一种盐酸洛拉曲克晶型Ⅲ及其制备方法与应用与流程
技术领域
本发明涉及一种化合物的新晶型,具体涉及一种盐酸洛拉曲克晶型Ⅲ及其制备方法与应用。
背景技术:
盐酸洛拉曲克国外最早由美国Agouron Pharmaceuticals公司开发。该公司根据酶活性中心三维结构特征,采用计算机模拟药物分子技术合成了一系列胸苷酸合成酶抑制剂,盐酸洛拉曲克为其中抑制胸苷酸合成酶活性最高的化合物之一。该公司与Roche Switzerland合作,将盐酸洛拉曲克其作为抗肿瘤药物开发,商品名为“Thymitaq”。该药从1994年起开始临床试验,1996年8月进入III期临床。1999年1月,AgouronPharmaceuticals公司进行产品战略调整,将Thymitaq的全部研发权独家转让给美国Zarix公司。但在已经公开的文献报道中,目前尚未见到关于盐酸洛拉曲克的多晶型方面的报道。
技术实现要素:
本发明的第一目的在于提供一种盐酸洛拉曲克晶型Ⅲ,该晶体形式是未见报道的,其轮廓分明,可完美再现,并且就过滤、干燥、稳定性、方法配制和可加工性而言,其显示有价值的特征,可用作抗肿瘤药物中的用途,具有广阔的应用前景。
为实现上述目的,本发明采用如下技术方案:
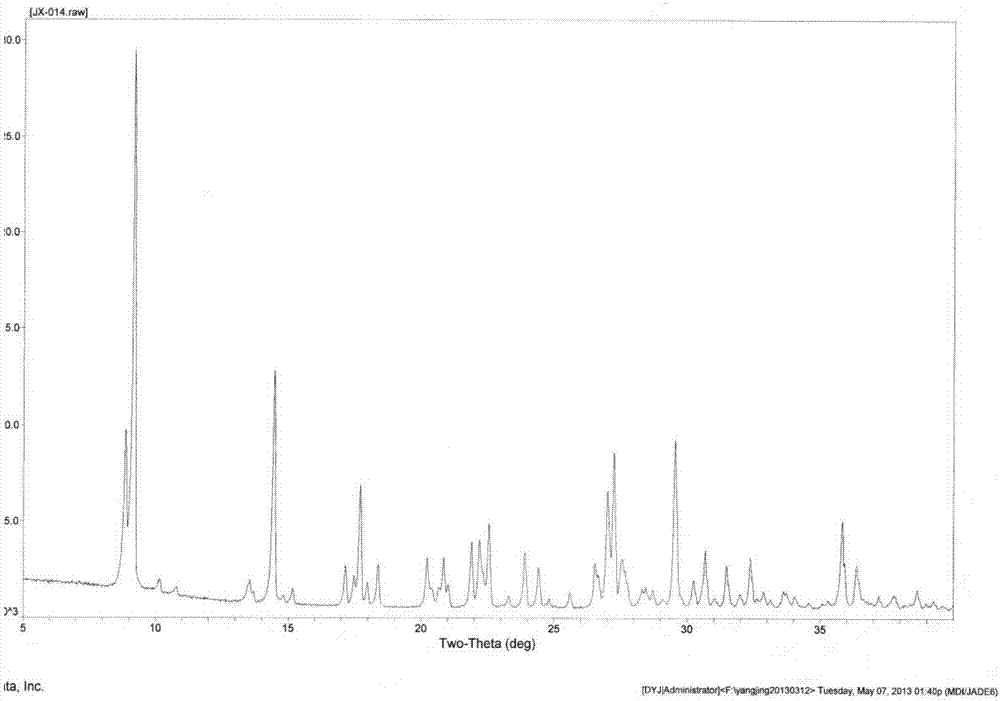
一种盐酸洛拉曲克晶型Ⅲ,其特征在于,所述盐酸洛拉曲克晶型Ⅲ用粉末X射线衍射测定法测定,以2θ±0.2°衍射角表示的X射线粉末衍射图谱在6.673°、8.838°、9.167°、12.748°、13.548°、14.443°、16.704°、17.014°、17.810°、21.807°、22.117°、22.527°、26.672°、27.005°、27.238°、29.051°和29.498°处显示出特征衍射峰。
具体地,本发明所述盐酸洛拉曲克晶型Ⅲ具有如图1所示的粉末X射线衍射图,所述衍射图使用日本理学RigakuD/MAXRC衍射仪测定,并根据谱线位置(布拉格角2θ,以度表示)、相对丰度和晶面间距d(以表示)表示,其中晶型Ⅲ表示如下:
所述盐酸洛拉曲克晶型Ⅲ的熔点为255-259℃。由X射线粉末衍射图和熔点可知,本发明得到了一种全新的盐酸洛拉曲克晶体。
本发明的第二目的在于提供上述盐酸洛拉曲克晶型Ⅲ的制备方法,所述制备方法包括如下步骤:
(1)将盐酸洛拉曲克粗品投入四口烧瓶中,加入乙腈和盐酸的复合溶剂,升温至回流,保持回流状态使形成均匀的蛋清色混悬液;
(2)停止回流后低速搅拌并缓慢降温至50-60℃,恒温低速搅拌60-100分钟,后继续缓慢降温至15-25℃,搅拌90-150分钟;
(3)过滤、干燥得到盐酸洛拉曲克晶型Ⅲ。
其中,本发明步骤1所述盐酸洛拉曲克粗品即为现有技术公开的市售产品,具体的获取和选择为本领域技术人员所掌握。本发明提供所述盐酸洛拉曲克粗品的一种制备方法,但可以理解的是,本发明所述盐酸洛拉曲克粗品的制备方法包括却并不局限于以下制备方法:
将洛拉曲克加入到水和乙腈的混合溶液中,加入少量浓盐酸,加热使完全溶解。滴加不良溶剂四氢呋喃至析出晶体,静置析晶后过滤得到盐酸洛拉曲克粗品。
其中,洛拉曲克与水/乙腈复合溶剂的质量体积比为1:8-1:10,水和乙腈的体积比为1:3-1:4;浓盐酸的体积以水体积的1/8-1/12为宜。此处的不良溶剂为四氢呋喃,其具体的添加量以晶体尽可能析出为准,而具体的静置析晶和过滤为本领域技术人员所能够理解的常规手段,本发明对此不作特别限定。
上述方法所得盐酸洛拉曲克粗品的熔点为224-227℃。
上述方法所得盐酸洛拉曲克粗品在相同的检测条件下,所得粉末X射线衍射图见图2,将其与本发明要求保护的盐酸洛拉曲克晶型Ⅲ粉末X射线衍射图(即图1)对比可知,本发明通过特定的重结晶方法得到了一种全新的晶型,且将该盐酸洛拉曲克晶型Ⅲ与盐酸洛拉曲克粗品或现有技术公开的盐酸洛拉曲克作药效学试验,彼此之间药效相当,无显著差异。
本发明所述盐酸洛拉曲克晶型Ⅲ的制备方法,步骤1复合溶剂中,乙腈的体积用量为盐酸洛拉曲克质量的15-25倍;盐酸的体积用量为盐酸洛拉曲克质量的0.03-0.08倍。
本发明所述的制备方法,所述步骤2中,停止回流后以50-70rmp的低速搅拌;并以0.5-1.5℃/min的速度缓慢降温至50-60℃至有大量晶体析出,恒温20-40rmp的低速搅拌75-85min,后以0.4-0.8℃/min的速度缓慢降温至15-25℃,养晶100-120分钟。
其中,所述的过滤干燥为在32-36℃、-0.085~-0.095MP下真空干燥6~7小时。
作为本发明的最佳实施方式,优选所述制备方法包括如下步骤:
将盐酸洛拉曲克粗品5.0克投入四口烧瓶中,加乙腈100ml、盐酸0.05ml,升温至回流,保持回流状态45分钟以形成均匀的蛋清色混悬液;
停止回流后以60rmp的低速搅拌,并以1℃/min的速度缓慢降温至55℃至有大量晶体析出;恒温并以30rmp的低速搅拌90min,后以0.6℃/min的速度继续缓慢降温至25℃,养晶120分钟;
过滤,湿粉在35℃、-0.092MP下真空干燥6.5小时,得盐酸洛拉曲克晶型Ⅲ。
除特殊说明外,本发明所述制备方法中,重量体积比的单位为g/ml。
本发明通过对溶剂的选择、温度、搅拌速度和养晶时间等方面的综合控制,得到了一种简单易行的制备方法,能快速得到本发明所述的高质量晶体,适宜推广应用在实际生产中。
此外,本发明的第三目的在于保护含有上述盐酸洛拉曲克晶型Ⅲ的药物组合物,具体该药物组合物包括但不限于为粉针、片剂、胶囊剂、冻干粉或输液等。本发明所述组合物可以进一步含有药学可接受的载体。譬如当为冻干粉时,所述药学可接受的载体可以为赋形剂;当所述组合物为片剂时,所述药学可接受的载体可以是崩解剂、赋形剂等。由于本发明所得到的盐酸洛拉曲克晶型Ⅲ具有理想的理化性能,因此,本领域技术人员可以预见将其制备成各种药物制剂后,也能进一步提供所述药物制剂的稳定性及疗效,而具体剂型及其处方的确定为本领域技术人员所掌握。
更优选地,本发明所述的药物组合物为粉针,所述粉针按400-500mg/支分装,优选按440mg/支分装。此外,本发明所述的粉针还可以进一步含有药学上可接受的各种载体,如甘露醇等。具体的选择和用量的确定为本领域技术人员所掌握。
此外,本发明所述的组合物还可以为冻干粉,该冻干粉按重量份计,由包括如下组分的原料制备成1000瓶:盐酸洛拉曲克晶型Ⅲ350-450g、甘露醇35-45g、注射用水加至3500-4500ml;其中优选盐酸洛拉曲克晶型Ⅲ400g、甘露醇40g、注射用水加至4000ml后除菌,分装,冻干。
本发明所述的冻干粉针制剂,可以采用现有技术公开或教导的各种冻干方法制备而成。本领域技术人员能够理解,由于本发明所述盐酸洛拉曲克晶型Ⅲ较现有技术公开的盐酸洛拉曲克具有更为理想的性能,将其制备成冻干粉针后同样也能获得更理想的稳定性、复溶性等以确保制剂的疗效,保证患者的用药安全。为了进一步验证上述推论,发明人将本发明要求保护的冻干粉针制剂与相同处方及制备方法得到的现有盐酸洛拉曲克制成的冻干粉针做质量对比,结果显示本发明所得冻干粉针剂的质量得到明显提高,进一步说明本发明所得盐酸洛拉曲克晶型Ⅲ用于具体的药物组合物后,本身的性质并未发生变化,依然具备理想的性能,从而提高了药物制剂的整体疗效。
但为了获得更高质量的冻干粉针,本发明还提供了一种冻干方法,具体为:将配制好的盐酸洛拉曲克制剂分装后放入冷冻干燥箱内,自室温起在5h内匀速降温至-30℃,冷冻10h;后升温至-10℃,保温10h;继续升温至22℃干燥,保温15h,结束冻干,自动压塞,盖铝盖、压盖。
此外,本发明还进一步要求保护上述盐酸洛拉曲克晶型Ⅲ在制备治疗抗肿瘤药物中的应用。本发明所述盐酸洛拉曲克晶型Ⅲ在药效学试验中表现出显著的抗肿瘤活性,能够广泛应用在抗肿瘤药物的制备中。
采用上述技术方案,本发明具有如下有益效果:
1、本发明盐酸洛拉曲克晶型Ⅲ具有显著优于盐酸洛拉曲克粗品的稳定性,且收率好,纯度高;
2、将本发明所得盐酸洛拉曲克晶型Ⅲ进一步制备成相关制剂后,能够显著提高药物制剂的稳定性,从而确保患者的用药安全;
3、本发明所述制备方法简单易行,能够稳定且批量化生产,有利于推广应用。
附图说明
图1是本发明盐酸洛拉曲克晶型Ⅲ的X射线粉末衍射图;
图2是盐酸洛拉曲克粗品的X射线粉末衍射图。
具体实施方式
下面结合附图和实施例对本发明做进一步描述。
实施例1盐酸洛拉曲克粗品的制备
将5g洛拉曲克加入到10ml水和33ml乙腈的混合溶液中,加入1.0ml浓盐酸,加热使完全溶解。滴加四氢呋喃至析出晶体,按常规方法静置析晶后过滤得到盐酸洛拉曲克粗品,其粉末X射线衍射图见图2。
实施例2盐酸洛拉曲克晶型Ⅲ及其制备
将盐酸洛拉曲克粗品(实施例1制备)5.0克投入四口烧瓶中,加乙腈100ml、盐酸0.4ml,升温至回流,保持回流状态45分钟以形成均匀的蛋清色混悬液;
停止回流后以60rmp的低速搅拌,并以1℃/min的速度缓慢降温至55℃至有大量晶体析出;恒温以30rmp的低速搅拌90分钟,后以0.6℃/min的速度继续缓慢降温至25℃,养晶120分钟;
过滤,湿粉在35℃、-0.092MP下真空干燥6.5小时,得盐酸洛拉曲克晶型Ⅲ4.81g。纯度为99.5%,收率为93.2%。
如图1所示,盐酸洛拉曲克晶型Ⅲ用粉末X射线衍射测定法测定,以2θ±0.2°衍射角表示的X射线粉末衍射图谱在6.673°、8.838°、9.167°、12.748°、13.548°、14.443°、16.704°、17.014°、17.810°、21.807°、22.117°、22.527°、26.672°、27.005°、27.238°、29.051°和29.498°处显示出特征衍射峰。
实施例3盐酸洛拉曲克晶型Ⅲ及其制备
将盐酸洛拉曲克粗品(实施例1制备或市售)5.0克投入四口烧瓶中,加乙腈75ml、盐酸0.15ml,升温至回流,保持回流状态60分钟以形成均匀的蛋清色混悬液;
停止回流后以50rmp的低速搅拌,并以0.5℃/min的速度缓慢降温至50℃至有大量晶体析出;恒温并以20rmp的低速搅拌60分钟,后以0.4℃/min的速度继续缓慢降温至15℃,养晶90分钟;
过滤,湿粉在34℃、-0.088MP下真空干燥6.5小时,得盐酸洛拉曲克晶型Ⅲ4.81g。纯度为99.4%,收率为92%。
实施例4盐酸洛拉曲克晶型Ⅲ及其制备
将盐酸洛拉曲克粗品(实施例1制备)5.0克投入四口烧瓶中,加乙腈125ml、盐酸0.4ml,升温至回流,保持回流状态50分钟以形成均匀的蛋清色混悬液;
停止回流后以70rmp的低速搅拌,并以1.5℃/min的速度缓慢降温至60℃至有大量晶体析出;恒温并以40rmp的低速搅拌100分钟,后以0.8℃/min的速度继续缓慢降温至25℃,养晶150分钟;
过滤,湿粉在36℃、-0.095MP下真空干燥7小时,得盐酸洛拉曲克晶型Ⅲ4.81g。纯度为99.3%,收率为89%。
实施例5盐酸洛拉曲克晶型Ⅲ及其制备
将盐酸洛拉曲克粗品(实施例1制备)5.0克投入四口烧瓶中,加乙腈120ml、盐酸0.2ml,升温至回流,保持回流状态55分钟以形成均匀的蛋清色混悬液;
停止回流后以65rmp的低速搅拌,并以0.8℃/min的速度缓慢降温至55℃至有大量晶体析出;恒温并以35rmp的低速搅拌85分钟,后以0.7℃/min的速度继续缓慢降温至22℃,养晶120分钟;
过滤,湿粉在32℃、-0.09MP下真空干燥6小时,得盐酸洛拉曲克晶型Ⅲ4.81g。纯度为99.4%,收率为88%。
实施例6含有盐酸洛拉曲克晶型Ⅲ的冻干粉
取400g实施例2所制备得到的盐酸洛拉曲克晶型Ⅲ、40g甘露醇,加注射用水至4000ml,溶解充分,除菌,分装后冻干,冻干方法具体为:将配制好的盐酸洛拉曲克制剂分装后放入冷冻干燥箱内,自室温起在5h内匀速降温至-30℃,保温10h;后升温至-10,保温10h;继续升温至22℃再干燥,保温15h,结束冻干,自动压塞,盖铝盖、压盖。
实施例7含有盐酸洛拉曲克晶型Ⅲ的冻干粉
取400g实施例3所制备得到的盐酸洛拉曲克晶型Ⅲ、35g甘露醇,加注射用水至4000ml,溶解充分,除菌,分装后冻干,冻干方法具体为:将配制好的盐酸洛拉曲克制剂分装后放入冷冻干燥箱内,自室温起在5h内匀速降温至-30℃,冷冻10h;后升温至-10℃,保温10h;继续升温至22℃干燥,保温15h,结束冻干,自动压塞,盖铝盖、压盖。
实施例8含有盐酸洛拉曲克晶型Ⅲ的冻干粉
取450g实施例4所制备得到的盐酸洛拉曲克晶型Ⅲ、45g甘露醇,加注射用水至4000ml,溶解充分,除菌,分装后冻干,冻干方法具体为:将配制好的盐酸洛拉曲克制剂分装后放入冷冻干燥箱内,自室温起在5h内匀速降温至-30℃,冷冻10h;后升温至-10℃,保温10h;继续升温至22℃再干燥,保温15h,结束冻干,自动压塞,盖铝盖、压盖。即得。实施例8含有盐酸洛拉曲克晶型Ⅲ的粉针
取350g实施例2所制备得到的盐酸洛拉曲克晶型Ⅲ,按440mg/支分装,即得。
实施例9含有盐酸洛拉曲克晶型Ⅲ的粉针
取350g实施例2所制备得到的盐酸洛拉曲克晶型Ⅲ,按420mg/支分装,即得。
实施例10含有盐酸洛拉曲克晶型Ⅲ的粉针
取350g实施例3所制备得到的盐酸洛拉曲克晶型Ⅲ,按440mg/支分装,即得。
实施例11含有盐酸洛拉曲克晶型Ⅲ的粉针
取350g实施例4所制备得到的盐酸洛拉曲克晶型Ⅲ,按500mg/支分装,即得。
试验例1稳定性试验
本发明还进一步提供如下试验例,以对本发明的技术方案进行说明。
本试验例检测了本发明所提供的盐酸洛拉曲克晶型Ⅲ的稳定性(试验结果均以各试验组盐酸洛拉曲克重量计算)。
试验对象:
实验组1为本发明实施例2所得盐酸洛拉曲克晶型Ⅲ;
实验组2为本发明实施例3所得盐酸洛拉曲克晶型Ⅲ;
对照组1为盐酸洛拉曲克粗品(本发明实施例1所得);
试验方法:
加速试验:将样品用小瓶包装,放入相对湿度为75%±5%的密闭容器(干燥器)中,将干燥器放入培养箱,于40℃±2℃放置6个月,分别于0,1,2,3,6月各取一个包装进行考察。
长期试验:将样品用小瓶包装,贮存于温度25℃±2℃,相对湿度60%±10%条件下,定期取样测试。
本试验结果见表1和表2:
表1加速试验结果
表2长期试验结果
试验例2
本试验例检测了本发明所提供的盐酸洛拉曲克晶型Ⅲ所制备的冻干粉的稳定性(试验结果均以各试验组盐酸洛拉曲克重量计算)。
试验对象:
实验组1为本发明实施例6所得盐酸洛拉曲克冻干粉;
实验组2为本发明实施例7所得盐酸洛拉曲克冻干粉;
对照组1为以盐酸洛拉曲克粗品(实施例1)为原料,按实施例6相同的处方和制备方法得到的冻干粉;
试验方法:
加速试验:将冻干粉样品,按拟市售包装,在温度40℃±2℃,相对湿度75%±5%的条件下放置6个月,分别于0,1,2,3,6个月取样进行检验。
长期试验:将供试品3批,按市售包装,贮存于温度25℃±2℃,相对湿度60%±10%的条件下,定期取样测试。
试验结果见表3和表4.
表3加速试验结果
表4长期试验结果
上述实施例中的实施方案可以进一步组合或者替换,且实施例仅仅是对本发明的优选实施例进行描述,并非对本发明的构思和范围进行限定,在不脱离本发明设计思想的前提下,本领域中专业技术人员对本发明的技术方案作出的各种变化和改进,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!