一种5‑氨基水杨酸药物共晶及其制备方法与流程
本发明涉及药物共晶技术领域,具体地说是一种5-氨基水杨酸与马来酸、5-氨基水杨酸与2,6-二羟基苯甲酸或5-氨基水杨酸与4-氨基吡啶药物共晶及其制备方法。
背景技术:
晶体工程是根据超分子化学的原理、方法以及控制分子间作用于晶体,用于设计和制造出奇特新颖、花样繁多、具有特定物理性质和化学性质的晶体。这些晶体通常是多维的、在空间上无限伸展的超分子聚集体,分子构筑单元在固态的排列状态决定了晶体材料的物理和化学性质。由此,通过选择合适的分子构筑基元,掌握这些分子间可采取的相互作用(例如配位键、氢键)的信息,使分子组分间的功能得到多方面配合,优化分子间不同强度的、定向的各种相互作用,就有可能得到具有特定构型和物理化学性质的材料,从而实现目标可控合成。
目前,药物共晶在修饰药物活性成分(activephaimaceuticalingredient,api)理化性质方面的应用近年来备受关注。药物活性分子(api)通常因含有各种官能团而具有不同的生物活性,这些官能团能够利用氢键或者其他非共价键作用而与其他有机分子通过分子间的识别作用生成超分子化合物,即药物共晶,而被引入的有机分子,被称为共晶试剂。共晶试剂的引入,可以改善药物本身的结晶性能、物化性质及药效,是药物固体试剂的一个新选择。常温下,药物活性成分(api)以多种固体形态存在,如多晶、溶剂化物、水合物、共晶、无定形以及盐等。一个药物的疗效很大程度上取决于api自身的理化性质及其选择的剂型,而api不同的固体形态对药物的溶解度、稳定性、溶出速率和生物利用度等均有影响。api固体形态的研究已成为新药开发中的一个重要方面,可为最佳药物固体形态的确定提供选择依据。药物共晶中各组分可以是中性分子,因而药物共晶可涵盖所有的api,包括酸、碱和非离子化分子,并可用来配合api形成共晶的分子很多,这些物质可能包括食品添加剂、防腐剂、药用辅料、维生素、矿物质、氨基酸以及其他活性分子,甚至其他的api,因此,对于给定的药物,可以生成数以百计的药物共晶,延长原有药物的市场周期,具有广阔的应用前景。
5-氨基水杨酸是一种用于治疗结核病的抗菌药,也被用于治疗炎症性肠病,通过抑制nf-κb及清除自由基而发挥作用,主要用于治疗结核病。同时5-氨基水杨酸是重要的医药和染料化工原料,也是一种治疗慢性结肠炎药物柳氮磺吡啶的主要原料,其本身也具有相似的治疗作用,临床上以5-氨基水杨酸(5-asa)为主要成分的各种制剂。马来酸又称顺丁烯二酸,它主要用于制造不饱和聚酯树脂和生产酒石酸、富马酸、琥珀酸、dl-苹果酸、染色助剂和油脂防腐剂等化合物,在医药、农药、食品等方面也有着比较广泛的应用。马来酸已成为食品饮料工业中的新型酸味剂。食品、饮料中添加适量马来酸可增强特殊果香味并改善口感。目前马来酸主要用于果汁、即饮茶、橘子汁、运动饮料及其他各种强化果味饮料与食品。2,6-二羟基苯甲酸和4-氨基吡啶是农药和医药中的重要中间体。
现有技术的药物共晶,操作影响因素多,对于成本和技术的要求都较高,而且产物易受溶剂影响,得到高纯度的晶体不太容易。
技术实现要素:
本发明的目的是针对现有技术的不足而提供的一种5-氨基水杨酸药物共晶及其制备方法,采用甲醇与丙酮混合溶剂的室温挥发,将5-氨基水杨酸与马来酸、2,6-二羟基苯甲酸或4-氨基吡啶通过氢键结合,得到三斜晶系p1空间群的5-氨基水杨酸与马来酸、2,6-二羟基苯甲酸或4-氨基吡啶药物共晶,合成选用的溶剂沸点较低,制备方法简便,不但保留了5-氨基水杨酸的药物特质外,而且溶解性有明显提高。
实现本发明目的的具体技术方案是:一种5-氨基水杨酸药物共晶,其特点是以5-氨基水杨酸作为药物活性成分,马来酸、2,6-二羟基苯甲酸或4-氨基吡啶为反应物,将药物活性成分与反应物研磨后在甲醇和丙酮的混合溶剂中,采用溶剂室温挥发法,使5-氨基水杨酸与马来酸、2,6-二羟基苯甲酸或4-氨基吡啶通过氢键结合,构成5-氨基水杨酸与马来酸、2,6-二羟基苯甲酸或4-氨基吡啶的药物共晶基元,所述5-氨基水杨酸与马来酸药物共晶为三斜晶系
所述5-氨基水杨酸与马来酸通过氢键结合是以5-氨基水杨酸分子中的n(n1)和o(o2)原子作为氢键的给体,马来酸分子中的o(o4)、o(o5)、o(o6)和o(o7)原子作为氢键的受体,所形成的五个分子间氢键为:[n1-h1a…o6(-x,-y+1,-z+1),
所述5-氨基水杨酸与2,6-二羟基苯甲酸通过氢键结合是以5-氨基水杨酸分子中的o(o2)、o(o5)、n(n1)和n(n2)原子作为氢键的给体,2,6-二羟基苯甲酸分子中的o(o7)、o(o8)、o(o10)原子以及水分子中的o(o3w)原子作为氢键的受体,所形成的五个分子间氢键为:[o2-h2b…o3w,
所述5-氨基苯甲酸与4-氨基吡啶通过氢键结合是以5-氨基苯甲酸中的n(n3)、n(n6)原子和4-氨基吡啶中的n(n1)、n(n4)原子作为氢键给体,5-氨基苯甲酸中的o(o2)、o(o4)、o(o5)、n(n3)原子和4-氨基吡啶中的n(n1)原子作为氢键受体,所形成的七个分子间氢键为:[n1-h1b…o2(-x,-y+1,-z),
一种5-氨基水杨酸药物共晶的制备方法,其特点是将5-氨基水杨酸与马来酸、2,6-二羟基苯甲酸或4-氨基吡啶混合,研磨均匀后加入甲醇研磨至溶剂挥干,然后在甲醇/丙酮混合溶剂中超声至溶解完全,静置3~10天,析出的块状淡黄色透明晶体,为5-氨基水杨酸与马来酸、5-氨基水杨酸与2,6-二羟基苯甲酸或5-氨基水杨酸与4-氨基吡啶药物共晶,所述5-氨基水杨酸与马来酸、2,6-二羟基苯甲酸或4-氨基吡啶、甲醇和甲醇/丙酮混合溶剂的重量体积比为:1mg:0.5~8mg:5~30μl:2~5ml;所述甲醇/丙酮混合溶剂中甲醇与丙酮的体积比为:0.5~2:1。
本发明与现有技术相比具有制备方法简单、可控,合成周期短,重复性好,合成的5-氨基水杨酸药物共晶,保留了5-氨基水杨酸的药物特质外,溶解性有明显提高,延长原有药物的市场周期,具有广阔的应用前景。
附图说明
图1为5-氨基水杨酸与马来酸药物共晶结构单元示意图:
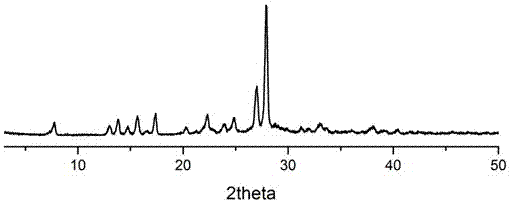
图2为5-氨基水杨酸与马来酸药物共晶的xrd图谱;
图3为5-氨基水杨酸与马来酸药物共晶热重图谱;
图4为5-氨基水杨酸与马来酸药物共晶在水溶液中的溶解曲线图;
图5为5-氨基水杨酸与马来酸药物共晶在乙醇溶液中的溶解曲线图;
图6为5-氨基水杨酸与2,6二羟基苯甲酸药物共晶结构单元示意图:
图7为5-氨基水杨酸与2,6二羟基苯甲酸药物共晶的xrd图谱;
图8为5-氨基水杨酸与2,6二羟基苯甲酸药物共晶热重图谱;
图9为5-氨基水杨酸与2,6二羟基苯甲酸药物共晶在水溶液中的溶解曲线图;
图10为5-氨基水杨酸与2,6二羟基苯甲酸药物共晶在乙醇溶液中的溶解曲线图;
图11为5-氨基水杨酸与4-氨基吡啶药物共晶结构单元示意图:
图12为5-氨基水杨酸与4-氨基吡啶药物共晶的xrd图谱;
图13为5-氨基水杨酸与4-氨基吡啶药物共晶热重图谱;
图14为5-氨基水杨酸与4-氨基吡啶药物共晶在水溶液中的溶解曲线图;
图15为5-氨基水杨酸与4-氨基吡啶药物共晶在乙醇溶液中的溶解曲线图;
具体实施方式
本发明以5-氨基水杨酸(c7h7o3n)为药物活性分子,马来酸(c4h4o4)为反应物,其5-氨基水杨酸与马来酸药物共晶的合成按如下结构反应式进行:
本发明以5-氨基水杨酸(c7h7o3n)为药物活性分子,2,6-二羟基苯甲酸(c7h6o4)为反应物,其5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶的合成按如下结构反应式进行:
本发明以5-氨基水杨酸(c7h7o3n)为药物活性分子,4-氨基吡啶(c5h6n2)为反应物,其5-氨基水杨酸与4-氨基吡啶药物共晶的合成按如下结构反应式进行:
以下通过具体实施例对本发明作进一步的详细说明。
实施例1
称取30mg5-氨基水杨酸与68.1mg马来酸置于玛瑙研钵中,研磨均匀后加入20μl甲醇,迅速研磨至溶剂挥干,然后将混合物移至玻璃试管中,加入2ml丙酮与2ml甲醇的混合溶剂,超声至溶解完全,封住试管口并扎小孔静置挥发6天,试管底部开始析出晶体,得到淡黄色透明块状晶体46mg为5-氨基水杨酸与马来酸药物共晶,其产率为89%。
实施例2
称取30mg5-氨基水杨酸与30.2mg2,6-二羟基苯甲酸置于玛瑙研钵中,研磨均匀后加入20μl甲醇,迅速研磨至溶剂挥干,然后将混合物移至玻璃试管中,加入2ml丙酮与2ml甲醇的混合溶剂,超声至溶解完全,封住试管口并扎小孔静置挥发3天,试管底部开始析出晶体,得到淡黄色透明块状晶体50mg为5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶,其产率为83%。
实施例3
称取30mg5-氨基水杨酸与18.4mg4-氨基吡啶置于玛瑙研钵中,研磨均匀后加入20μl甲醇,迅速研磨至溶剂挥干,然后将混合物移至玻璃试管中,加入2ml丙酮与2ml甲醇的混合溶剂,超声至溶解完全,封住试管口并扎小孔静置挥发5天,试管底部开始析出晶体,得到淡黄色透明块状晶体27mg为5-氨基水杨酸与4-氨基吡啶药物共晶,其产率为56%。
上述各实施例制备的药物共晶采用下述仪器对其进行共晶结构及其性能的检测:
(1)、共晶结构由x-射线单晶衍射仪测定,全称为brukersmartapexⅱdiffractometer,mo-kα
(2)、pxrd衍射是由粉末衍射仪测定,型号为rigaku(d/max-ultimaⅳ)diffractometer,cu-kα
(3)、热重曲线是由tg-dsc分析仪测试的,型号是netzschsta449f3,氮气气氛下,升温速率10℃/min。
参阅附图1,实施例1制备的5-氨基水杨酸与马来酸药物共晶经检测,其共晶分子中的5-氨基水杨酸分子与马来酸分子通过氢键结合,构成5-氨基水杨酸药物共晶的基本单元。其中,5-氨基水杨酸分子中的n(n1)、o(o2)原子作为氢键的给体,马来酸分子中的o(o4)、o(o5)、o(o6)、o(o7)原子作为氢键的受体,所形成的五个分子间氢键为:[n1-h1a…o6(-x,-y+1,-z+1),
所述5-氨基水杨酸与马来酸的药物共晶为三斜晶系
参阅附图2,从x-射线衍射峰中可以看出,实施例1制备的5-氨基水杨酸与马来酸药物共晶的特征峰值出现在:7.76°、13.82°、14.68°、15.74°、16.48°、17.4°、20.28°、21.18°、22.32°、23.92°、24.8°、27.02°、27.96°、31.24°、32.92°、33.08°、33.2。、38.2°、38.96°和40.02°。
参阅附图3,实施例1制备的5-氨基水杨酸与马来酸药物共晶在氮气氛围测试条件下,其热重曲线在141~226℃开始分解,共失重57%。
对实施例1制备的5-氨基水杨酸与马来酸药物共晶进行如下溶解度测试:
(1)、称取52.7mg5-氨基水杨酸与马来酸药物共晶(由单晶结构得到共晶中5-氨基水杨酸和马来酸的计量比为1:1,因此52.7mg共晶中5-氨基水杨酸的质量为30mg,已保证5-氨基水杨酸的量与空白组相同)作为实验组分别溶解于4ml水和4ml乙醇中,配得共晶溶液。
(2)、称取5-氨基水杨酸30mg分别溶解于4ml水和4ml乙醇中,5-氨基水杨酸溶液作为空白溶液作对比。
(3)、用紫外分光光度计分别测定共晶水溶液,共晶乙醇溶液,5-氨基水杨酸水溶液,5-氨基水杨酸乙醇溶液的吸收情况,通过和空白溶剂的最大吸收峰对比,观察药物形成药物共晶后其溶解度的变化情况。
参阅附图4,图中a曲线为药物共晶的紫外吸收曲线,b曲线为空白溶液的紫外吸收曲线,从实验结果可以看出,药物共晶在水溶液中的溶解度相较于空白溶液明显增大。
参阅附图5,图中a曲线为药物共晶的紫外吸收曲线,b曲线为空白溶液的紫外吸收曲线,从实验结果可以看出,药物共晶在乙醇溶液中的溶解度相较于空白溶液明显增大。
参阅附图6,实施例2制备的5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶经检测,其共晶分子中的5-氨基水杨酸分子与2,6-二羟基苯甲酸分子以及水分子通过氢键结合,构成5-氨基水杨酸药物共晶的基本单元。其中,5-氨基水杨酸分子中的o(o2)、o(o5)、n(n1)、n(n2)原子作为氢键的给体,2,6-二羟基苯甲酸分子中的o(o7)、o(o8)、o(o10)原子以及游离水分子中的o(o3w)原子作为氢键的受体,所形成的五个分子间氢键为:[o2-h2b…o3w,
所述5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶为三斜晶系
参阅附图7,从x-射线衍射峰中可以看出,实施例2制备的5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶的特征峰值出现在:11.84°、12.4°、14.6°、15.24°、19.32°、20.7°、22.04°、23.5°、23.82°、24.9°、26.04°、27.12°、27.72°和34.54°。
参阅附图8,实施例2制备的5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶在氮气氛围测试条件下,其热重曲线在163~218℃开始分解,共失重37%。
对实施例2制备的5-氨基水杨酸与2,6-二羟基苯甲酸药物共晶进行如下溶解度测试:
(1)、称取60.2mg5-氨基水杨酸与2,6二羟基苯甲酸药物共晶(由单晶结构得到共晶中5-氨基水杨酸和2,6-二羟基苯甲酸的计量比为1:1,因此60.2mg共晶中5-氨基水杨酸的质量为30mg,已保证5-氨基水杨酸的量与空白组相同)作为实验组分别溶解于4ml水和4ml乙醇中,配得共晶溶液。
(2)、称取5-氨基水杨酸30mg分别溶解于4ml水和4ml乙醇中,5-氨基水杨酸溶液作为空白溶液作对比。
(3)、用紫外分光光度计分别测定共晶水溶液,共晶乙醇溶液,5-氨基水杨酸水溶液,5-氨基水杨酸乙醇溶液的吸收情况,通过和空白溶剂的最大吸收峰对比,观察药物形成药物共晶后其溶解度的变化情况。
参阅附图9,图中a曲线为药物共晶的紫外吸收曲线,b曲线为空白溶液的紫外吸收曲线,从实验结果可以看出,药物共晶在水溶液中的溶解度相较于空白溶液明显增大。
参阅附图10,图中a曲线为药物共晶的紫外吸收曲线,b曲线为空白溶液的紫外吸收曲线,从实验结果可以看出,药物共晶在乙醇溶液中的溶解度相较于空白溶液明显增大。
参阅附图11,实施例3制备的5-氨基水杨酸与4-氨基吡啶药物共晶经检测,其共晶分子中的5-氨基水杨酸分子与4-氨基吡啶分子通过氢键结合,构成5-氨基水杨酸药物共晶的基本单元。其中5-氨基苯甲酸中的n(n3)、n(n6)原子和4-氨基吡啶中的n(n1)、n(n4)原子作为氢键给体,5-氨基苯甲酸中的o(o2)、o(o4)、o(o5)、n(n3)原子和4-氨基吡啶中的n(n1)原子作为氢键受体,所形成的七个分子间氢键为:[n1-h1b…o2(-x,-y+1,-z),
所述5-氨基水杨酸与4-氨基吡啶药物共晶为三斜晶系
参阅附图12,从x-射线衍射峰中可以看出,实施例3制备的5-氨基水杨酸与4-氨基吡啶药物共晶的特征峰值出现在:7.86°、15.92°、17.5°、20.3°、21.28°、22.48°、24.02°、24.9°、27.12°、28.06°和32.34°。
参阅附图13,实施例3制备的5-氨基水杨酸与4-氨基吡啶药物共晶在氮气氛围测试条件下,其热重曲线在216~275℃开始分解,共失重89%。
对实施例3制备的5-氨基水杨酸与4-氨基吡啶药物共晶进行如下溶解度测试:
(1)、称取48.4mg5-氨基水杨酸与4-氨基吡啶药物共晶(由单晶结构得到共晶中5-氨基水杨酸和4-氨基吡啶的计量比为1∶1,因此48.4mg共晶中5-氨基水杨酸的质量为30mg,已保证5-氨基水杨酸的量与空白组相同)作为实验组分别溶解于4ml水和4ml乙醇中,配得共晶溶液。
(2)、称取5-氨基水杨酸30mg分别溶解于4ml水和4ml乙醇中,5-氨基水杨酸溶液作为空白溶液作对比。
(3)、用紫外分光光度计分别测试共晶水溶液,共晶乙醇溶液,5-氨基水杨酸水溶液,5-氨基水杨酸乙醇溶液的吸收情况,通过和空白溶剂的对比得到药物共晶对于药物溶解度的改变状况。
参阅附图14,图中a曲线为药物共晶的紫外吸收曲线,b曲线为空白溶液的紫外吸收曲线,从实验结果可以看出,药物共晶在水溶液中的溶解度相较于空白溶液明显增大。
参阅附图15,图中a曲线为药物共晶的紫外吸收曲线,b曲线为空白溶液的紫外吸收曲线,从实验结果可以看出,药物共晶在乙醇溶液中的溶解度相较于空白溶液明显增大。
本发明制备的药物共晶不但具有5-氨基水杨酸药物特质外,而且溶解性有明显提高,延长原有药物的市场周期。以上各实施例只是对本发明做进一步说明,并非用以限制本发明专利,凡为本发明等效实施,均应包含于本发明专利的权利要求范围之内。
- 还没有人留言评论。精彩留言会获得点赞!