一种立达霉代谢产物16的合成方法与流程
本发明涉及化学合成技术领域,具体地说,是一种立达霉代谢产物16:N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的合成方法。
背景技术:
立达霉(Ridomil)为F.Scwinn于1977年首先报道的新型高效内吸杀菌剂,又名甲霜灵,其性能较稳定,对pH的适应范围广泛(1.0~9.0),对高温度和强光耐受性好。作为一种新型高效内吸杀菌剂,其作用机理是通过作用于菌丝体、孢子的细胞壁,以使其选择通透性,影响水霉菌对营养物质的吸收利用,使菌丝体细胞死亡、孢子失去萌发力,阻断其生活史,现有研究证明它对鱼体和鱼卵的水霉病具有良好的防治效果,如复方立达霉粉——“美婷”的泼洒浓度为5~10g/m3、浸浴浓度为20g/m3时,可有效地防治鱼卵、鱼苗和成鱼的水霉病。
目前,国内主要使用的含立达霉的剂型有35%拌种剂、25%可湿性粉剂、5%颗粒剂和25%乳油,常见混剂有58%甲霜灵·锰锌可湿性粉剂(立达霉+代森锰锌)、60%甲霜灵·锰锌可湿性粉剂、72%甲霜灵·锰锌可湿性粉剂等。
2002年农药残留联席会议(JMPR)建立了立达霉的毒理学,并确立了立达霉的每日容许摄入量(ADI)。同时,经研究表明,立达霉在动物体内代谢主要有16种代谢产物,其中,根据在不同动物中检测出的含量,较多的(>0.1mg/kg)的代谢产物为代谢物6、8、12、16。而其中,代谢产物16为N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸,目前国内外尚未有合成获得该种立达霉主要代谢产物的相关研究。
技术实现要素:
本发明的目的是针对现有技术中的不足,提供一种立达霉代谢产物16:N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的合成方法。
为实现上述目的,本发明采取的技术方案是:一种合成N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的方法,所述的方法的合成路线为:
所述的方法包括以下步骤:
1)取立达霉(Ⅰ)为原料药,溶解于酸溶液中,得到化合物(Ⅱ);
2)将化合物(Ⅱ)加还原剂还原得到化合物(Ⅲ);
3)化合物(Ⅲ)酸化,得到化合物(Ⅳ);
4)化合物(Ⅳ)再加入碱溶液得到产物(Ⅴ)。
步骤1)中的酸溶液为65%硝酸和98%浓硫酸的混合酸溶液。
步骤3)中使用硫酸酸化。
一种合成N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的方法,所述的方法包括以下步骤:
1)将65%硝酸10mL和98%浓硫酸30mL在0度混合得到混酸,反应容器中加入二氯甲烷150mL,再加入27.9g甲霜灵搅拌溶解透明,在0度滴加所述的混酸,保持温度在5度,滴加完毕搅拌过夜,原料反应完全后加入冰水中分层,水层用二氯甲烷50mL提取两次,合并二氯甲烷层,用水洗两次,二氯甲烷层无水硫酸钠干燥活性炭脱色,抽滤,滤液回收二氯甲烷,得到固体,固体用柱层析纯化,洗脱剂PE:EA5:1----3:1,得到产品;
2)取5g上述产品加入50mL甲醇溶解,加入10%Pd/C0.5g,通入氢气氢化还原过夜得到氨基化合物3g;
3)3g氨基化合物溶解到20%硫酸溶液中,然后冷却到5度,滴加亚硝酸钠溶液1.5mL,低温下重氮化3小时,然后将重氮化溶液加入到90度的水中,水解半小时,水解液用乙酸乙酯20mL提取两次,合并乙酸乙酯层,干燥脱色,抽滤,回收乙酸乙酯,产品用柱层析纯化,洗脱剂PE:EA3:1----1:1.得到产品0.6g。
本发明优点在于:
本发明提供的立达霉中代谢产物16的合成方法,能够获得高纯度的N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸,且该方法获得的代谢产物具有较好的回收率,具有非常可观的应用前景。
附图说明
附图1是立达霉代谢产物16:N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的核磁共振氢谱。
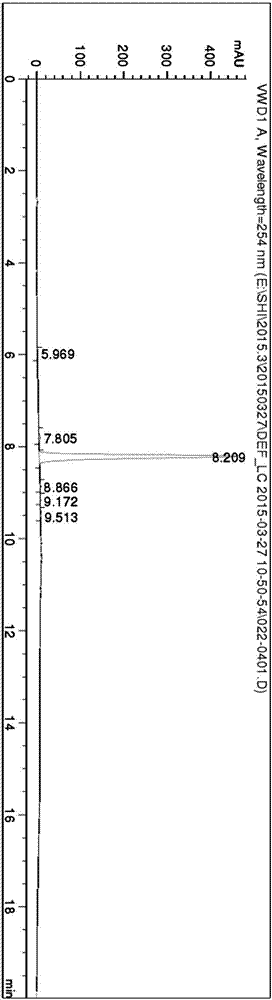
附图2是立达霉代谢产物16:N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的HPLC图谱。
具体实施方式
下面结合实施例对本发明提供的具体实施方式作详细说明。
本发明提供一种立达霉代谢产物16的合成方法,包括以下步骤:
1)取立达霉(Ⅰ)为原料药,溶解于酸溶液中,得到化合物(Ⅱ)。
2)将化合物(Ⅱ)加还原剂还原得到化合物(Ⅲ)。
3)化合物(Ⅲ)酸化,得到化合物(Ⅳ)。
4)化合物(Ⅳ)再加入碱溶液得到代谢产物16(Ⅴ)。
以下为反应步骤:
下列实施例中未具体注明的工艺设备或装置均采用本领域内的常规设备或装置;所有压力值和范围都是指相对压力。以下实施例中使用的试剂如硝酸、硫酸、二氯甲烷、无水硫酸钠、活性炭、洗脱剂PE、EA、甲醇、Pd、碳、氢气、亚硝酸钠、重氮化溶液、乙酸乙酯、乙腈等均为本领域常规使用的试剂。以下实施例中使用的立达霉为甲霜灵分析标准品,由国药集团化学试剂有限公司生产。薄层色谱板(TLC)、高效液相色谱分析仪(HPLC)、核磁共振氢谱仪(HNMR)均为本领域常规使用的仪器。
此外应理解,本发明中提到的一个或多个方法步骤并不排斥在所述组合步骤前后还可以存在其他方法步骤或在这些明确提到的步骤之间还可以插入其他方法步骤,除非另有说明;还应理解,本发明中提到的一个或多个设备/装置之间的组合连接关系并不排斥在所述组合设备/装置前后还可以存在其他设备/装置或在这些明确提到的两个设备/装置之间还可以插入其他设备/装置,除非另有说明。而且,除非另有说明,各方法步骤的编号仅为鉴别各方法步骤的便利工具,而非为限制各方法步骤的排列次序或限定本发明可实施的范围,其相对关系的改变或调整,在无实质变更技术内容的情况下,当亦视为本发明可实施的范畴。
实施例1
将65%硝酸10mL和98%浓硫酸30mL在0度左右混合得到混酸待用。250mL三口瓶中加入二氯甲烷150mL,再加入27.9g甲霜灵搅拌溶解透明,在0度左右滴加上述混酸,保持温度在5度左右。滴加完毕搅拌过夜,原料反应完全后加入冰水中分层,水层用二氯甲烷50mL提取两次,合并二氯甲烷层,用水洗两次,二氯甲烷层无水硫酸钠干燥活性炭脱色,抽滤,滤液回收二氯甲烷,得到固体26g。固体用柱层析纯化,洗脱剂PE:EA5:1----3:1,得到产品10g,取5g上述产品加入50mL甲醇溶解,加入10%Pd/C0.5g,通入氢气氢化还原过夜得到氨基化合物3g,3g产品溶解到20%硫酸溶液中,然后冷却到5度,滴加亚硝酸钠溶液1.5mL,低温下重氮化3小时,然后将重氮化溶液加入到90度的水中,水解半小时,水解液用乙酸乙酯20mL提取两次,合并乙酸乙酯层,干燥脱色,抽滤,回收乙酸乙酯,产品用柱层析纯化,洗脱剂PE:EA3:1----1:1.得到产品0.6g。总回收率为10%。
所述摩尔收率的计算公式(i)如下。
收率=(目的产物生成量/关键组分起始量)×100%(i)
计算公式(i)中,根据原料的分子量算出摩尔量,然后在根据摩尔量和产物的分子量算出产物的理论量,实际量除以理论量就是收率。
实施例2
通过高效液相色谱法和核磁分析的方法对代谢物的纯度进行鉴定。
高效液相色谱法:取5mg样品加乙腈溶解,并滴加少量含有0.1%三氟乙酸的乙腈,进样10μL。色谱条件:Waters Xbridge C18柱(4.6*250*5μm),流动相:乙腈/水=40/60(v/v),流速:1mL/min,检测波长:220nm,温度:35℃。
核磁分析方法:取样品5mg加入合适的氘代试剂0.5mL,超声5分钟,溶解均匀澄清后进入核磁共振仪器,氢谱扫谱次数64次。
将实施例1中获得的代谢产物16分别进行HNMR和HPLC检测。
其中,如附图1所示,通过HNMR检测可知,产品在氘代氯仿中不溶解,所以换用氘代试剂(DMSO-d6)溶剂残留峰为2.5ppm和3.3ppm峰形列分不好。0.89ppm为与次甲基相连的甲基的化学位移,1.91-2.22ppm为芳环中甲基的位移,3.4-3.5ppm处为甲醚中甲基化学位移和甲醚与羰基相连的亚甲基中两个氢的重叠区,共计5个氢,4.26ppm处为与甲基相连的次甲基的化学位移。6.8-6.9ppm处双重峰为芳环中两个氢化学位移,9.4ppm处为酚羟基的化学位移,12.4ppm处为羧酸化学位移,可见在DMSO中虽然峰形列分不好,但是活泼氢都能清楚的看到,而且从图谱上看不到明显的杂质峰,可以判定立达霉代谢物16:N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的纯度大于95%。
如附图2所示,通过HPLC检测,采用HPLC面积归一法测定获得代谢产物立达霉代谢物16:N-(3-羟基-2,6-二甲基苯基)-N-(甲氧乙酰基)-丙氨酸的保留时间约为8.21min,HPLC面积归一法也表明含量为98.5%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!