β‑1,4‑异木聚糖、其硫酸化衍生物及其制备方法和用途与流程
本发明涉及一种中草药多糖及其制备方法,更具体地说,涉及一种铁皮石斛β-1,4-异木聚糖、其硫酸化衍生物以及它们的制备方法及所述硫酸化衍生物在抗血管生成方面的应用。
背景技术:
道家经典《道藏》曾把铁皮石斛列为“中华九大仙草之首”,主要分布在浙江、安徽、云南等地。它的干燥茎具有滋阴养胃,增强免疫力,调节血糖水平等的功效。早期研究表明,多糖是一种重要的活性物质,多糖的含量也间接反映药材品质。因此,对石斛多糖的定性研究不仅可以为石斛品种鉴别提供依据还可以规范中药市场。
前期研究主要集中在水提多糖,因为铁皮石斛多糖的分离纯化及结构解析仍然面临很多挑战,迄今为止从铁皮石斛中只获得少数水提均一多糖。然而,对铁皮石斛碱提多糖知之甚少。作为碱提多糖中重要的组成成分之一的木聚糖是一种重要的复杂多糖,不同来源的木聚糖之间也存在着很大的区别。因此,它的结构也可以作为品种鉴定的参考指标之一。更重要的是,功能性木聚糖衍生物作为新一代的生物制品具有广泛的药理活性。与其它衍生物相比,硫酸化多糖则具有更强的生物活性。硫酸化多糖具有多种生物活性,如抗凝、抗补体、抗肿瘤和抗血管生成等活性。
血管生成在机体中发挥着重要的作用。研究表明,血管生成不直接引发肿瘤,但是可以促进恶性肿瘤发展和转移。血管生成虽然有益于机体器官的生长和修复,但是也可以为肿瘤细胞的生长提供条件并引起恶性疾病和炎症。如今,越来越多的证据表明,抗血管生成的化合物也可以阻止肿瘤的发展。抗血管生成方面的研究很有可能改变临床抗肿瘤药物的现状,肿瘤患者也将受益于抗血管生成疗法。在大量生物活性筛选的基础上,出现越来越多抗血管生成化合物。研究发现,硫酸化多糖具有较强的抑制血管生成的活性。虽然一些硫酸化多糖具有良好的生物活性,但同时也表现出不可忽略的细胞毒性。因此,具有合适的取代度,无细胞毒性的抗血管生成硫酸化多糖有望被开发成为抗血管生成类抗肿瘤药物的候选药物,在肿瘤治疗候选药物上方面具有巨大的应用前景。
技术实现要素:
本发明的目的在于提供一种无细胞毒性并且具有抗血管生成活性的药物。
一方面,本发明提供一种β-1,4-异木聚糖(下文中称为:s32),其结构式如下:
其中,rx为α-l-araf-(1→4)-α-d-glcp-(1→;β-d-xylp-(1→;α-l-araf-(1→,或β-d-galp-(1→3)-α-l-rhap-(1→,n=3-42,并且优选地,n=19-21。
其中,所述β-1,4-异木聚糖的尖峰分子量为:3.7×104da。
另一方面,本发明提供所述β-1,4-异木聚糖的制备方法,该方法包括以下步骤:
a.多糖提取:铁皮石斛干燥茎经脱脂后,用碱提取,经醇沉洗涤后得到粗多糖doa;
b.多糖纯化:得到的粗多糖doa经阴离子交换柱分离、去除淀粉后,经凝胶柱纯化得到s32。
具体地,所述方法包括以下步骤:
a.多糖提取:铁皮石斛干燥茎经95%乙醇浸泡脱脂,风干,粉碎后沸水反复提取10次。过滤后残渣用浓度为0.1-2m,优选的浓度值为1m的氢氧化钠溶液提取,滤液用盐酸中和后浓缩,透析,透析内液经离心,5倍体积的95%乙醇沉淀,离心后经无水乙醇和丙酮交替洗涤3次后干燥得到铁皮石斛碱提粗多糖doa;
b.多糖纯化:取铁皮石斛碱提粗多糖,去离子水溶解,离心,上清液经deaesepharosefastflow阴离子交换柱分离;采用硫酸-苯酚法检测,收集0.1m氯化钠洗脱组分doa1;进一步采用α-淀粉酶除去doa1中的淀粉得到doa1a,产物进一步用sephacryltms-300纯化得到s32。
对通过以上方法得到的β-1,4-异木聚糖进行结构鉴定:
经高效凝胶渗透色谱(highperformancegelpermeationchromatography,hpgpc)法测定,s32多糖相对分子质量为3.7×104da。间苯基苯酚法测定结果显示s32的糖醛酸含量为15.0%。采用0.05m三氟乙酸(tfa)将s32部分酸水解后得到次级多糖s3205i。还原前后及水解前后对多糖的单糖组成和甲基化分析。综合对s32多糖、还原的以及部分酸水解的次级多糖的单糖组成,甲基化、红外以及核磁共振数据解析,确定s32含有木糖、4-甲氧基-葡萄糖醛酸、阿拉伯糖、葡萄糖以及痕量的半乳糖和鼠李糖,其对应的比例为62.7:12.3:8.9:8.5:3.7:3.9。s32以1,4-β-d-吡喃木糖为主链结构,c-2位上有分支。分支主要由末端连接的α-l-呋喃阿拉伯糖、4-甲氧基-α-d-吡喃葡萄糖醛酸、少量β-d-吡喃半乳糖-(1→3)-α-l-吡喃鼠李糖、末端连接的β-d-吡喃木糖以及α-l-呋喃阿拉伯糖-(1→4)-α-d-吡喃葡萄糖组成。
再一方面,本发明提供所述β-1,4-异木聚糖的硫酸化衍生物(下文中称为:s32s)的制备方法,该方法包括以下步骤:
1)采用氯磺酸与吡啶制备硫酸化试剂;
2)使制得的硫酸化试剂与s32反应,得到s32s。
在上述步骤1)中,氯磺酸与吡啶的体积比可为1:1-1:5,例如可为1:3。在上述步骤2)中,反应温度可为40-80℃,例如可为60℃。
在一个具体的实施方式中,所述β-1,4-异木聚糖的硫酸化衍生物的制备方法包括以下步骤:向吡啶中逐滴加入氯磺酸制备硫酸化试剂。将s32溶解于甲酰胺中并加入硫酸化试剂使反应。反应结束后用氢氧化钠溶液于冰浴中中和,再用饱和碳酸氢钠透析,接着用去离子水透析。透析内液冷冻干燥后得到淡黄色蓬松硫酸化产物s32s。其中,得到的s32s硫酸化的位点主要为1,4-β-d-吡喃木糖的c-2位或者c-3位,无区域选择性。1,4-β-d-吡喃葡萄糖的硫酸化位点主要发生在c-6位。大部分的4-甲氧基-α-d-吡喃葡萄糖醛酸在c-3位发生了硫酸化取代。少量的1,2,4-β-d-吡喃木糖的c-3位也发生取代。
进一步,本发明提供一种根据如上所述的方法制备的β-1,4-异木聚糖的硫酸化衍生物。
更进一步,本发明提供一种药物组合物,其包含治疗有效量的所述β-1,4-异木聚糖的硫酸化衍生物和药学上可接受的载体。
更进一步,本发明提供所述β-1,4-异木聚糖的硫酸化衍生物在制备抗血管生成药物中的用途。
再进一步,本发明提供所述β-1,4-异木聚糖的硫酸化衍生物在制备用于抑制恶性肿瘤的发展和转移的药物中的用途。
本发明通过以下附图和实施例作进一步阐述,但并不限制本发明的内容。
附图说明
图1为s32的特征高效液相色谱图;
图2为s32的特征红外光谱图;
图3为s32s的特征红外光谱图;
图4为s32的特征13cnmr图谱;
图5为s32的特征hmbc图谱;
图6为s32s与s32的特征dept135nmr图谱,其中,a为s32s的特征dept135nmr图谱,b为s32的特征dept135nmr图谱;
图7为s32及s32s在不同浓度下对人表皮血管内皮细胞hmec-1在基质胶管腔形成抑制作用的示意图;
图8为s32s在不同浓度下对人表皮血管内皮细胞hmec-1迁移抑制作用的示意图;
图9为s32s在不同浓度以及不同处理时间下对lo2细胞(图9a)和hmec-1细胞(图9b)生长的影响。
具体实施方式
下面结合具体实施例进一步阐述本发明,但这些实施例仅用于说明本发明,而不用于限制本发明的范围。
在下文中,术语“原糖”指的是未经衍生化的多糖。
实施例1:用碱提取铁皮石斛制备s32
1、多糖提取
铁皮石斛干燥茎1.5kg,用95%的乙醇脱脂2周后风干。铁皮石斛干燥茎经粉碎后用沸水提取10次(20l/次),每次提取4h,至硫酸-苯酚法检测无明显反应。滤渣用1mnaoh于冰浴中提取(12.5l×2),不时搅拌。3h后用2mhcl中和,合并滤液并浓缩至10l,用流水透析2天。透析内液浓缩至4l,离心。向上清液中边搅拌边加入5倍体积的95%乙醇,静置过夜。离心后沉淀用无水乙醇和丙酮交替洗涤3次后,得铁皮石斛粗多糖14.0g。
2、多糖纯化
将上述制备的铁皮石斛粗多糖5.0g溶解于80ml去离子水中,以4000r/min离心10min除去不溶物,上清液上样于deaesepharosefastflow弱阴离子交换柱,依次用水,0.1m、0.2mnacl溶液洗脱,收集器收集洗脱液。硫酸-苯酚法检测并绘制洗脱曲线,根据洗脱曲线收集0.1mnacl溶液洗脱部分。洗脱液经浓缩、透析、冷冻干燥后得到初步纯化的多糖doa1。取doa1溶解于磷酸盐缓冲液中(0.05m,ph=6.0)于37℃加入淀粉酶(底物和酶的浓度均为10mg/ml),反应1h后,100℃、5min灭活,离心后取上清液浓缩、透析、冷冻干燥后得doa1a。取200mgdoa1a溶解于3ml去离子水中,4000r/min离心10min。取上清液上样于sephacryltms-300凝胶柱,以0.2mnacl洗脱并控制流速0.3ml/min。硫酸-苯酚法检测并绘制洗脱曲线,根据洗脱曲线收集洗脱液,将洗脱液浓缩、透析、冷冻干燥后得到纯化的多糖s32。
s32在串连ultrahydrogeltm2000和ultrahydrogeltm500多糖分析凝胶柱上的特征图谱如图1所示(使用agilent1260液相色谱仪,色谱条件为:流动相:0.1mnano3;流速:0.5ml/min;柱温:25℃。检测器:示差检测器和紫外检测器)。
实施例2:s32的结构鉴定
1、多糖的部分酸水解
取s32400mg溶解于0.05m的三氟乙酸中,密封后于100℃水解1h。反应完毕后,去离子水透析(1l×5),透析内液浓缩、冷冻干燥得到部分酸水解产物s32-05i。
2、糖醛酸含量测定(间苯基苯酚法)
a.试剂
试剂a:取硼砂(nab4o7·10h2o)1.1925g,加入浓硫酸40ml,搅拌溶解(可稍加热),冷却后,用浓硫酸定容至250ml。
试剂b:取0.25gnaoh,加适量水溶解,定容至50ml,加入间苯基苯酚75mg,搅拌溶解。
糖醛酸标准溶液(150μg/ml):取葡萄糖醛酸7.5mg,溶于水中,加去离子水定容至50ml。
样品溶液(150μg/ml):取样品按上述标准溶液方法配制。
b.方法
表1
注:以上表1中,1-5号管中为标准液,6号管中为样液。
取10ml试管,按表1中的量加入标准溶液/样液以及水,冰浴下加入试剂a,摇匀,100℃水浴5min,然后在冰水中冷却,加入试剂b,混合均匀,室温放置15min,测定od520。
检测结果:由上述方法测得s32中的糖醛酸含量为15.0%。由此推测,该多糖含有糖醛酸,其含量约为15.0%。
3、红外光谱分析
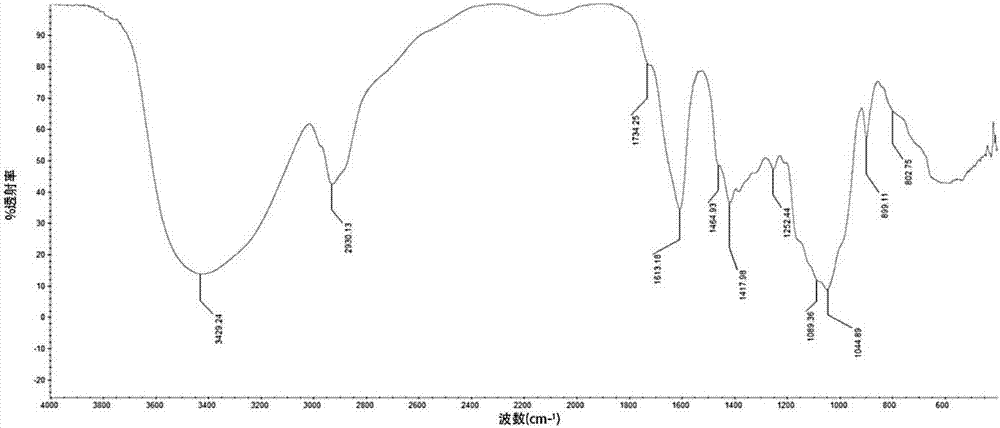
取s32多糖样品约2mg,采用溴化钾压片法测定原糖s32及其硫酸化衍生物s32s的红外光谱,结果如图2和图3。
4、糖醛酸还原(conrad法)
a)多糖样品40mg溶于40ml去离子水中,搅拌状态下加入cmc600mg,ph计监控下滴加0.01mhcl维持ph值在4.75,反应2.5小时。
b)滴加硼氢化钠溶液(2m)16ml,其间用4m盐酸控制ph值在7.0左右。硼氢化钠于45min内加完。硼氢化钠加完后,室温继续反应2小时(ph=7.0)。
c)反应液用去离子水透析2天,2l×6。透析袋内液减压浓缩,冻干。
5、nmr分析
取多糖s3230mg,加d2o0.45ml溶解,加2.5μl丙酮为内标(δh=2.23ppm,δc=31.5ppm),于brukeravanceiii500m核磁共振仪上,25℃分别测定一维和二维核磁共振谱,并参考核磁图谱对s32以及s32s进行结构确证,结果如图4,图5、图6a和图6b所示。
6、s32结构综合解析
hpgpc法分析表明s32为均一多糖,分子量为3.7×104da。
对s32还原前后糖组成、糖醛酸含量测定以及13cnmr图谱的综合分析表明s32含有木糖、4-甲氧基-葡萄糖醛酸、阿拉伯糖、葡萄糖以及痕量的半乳糖和鼠李糖,其对应的比例为62.7:12.3:8.9:8.5:3.7:3.9。对s32原糖以及水解和还原后的多糖进行甲基化分析,综合分析结果可知s32的连接方式主要有含有1,4-木糖(42.8%)、1,2,4-木糖(21.4%)、1,4-葡萄糖(5.4%)、末端连接的4-甲氧基-葡萄糖醛酸(15.1%)、末端连接的阿拉伯糖(6.2%),极少量的末端半乳糖(1.6%),1,3-鼠李糖(2.0%)和末端连接的木糖(5.5%)。
红外图谱(图2)显示,3429.24cm-1为o-h伸缩振动吸收峰,2930cm-1为c-h伸缩振动吸收峰,1464.93–1044.89cm-1附近为c-o和糖环振动信号,1734.25cm-1为羧基c=o伸缩振动,表明该多糖含有糖醛酸。
13cnmr谱中(图4),位于109.63ppm的端基碳信号,可归属为末端α-l-呋喃阿拉伯糖的c-1信号。位于103.03ppm和102.88ppm的碳信号均属于不同化学环境下的1,4-β-d-吡喃木糖的c-1信号。102.59ppm处的端基碳信号归属于1,2,4-β-d-吡喃木糖的c-1信号。1,4-α-d-吡喃葡萄糖的c-1信号则可归属到101.26ppm处的端基碳信号。105.00ppm处的端基碳信号则可归属为末端的β-d-半乳糖的c-1信号,99.79ppm处的信号峰则归属为1,3-α-l-吡喃鼠李糖的c-1信号,其甲基碳的信号峰则出现在18.05ppm处。而末端的木糖的c-1信号则被102.4ppm处的信号峰掩盖。末端连接的4-甲氧基-α-d-葡萄糖醛酸的c-1信号则可在98.78ppm处观察到,而位于61.10ppm的甲氧基碳信号以及177.74ppm的羧酸c=o信号则进一步证实了甲氧基和糖醛酸的存在。
在hmbc图谱中(图5),以a,b、c、d、e、f、g和h分别代表1,4-β-d-吡喃木糖,1,2,4-β-d-吡喃木糖、末端连接的4-甲氧基-α-d-吡喃葡萄糖醛酸、1,4-β-d-吡喃葡萄糖、末端连接的α-l-呋喃阿拉伯糖、β-d-吡喃半乳糖、末端连接的β-d-吡喃木糖和1,3-连接的-α-l-吡喃鼠李糖。远程相关峰109.63/3.43ppm、102.40/3.43ppm、101.26/3.43ppm、99.79/3.43ppm和98.78/3.43ppm,分别表示e的c-1和b的h-2远程相关,g的c-1和b的h-2远程相关,d的c-1和b的h-2远程相关,h的c-1和b的h-2远程相关,c的c-1和b的h-2远程相关,说明e、g、d、h、c通过糖苷键与b的c-2相连,而d的c-4(79.16ppm)与e的h-1(5.27ppm)远程相关,说明部分e也通过糖苷键与d的c-4相连。相关峰61.10/3.23ppm和83.59/3.45ppm为-och3的c和4-甲氧基-α-d-吡喃葡萄糖醛酸的h-4远程相关,4-甲氧基-α-d-吡喃葡萄糖醛酸的c-4和-och3的h远程相关信号。进一步证实了甲氧基连接于葡萄糖醛酸的c-4位。综上所述,该多糖是以1,4-β-d-吡喃木糖为主链,其它糖残基为支链直接或者间接连接在主链糖残基的c-2位。
以上结果表明s32的结构为:
rx为α-l-araf-(1→4)-α-d-glcp-(1→;β-d-xylp-(1→;α-l-araf-(1→,或β-d-galp-(1→3)-α-l-rhap-(1→。
实施例3:s32s的制备及结构鉴定
1、s32s的制备
向6ml吡啶中逐滴加入2ml氯磺酸制备成硫酸化试剂。将104mgs32溶于5ml甲酰胺中,搅拌溶解,将上述硫酸化试剂于60℃加入s32多糖溶液中反应3h。反应结束后,于冰浴中加入5m氢氧化钠中和后用饱和碳酸氢钠溶液透析24h后并用去离子水继续透析72h。透析内液浓缩,冷冻干燥后得224mg淡黄色蓬松硫酸化多糖s32s。
2、s32s结构鉴定及解析
1)硫酸基含量测定(氯化钡-明胶法)。
试剂配制:
(1)1mol/lhcl溶液:8.3ml浓盐酸(12n)加水定容至100ml。
(2)氯化钡-明胶试剂:1.25g明胶溶于50ml水中,加热溶解,定容至250ml;取250ml该溶液,加入2.5g氯化钡,溶解后4℃下放置。
(3)3%三氯乙酸溶液(w/v):取三氯乙酸(ar)30.0g溶于水中,加水定容至1000ml。
(4)标准硫酸基溶液:精确称取无水硫酸钠86.7mg,置于100ml容量瓶中,加入1mol/lhcl溶液至刻度。
测定方法:
(1)标准曲线测定:精确量取0~0.2ml标准溶液(每管按0.04ml递增)于具塞试管中,补加1mol/lhcl至体积为0.2ml,分别加入3.8ml3%三氯乙酸和1ml氯化钡-明胶试液,混合后在室温下静置15~20min,以0号(不含标准液)作参比测定在360nm的吸光度od360。
(2)样品测定:精确称取样品4.5mg左右,置于试管中,加入1mol/lhcl4.5ml,封管后,振荡器混匀,加热辅助溶解若干分钟,于100℃烘箱中加热水解2.5h,冷却至室温后取0.2ml进行测定,其余操作同标准曲线各管。
检测结果:由上述方法测得s32s硫酸基的取代度为0.90。由此推算平均每个糖残基单元上连有一个硫酸基团。
2)s32s的结构综合解析
经hpgpc分析s32s的尖峰分子量为5.4×104da。氯化钡-明胶比浊法测得多糖硫酸基取代度约为0.90。s32s红外图谱(图3)与原糖s32红外图谱(图2)对比,1267.26cm-1出现的较强的吸收峰可归属为s=o的特征伸缩振动吸收峰,表明硫酸化成功。图6a为s32s的dept135nmr图谱,对比原糖图谱(图6b),硫酸化的位点主要无区域选择性地发生在1,4-β-d-吡喃木糖的c-2位或者c-3位,1,4-β-d-吡喃葡萄糖的硫酸化位点主要发生在c-6,大部分的4-甲氧基-α-d-吡喃葡萄糖醛酸在c-3位发生了硫酸化取代,少量的1,2,4-β-d-吡喃木糖的c-3位也发生取代。
药理学实施例:
1、s32及s32s抑制hmec-1细胞管腔形成实验
将在含有15%胎牛血清(fbs),2mml-谷氨酰胺,10ng/ml表皮生长因子(egf)的mcdb131培养基中培养的hmec-1细胞置于5%co2培养箱37℃孵育,2-3天换培养液1次。
将基质胶(50μl)均匀涂布于96孔板中并置于37℃培养箱中固化。半个小时后,将50μlhmec-1细胞以5×104/孔的密度接种在96孔板内并加入50μl不同浓度的s32s(调整终浓度为0.14,0.29,0.58μm),以空白组作为对照。12h后采用显微镜在20×物镜下随机挑选3个视野并记录(olympusix51digitalcamera(tokyo,japan))。
由图7可观察到,空白组在单独孵育12h后,细胞变成长梭形并向基质中延伸生长,线形排列形成管状结构,多个管状结构相互连接形成完整三维网状结构。s32对hmec-1细胞管腔形成并无明显抑制作用,而与低浓度(0.14μm)的硫酸化多糖s32s共孵育12h后,管腔形成受到抑制,在浓度0.29μm时,hmec-1细胞基本不形成完整的管腔,管腔形成受到明显抑制。证明硫酸化多糖s32s具有抑制血管生成的活性。
2、s32s抑制hmec-1细胞迁移实验
将hmec-1细胞(5×105细胞/孔)培养于六孔板中。24h后,用100μl的枪头末端在六孔板的中央作划痕。磷酸缓冲溶液(pbs)轻轻吹洗3遍以除去划落的细胞碎片,分别加入含s32s新的mcdb131培养液(调整终浓度依次为0,0.14,0.29,0.58μm),置于培养箱中培养12h后,采用显微镜在20×物镜下记录在不同时间段不同浓度s32s对划痕愈合影响情况。应用image-j软件定量计算细胞迁移的面积。图8可观察到,空白组在培养12h后基本愈合,而s32s实验组可以在不同浓度下均抑制hmec-1细胞的迁移。与空白组细胞迁移率(73.6±2.5%)相比,在浓度为0.14μm时,hmec-1细胞迁移率仅为21.8±2.4%,随着浓度升高到0.29μm和0.58μm,对应的hmec-1的细胞迁移率分别为44.8%(±2.2)及37.2%(±1.5)。综上所述,s32s能在低浓度下抑制hmec-1细胞迁移,但不呈现明显的剂量依赖性。
3、s32s对正常hmec-1细胞/lo2细胞的细胞毒性
将hmec-1细胞/lo2细胞50μl(5×103细胞/孔)种入96孔板内,置于5%co2的培养箱中37℃孵育过夜后加入50μl含有s32s的mcdb131/rpmi1640(含10%fbs,100u/ml青霉素和100μg/ml链霉素)培养液,使之终浓度为0-18.52μm,每个浓度设置三个副孔。继续于37℃、5%co2的培养箱中培养,分别于24h,48h,72h后,每孔加入10μl5mg/mlmtt,继续孵育4h后小心弃去培养液上清。加入150μldmso溶解甲瓒晶体。置于摇床快速摇20分钟使其充分溶解,之后于490nm处测定其吸光度值。记录结果并计算细胞的存活率。
如图9所示,s32s即使在较高浓度(1mg/ml)时,对正常hmec-1细胞和lo2细胞于72h内无明显细胞毒性。
以上药理实验表明s32s在0.29μm的浓度下,在体外可完全抑制血管生成,并在细胞水平上可抑制hmec-1细胞的迁移。细胞毒性实验表明s32s对正常hmec-1和lo2细胞几乎无毒性。因此,s32s有望被开发成为一种抗血管生成类抗肿瘤药物。
最后有必要说明的是,以上实施例仅用于进一步详细地说明本发明的技术方案,不能理解为对本发明保护范围的限制,任何根据本发明的上述内容作出的一些非本质的调整和改进均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!