcaatgc catcatgaac 120
ggaggaaagc ttgtcattgt gccacaagaa ttacgaggag actcgattga aattgcaaga 180
attatccgcg atcagaaggt gactttcacg ctagccactc cgtccgaata cctcgtcatg 240
ctccaacatg gtcgtgaata cctcaacaat tacaccggat ggcggcacgc ctgccttggg 300
ggtgagccat tcacagacca gctcaagagg gagtttgtga ggctgggaaa gaactgccct 360
gtggtgcagg actcctacgg tgtgaccgag atttccgcct gcacaacctt tgagacgatg 420
tctgcctcgc agctggaaga c 441
<210> 2
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> A-1
<400> 2
gcactgctga gtcac 15
<210> 3
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> A-2
<400> 3
tgattgctga gtcac 15
<210> 4
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> 长链接头(通用引物)
<400> 4
tcgtcactgc acggtgactc agca 24
<210> 5
<211> 21
<212> DNA
<213> Aspergillus flavus
<400> 5
cgctgccaag aggaaagaac t 21
<210> 6
<211> 24
<212> DNA
<213> Aspergillus flavus
<400> 6
cttgtggcac aatgacaagc tttc 24
<210> 7
<211> 444
<212> DNA
<213> Vibrio cholerae
<400> 7
tgccttcggg agctctgaga caggtgctgc atggctgtcg tcagctcgtg ttgtgaaatg 60
ttgggttaag tcccgcaacg agcgcaaccc ttatccttgt ttgccagcac gtaatggtgg 120
gaactccagg gagactgccg gtgataaacc ggaggaaggt ggggacgacg tcaagtcatc 180
atggccctta cgagtagggc tacacacgtg ctacaatggc gtatacagag ggcagcgatc 240
ccgcgaggtg gagcgaatct cacaaagtac gtcgtagtcc ggattggagt ctgcaactcg 300
actccatgaa gtcggaatcg ctagtaatcg caaatcagaa tgttgcggtg aatacgttcc 360
cgggccttgt acacaccgcc cgtcacacca tgggagtggg ctgcaaaaga agcaggtagt 420
ttaaccttcg ggaggacgct tgcc 444
<210> 8
<211> 16
<212> DNA
<213> Artificial Sequence
<220>
<223> A-3
<400> 8
aggcatgctg agtcac 16
<210> 9
<211> 16
<212> DNA
<213> Artificial Sequence
<220>
<223> A-4
<400> 9
acttttgctg agtcac 16
<210> 10
<211> 42
<212> DNA
<213> Artificial Sequence
<220>
<223> FIP
<400> 10
gctgccctct gtatacgcca ttgtcaagtc atcatggccc tt 42
<210> 11
<211> 40
<212> DNA
<213> Artificial Sequence
<220>
<223> BIP
<400> 11
cgcgaggtgg agcgaatctc atggagtcga gttgcagact 40
<210> 12
<211> 20
<212> DNA
<213> Vibrio cholerae
<400> 12
gataaaccgg aggaaggtgg 20
<210> 13
<211> 20
<212> DNA
<213> Vibrio cholerae
<400> 13
cgcaacattc tgatttgcga 20
<210> 14
<211> 346
<212> DNA
<213> Salmonella typhimurium
<400> 14
gcctgtgcgg gtgcgcaggc cgccagcgag aaagtagaga tgaatctggt gacggcgcaa 60
ggcgtaggac agtctatcgg caccgtcgtc atcgatgaaa ccgaaggcgg cttaaaattt 120
accccacacc ttaaagcgtt gccgccgggc gagcatggtt ttcacattca tgccaacggt 180
agctgccagc ccgcgattaa agacggcaaa gcggttgccg cagaagccgc tggtggtcat 240
ctggacccac aaaataccgg caagcatgaa ggaccggaag gccaggggca tctgggcgac 300
ctcccggtgt tagtcgttaa taatgatggt atcgccagcg aaccgg 346
<210> 15
<211> 16
<212> DNA
<213> Artificial Sequence
<220>
<223> A-5
<400> 15
caggctgctg agtcac 16
<210> 16
<211> 16
<212> DNA
<213> Artificial Sequence
<220>
<223> A-6
<400> 16
ttacttgctg agtcac 16
<210> 17
<211> 38
<212> DNA
<213> Artificial Sequence
<220>
<223> FIP
<400> 17
tgaaaaccat gctcgcccgg gaaaccgaag gcggctta 38
<210> 18
<211> 38
<212> DNA
<213> Artificial Sequence
<220>
<223> BIP
<400> 18
atgccaacgg tagctgccag gggtccagat gaccacca 38
<210> 19
<211> 18
<212> DNA
<213> Salmonella typhimurium
<400> 19
ctatcggcac cgtcgtca 18
<210> 20
<211> 18
<212> DNA
<213> Salmonella typhimurium
<400> 20
ttcatgcttg ccggtatt 18
<210> 21
<211> 511
<212> DNA
<213> Klebsilla Phage P13
<400> 21
aacctgttaa cttgggctgg tgcaagtgac tactccatct ctcgcatggg tactaccatt 60
gtcatatcca gccttagtgg tgcaagcttc acggtggaca cggaggatgg ctctaaaggt 120
aaagaccttg tagccatcca gtacaaggtg acttcaactg acctgcttcc aagcaaggca 180
cctgttggtt acctggtcca ggtatggcct accggaagca agcctgagtc tcgctactgg 240
ctcaaggcgg aggcggcgga tggcaacctt gttacctggc aggaaaccct tggggcagat 300
gaggtgctgg gttttaatgg gacgactatg ccatatatca ttgagcgaac caatattgtt 360
ggtggtatcg ctcagttcac cattaagcag ggctattggg atgaccgggc ggtgggtgat 420
gagctaacca accctatgcc ttcctttatt gaccaaagcc tcagtgatat cttcatggtg 480
caaaacaggt tatgcctggc agctggagaa t 511
<210> 22
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> A-7
<400> 22
ggtttgctga gtcac 15
<210> 23
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> A-8
<400> 23
cttgtgctga gtcac 15
<210> 24
<211> 21
<212> DNA
<213> Klebsilla Phage P13
<400> 24
tgggctggtg caagtgacta c 21
<210> 25
<211> 22
<212> DNA
<213> Klebsilla Phage P13
<400> 25
gcccggtcat cccaatagcc ct 22
<210> 26
<211> 20
<212> DNA
<213> Vibrio cholerae
<400> 26
ctctgagaca ggtgctgcat 20
<210> 27
<211> 18
<212> DNA
<213> Vibrio cholerae
<400> 27
tgcttctttt gcagccca 18
- 还没有人留言评论。精彩留言会获得点赞!