具有因子VII活性的融合蛋白质的制作方法
本申请是申请号为201180027707.6的中国专利申请的分案申请,原申请是2011年06月07日提交的pct国际申请pct/kr2011/004131于2012年12月04日进入中国国家阶段的申请。本发明涉及具有因子vii(fvii)活性的融合蛋白质;以及,更特别地,涉及包含fvii和转铁蛋白并且与未融合天然型fvii相比具有0.7或更高的fvii比活性(specificactivity)的融合蛋白质、为其编码的dna、包含所述dna的重组载体、以及包含所述重组载体的宿主细胞。
背景技术:
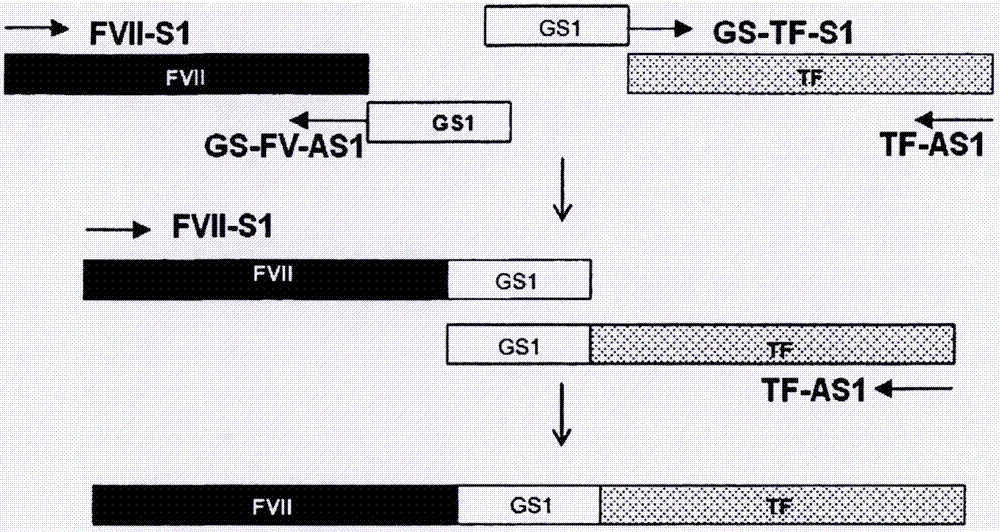
:多种出血性疾病由缺乏凝血因子引发。最常见的疾病是分别由凝血因子viii和ix缺乏或异常引发的血友病a和b。血友病a是由有缺陷的因子viii基因的x连锁隐性特征(x-linkedrecessivetrait)所引起的遗传性出血性疾病。血浆源性或重组的因子viii的浓缩物已被用于治疗血友病a。血友病b是由因子ix缺乏或功能不良而引起的,通过使用血浆源性或重组的因子ix的浓缩物对其进行治疗。但是,出现针对替代因子(replacementfactor)的同种抗体仍是血友病a和b的治疗中严重的医学问题。在高至30%的血友病a患者中产生针对因子viii的抗体。尽管针对因子ix的抗体产生的较少,但它们对免疫耐受诱导治疗较不敏感,导致更严重的结果。凝血由血管壁损伤之后暴露于循环血液的组织因子与激活形式的因子vii(fviia)之间形成复合物而引发。这样的复合物激活因子ix和因子x,并且所产生的因子xa产生有限量的凝血酶。在正反馈环中,凝血酶激活凝血级联的多种因子(例如,因子viii、因子v、因子xi等),并且所激活的因子构成因子x酶复合物(factorxasecomplex)或前凝血素酶复合物。这些复合物还扩增其自身的生成并且产生凝血酶。这种被称为“凝血酶爆发(thrombinburst)”的足够量的凝血酶将出血部位的纤维蛋白原转化为纤维蛋白,从而实现完全止血。但是,在具有针对因子viii或因子ix的高浓度中和抗体的血友病患者的情况中,因为不能产生上述因子x酶复合物,所以达不到足够的止血。fviia已用作主要的治疗剂,用于具有针对因子viii或因子ix的中和抗体的患者,因为fviia可激活因子x(甚至在不存在因子viii和因子ix时),从而最终产生足够量的凝血酶以实现期望的治疗效果。fvii是由406个氨基酸组成的单链糖蛋白,分子量为50kda,并且作为酶原而被分泌到血流中。fvii由四个不同的结构域组成,即,氨基末端-羧基谷氨酸(gla)结构域、两个表皮生长因子(egf)样结构域、以及丝氨酸蛋白酶结构域(hagenfs等,proc.natl.acad.sci.usa,83(8):2412-2416,1986)。经过蛋白水解位于arg152-ile153的单个肽键,通过形成由二硫键连接的两个多肽链(即,n末端轻链(24kda)和c末端重链(28kda)),fvii被转化为其激活形式fviia。fvii以500ng/ml的浓度存在于血浆中,并且1%(即,5ng/ml)的fvii作为fviia而存在。同时,已报道血浆中fvii的半衰期为约4小时(3~6小时),而fviia的半衰期为约2.5小时。由于半衰期短,需要通过多次静脉内注射或连续注射来施用fviia。然而,就高治疗费用和使患者不适而言,这会限制fviia的治疗应用。为了克服这些问题,已提供方法用于制备包含fvii以及与其相连的融合伴侣的融合蛋白质,但是所产生的蛋白质具有丧失其生物活性的问题,尽管与非融合蛋白质相比,较短的体内半衰期有所提高。因此,有需要提供和获得具有提高的体内半衰期同时保持天然型fvii的生物活性的fvii融合蛋白质。技术实现要素:技术问题本发明的一个目的是提供具有天然型fvii的生物活性的融合蛋白质。本发明的另一个目的是提供编码所述融合蛋白质的基因。本发明的另一个目的是提供包含所述基因的重组载体。本发明的另一个目的是提供包含所述重组载体的宿主细胞。问题的解决方案根据本发明的一个方面,提供了包含因子vii(fvii)和转铁蛋白的融合蛋白质,其中所述转铁蛋白与所述fvii的c末端相连接。根据本发明的另一个方面,提供了编码所述融合蛋白质的dna。根据本发明的另一个方面,提供了包含所述dna的重组载体。根据本发明的另一个方面,提供了包含所述重组载体的宿主细胞。发明的有益效果根据本发明的融合蛋白质具有与天然型fvii相比提高的体内半衰期同时保持fvii的高生物活性,因此可有效地用于使用fvii的治疗。附图说明当与附图结合时,通过以下对发明的描述,本发明的上述和其他目的及特征将变得显而易见,所述附图分别示出:图1是示意图,其示出用于由包含编码fvii序列的cdna的载体以及包含编码转铁蛋白(tf)序列的cdna的载体构建fvii-tf表达载体的克隆流程;图2是示意图,其示出用于通过重叠pcr构建fvii-gs1(接头)-tf表达载体的流程;图3是示意图,其示出包含gs3、gs5、gs7、gs9、gs11、gs13、gs15或gs-1-t作为接头的fvii-gs接头-tf表达载体的构建流程;图4显示了本发明的vii-tf、fvii-gs1-tf、fvii-gs3-tf、fvii-gs5-tf、fvii-gs7-tf、fvii-gs9-tf、fvii-gs11-tf、fvii-gs13-tf、fvii-gs15-tf、fvii-gs1-t-tf和fvii-螺旋-tf(fvii-helix-tf)融合蛋白质,fvii-白蛋白融合蛋白质(fvii-alb)和fvii(诺其tm(novoseventm))的western印迹结果;图5是图表,其示出本发明的fvii-tf、fvii-gs1-tf、fvii-gs3-tf、fvii-gs5-tf、fvii-gs7-tf、fvii-gs9-tf、fvii-gs11-tf、fvii-gs13-tf、fvii-gs15-tf、fvii-gs1-t-tf和fvii-螺旋-tf融合蛋白质、以及fvii-白蛋白融合蛋白质(fvii-alb)的比活性(specificactivity);图6呈现接头和fvii-gs1-t-tf融合蛋白质中两个末端处的限制性识别序列的结构;以及图7示出经纯化的本发明fvii-tf、fvii-gs1-tf、fvii-gs1-t-tf、fvii-gs3-tf以及fvii-gs15-tf融合蛋白质,novoseventm和fvii的western印迹结果。具体实施方式发明的最佳实施方式本发明提供包含因子vii(fvii)和转铁蛋白的融合蛋白质。本发明的融合蛋白质的fvii和转铁蛋白可源自任何哺乳动物,优选人。更具体地,本发明中所使用的fvii和转铁蛋白可分别具有与见于人血液中那些天然型蛋白质不少于95%的序列同源性。最优选地,fvii具有seqidno:1的氨基酸序列并且转铁蛋白具有seqidno:2的氨基酸序列。此外,用于本发明的融合蛋白质的fvii或转铁蛋白可以是其天然型的功能等价物或功能衍生物,其具有基本等价的功能活性。示例性的功能等价物包括由分别在seqidno:1和2所示的氨基酸序列中缺失、插入或者非保守性或保守性替换任何氨基酸残基或者其组合所诱导的突变体,其中这样的改变基本不改变提供对fvii的生物活性的活性位点或结构域。在一些情况中,可例如通过磷酸化(phosphorylation)、硫酸化(sulfation)、丙烯酰化、糖基化、甲基化、法尼基化、乙酰化、酰胺化等来修饰本发明的融合蛋白质,用于提高或降低其物理或化学特性,并且只要基本保持fvii的生物活性,这样的功能衍生物也落入本发明的范围之内。在本发明的融合蛋白质中,转铁蛋白优选地与fvii的c末端相连接。采用fvii-转铁蛋白顺序的融合蛋白质优于采用转铁蛋白-fvii顺序的融合蛋白质,可能是由于fvii的n末端的暴露(见表3)。本发明的融合蛋白质还可在fvii与转铁蛋白之间包含限制酶的识别序列以利于插入以下所描述的接头。限制性识别序列可以是本领域技术人员已知的任何限制性识别序列,并且可优选地使用agei识别序列(a/ccggt)。换句话说,融合蛋白质(其中限制性识别序列与fvii的c末端相连接并且转铁蛋白与限制性识别序列相连接)包括在本发明的范围之内。本发明提供在fvii与转铁蛋白之间包含接头的融合蛋白质。所述接头可具有1至100个氨基酸,优选1至75个氨基酸,更优选5至25个氨基酸,并且所述接头可以是可将fvii与转铁蛋白功能性分离的任何肽。所述接头可具有稳定的二级结构(例如螺旋)或者可源自igg铰链区。优选地,所述接头在水溶液中可自由旋转,并且不具有固定结构,因此,所述接头可以是非免疫原性的并且可通过使两个融合伴侣之间的潜在干扰最小化而提高融合蛋白质的fvii活性。例如,这样的接头可以是氨基酸序列seqidno:11所示的螺旋接头。此外,这样的柔性接头可包含重复或随机模式的甘氨酸(g)和丝氨酸(s)。例如,所述接头包含(ggggs)n(其中n是1至20的整数),并且优选具有选自seqidno:3至11的任一氨基酸序列(见表1)。此外,具有与所述接头不低于80%同源性(优选具有不低于85%同源性)的任何氨基酸序列也可用于本发明的融合蛋白质。此外,所述接头还可包括蛋白酶切割位点,其可被损伤组织中丰富的蛋白酶识别。所述切割位点可被选自凝血酶、因子xa、因子ixa和因子viia的蛋白酶切割。具有这种蛋白酶切割位点的融合蛋白质在工作位点处被切割以产生每种蛋白质(即,fvii和转铁蛋白),并且所产生的蛋白质作为个体蛋白质而发挥功能。优选地,所述接头具有seqidno:12的氨基酸序列(见表1)。可更容易地通过位于fvii与转铁蛋白之间的限制酶识别序列将所述接头插入到融合蛋白质中。因此,所述限制酶识别序列可存在于所述接头的任一端或两端,并且继而被翻译成由所述序列编码的氨基酸。例如,当使用agei限制酶识别序列时,thr可存在于所述接头的n末端,并且thr-gly可存在于所述接头的c末端。即,当使用接头(ggggs)3时,所述识别序列和接头可以以--t(ggggs)3tg--的形式存在。在所述接头的n末端和c末端处翻译的氨基酸可根据所使用的限制酶识别序列而有所不同,但其存在并不影响融合蛋白质的活性(见表5)。本发明的融合蛋白质表现出不低于0.7的与未融合天然型fvii相比的fvii比活性。在本发明的一个方面中,所述融合蛋白质(其包含由seqidno:1的氨基酸序列所示的fvii和由seqidno:2的氨基酸序列所示的转铁蛋白)具有约0.82至约0.92的与未融合天然型fvii相比的fvii比活性(见表2和表3)。此外,所述融合蛋白质(其包含由seqidno:1的氨基酸序列所示的fvii、由seqidno:3的氨基酸序列所示的接头以及由seqidno:2的氨基酸序列所示的转铁蛋白)具有约0.97的与未融合天然型fvii相比的fvii比活性(见表2)。根据本发明的融合蛋白质(其中另一些接头插入fvii与转铁蛋白之间)也具有约0.74至约1的与未融合天然型fvii相比的fvii比活性(见表2)。此外,本发明的融合蛋白质具有比其上没有连接转铁蛋白的fvii长3~4倍的半衰期(见表6)。本发明还提供编码所述融合蛋白质的dna。由于密码子简并性或者考虑生物体中优选的密码子,可对编码融合蛋白质的dna进行多种改变和修改以表达所述融合蛋白质,除非所述融合蛋白质的氨基酸序列实质地改变,经修改的dna也包括在本发明的范围之内。在本发明中,编码所述融合蛋白质的dna可优选地由seqidno:13至24的任一核苷酸序列所示。可通过用于表达所述dna的载体来提供本发明编码所述融合蛋白质的dna。本发明提供包含编码所述融合蛋白质的dna的重组载体。本文中使用的术语“载体”是指用于将编码所述融合蛋白质的dna引入宿主细胞并在其中表达所述融合蛋白质的工具(means)。所述载体可包括常规载体,例如质粒载体、粘粒(cosmid)载体、噬菌体载体、病毒载体等,优选质粒载体。合适的表达载体包含表达调节元件(例如,启动子、起始密码子、终止密码子、聚腺苷酸化信号和增强子),以及用于膜导向或分泌的信号序列或前导序列,并且可根据目的而多样地制备。应确保所述起始密码子和终止密码子在施用基因构建体的生物体中运作,并且处于编码序列的阅读框中(in-frame)。此外,所述表达载体包含用于选择含有所述载体的宿主细胞的选择标记,以及复制起点(如果所述表达载体是可复制的)。所述载体可自身复制或可被整合到宿主细胞的dna中。具体地,可通过将编码所述融合蛋白质的dna插入到pcdna3.1-hygro载体而制备根据本发明的重组表达载体。此外,本发明提供通过用所述重组表达载体转化而产生所述融合蛋白质的宿主细胞。因为,所述蛋白质的表达水平和修饰根据宿主细胞的类型而有所不同,优选选择最适于目的的宿主细胞。宿主细胞的实例包括哺乳动物细胞,例如中国仓鼠卵巢(cho)细胞、人胚肾细胞(hek293)、仓鼠肾细胞(bhk21)、人肝癌细胞(hepg2)等,但不限于此。为了用根据本发明的重组表达载体转化宿主细胞,可使用本领域技术人员已知的任何方法,此类方法的实例包括但不限于电穿孔、原生质体融合、磷酸钙(capo4)沉淀、氯化钙(cacl2)沉淀等。发明的实施方式以下实施例仅为举例说明的目的而给出,并不旨在限制本发明的范围。实施例1:制备因子vii(fvii)质粒载体(pcdna3.1-hygro-fvii)将从hepg2细胞(kclbno.88065)纯化的总rna用作模板用于逆转录。通过使用fvii基因特异性引物fvii-f和fvii-r(seqidno:25和26)的pcr扩增互补dna(cdna)以获得人fvii-f基因的开放阅读框。在以下条件下通过处理50μl的反应溶液(0.5μlcdna、0.4μm(10pmol/μl)seqidno:25和26的引物、0.2mmdntp、5单位的taqdna聚合酶和水)而进行pcr:在94℃下5分钟的1个变性循环,在94℃下1分钟、60℃下1分钟、以及在72℃下2.5分钟的35个扩增循环,以及在72℃下5分钟的1个最终延伸循环。将经纯化的pcr产物克隆到pgem-t易载体(pgem-teasyvector)(promega,cat#:a1360)中。通过使用ecori和ncoi的限制性酶切选择阳性克隆。通过dna测序进一步核实所选择的克隆。为了将fvii的orf(fvii-orf)转移至表达载体,通过t4dna聚合酶使以noti切割的fvii-orf成平末端(blunt),并与以hindiii/xbai酶切并且成平末端的pcdna3.1-hygro载体(invitrogen)相连接。通过用apai、xbai、ecori、ncoi、psti限制性酶切和dna测序而确证经连接的载体。该载体被定名为“pcdna3.1-hygro-fvii”。实施例2:构建fvii-tf表达载体(pcdna3.1-hygro-fvii-tf)将实施例1中制备的fviicdna与人转铁蛋白(tf)cdna融合以作为动物细胞中的单一酶原而表达。人转铁蛋白cdna购自origene(cat#:sc322130)并且核实其与genbank登录号:nm_001063.2的序列是否相同。设计融合中所使用的引物以除去fvii的终止密码子以及转铁蛋白的信号肽。为了利于在fvii与tf之间插入多种大小的接头,向连接引物中添加agei位点(accggt),其将被翻译成苏氨酸(thr)和甘氨酸(gly)。所产生的融合蛋白质可具有以下结构:(前导肽)-(成熟fvii)-(thr-gly)-(成熟tf)(其中前导肽由成熟fvii中不存在的信号肽(前肽(prepeptide))和待通过加工酶切割的肽原(propeptide)组成,由38个氨基酸构成,并且与seqidno:1氨基酸序列中的1至38位置的氨基酸相对应)。通过使用引物fvii-s1、fvii-as1、tf-s1和tf-as1(seqidno:27至30)扩增fvii以及tf的cdna,并且使用实施例1中所描述载体。seqidno:27和30的引物分别包含nhei和xhoi位点。图1中描述了用于将fviicdna与tfcdna相连接的克隆策略。首先,通过pcr从pcdna3.1-hygro-fvii载体扩增fviicdna。在以下条件下通过处理50μl的反应溶液(1μl载体模板、1μl引物对、fvii-s1和fvii-as1(10μm)、10μl5xphusionhf缓冲液、200μmdntp、0.5μlphusiondna聚合酶(finnzymes,#f-530s,2单位/μl)和35.5μl水)而进行pcr:在98℃下30秒的1个变性循环,在98℃下10秒、60℃下45秒、以及在72℃下30秒的30个扩增循环,以及在72℃下7分钟的1个最终延伸循环。接下来,使用转铁蛋白cdna作为模板扩增tf。重复上述pcr操作,不同之处在于使用引物对(tf-s1:10μm;tf-as1:10μm)。通过一系列的限制性酶切(restriction)和连接而将经扩增的fvii和tfcdna相连接。用agei和xhoi或者用nhei酶切通过pcr扩增的每种dna。将经酶切的dna纯化并以1∶1的摩尔比连接。将经连接的dna亚克隆到用nhei/xhoi酶切的pcdna3.1-hygro载体(invitrogen)中。通过dna测序进一步核实插入物的大小和序列。实施例3:构建fvii-gs接头-tf表达载体使用包含甘氨酸和丝氨酸的由5个氨基酸组成的肽作为基本接头单位。所述基本接头单位包含四个甘氨酸和一个丝氨酸,具有以下序列:“ggggs”。应用所述基本接头单位(下文中称为“gs-x接头”,其中x是重复数目的基本gs接头单位)以构建更长的gs接头。在该实施例中,构建gs-1至gs-15的接头。1)构建fvii-gs-1接头-tf表达载体合成一组包含基本gs接头单位的引物gs-fv-as1和gs-tf-s1(seqidno:31和32),并通过重叠pcr将其插入到fvii与tf之间(见图2)。通过使用一组引物fvii-s1和gs-fv-as1(seqidno:27和31)以及phusiondna聚合酶(finnzymes,#f-530s)的pcr将所述gs-1接头与fvii相连接。通过在以下条件下处理50μl的反应溶液(1μlpcdna3.1-hygro-fvii-tf载体、1μlfvi1-s1(10pmole/1μl)、1μlgs-fv-as1(10pmole/μl)、1μl10mmdntp、10μl5xphusionhf缓冲液、35.5μl水和0.5μlphusiondna聚合酶(2单位/μl))而进行pcr:在98℃下30秒的1个变性循环,在98℃下10秒、64℃下30秒、以及在72℃下45秒的35个扩增循环,以及在72℃下7分钟的1个最终延伸循环。同时,为了将gs-1接头与tf相连接,重复上述pcr操作,不同之处在于使用一组引物gs-tf-s1和tf-as1(seqidno:32和30)。使用经扩增的pcr产物作为重叠pcr的模板。通过在以下条件下处理反应溶液(1μl经扩增的pcr产物、1μlfvii-s1(10pmole/μl,seqidno:27),1μl反义引物(tf-as110pmole/μl,seqidno:30)、10μl5xphusionhf缓冲液、1μl10mmdntp、34.5μl水、0.5μlphusiondna聚合酶(2单位/μl))而进行重叠pcr:在98℃下1分钟的1个变性循环,在98℃下10秒、66/68℃下30秒、以及在72℃下45秒的45个扩增循环,以及在72℃下7分钟的1个最终延伸循环。将进扩增的重叠pcr产物克隆到用nhei和xhoi酶切的pcdna3.1-hygro-lacz中。2)构建fvii-gs-3接头-tf表达载体合成包含gs-3和agei位点的引物对gs3-s和gs3-as(seqidno:33和34)。为了制造gs-3双链接头,通过在98℃下加热混合物(5μlgs3-s(100pmole/μl)、5μlgs3-as(100pmole/μl)、2μl10x退火缓冲液(100mmtris-cl[ph8.0]、1mnacl、10mmedta)和8μl水)10分钟并在25℃下冷却1小时而使引物退火。用agei酶切经退火的接头,并且还用agei酶切实施例2中制备的pcdna3.1-hygro-fvii-tf载体。在37℃下用1μlcip(小牛肠磷酸酶;neb,#m0290s)处理经酶切的载体1小时,并对其进行凝胶提取操作(qiagen,#28704),随后使用t4dna连接酶(takara,#2011a)以1∶3的摩尔比(载体∶插入物)连接。3)构建fvii-gs-5接头-tf至fvii-gs-15接头-tf的表达载体为了构建包含gs-5接头的融合蛋白质表达载体,实施了新的策略。通过以下两个步骤构建包含接头经延长的fvii-tf融合载体。第一步是向以前获得的接头中添加合成的双链(ds)gs2接头。确保延长接头之后,将所述接头切出并将其插入pcdna3.1-hygro-fvii-tf载体中的fvii基因与tf基因之间。例如,为了将gs-3接头延长至gs-5接头,用bglii酶切seqidno:35的经合成dsgs-2接头单位,并与用bamhi和stui处理的pcdna3.1-hygro-fvii-gs3-tf载体连接。接下来,在通过bamh1和agei酶切确证延伸所述接头之后,用agei将经延长的接头切出,并将其克隆到用agei和cip处理的pcdna3.1-hygro-fvii-tf载体中。通过相同的策略构建包含gs-7、gs-9、gs-11、gs-13和gs-15接头的fvii-tf融合表达载体(见图3)。实施例4:构建含有包含凝血酶切割位点的接头的fvii-tf表达载体(pcdna3.1-hygro-fvii-gs1-t-tf)通过使gs-1单位与凝血酶识别序列(下文中称为“gs1-t接头”)的两端邻接而制备包含凝血酶切割位点的接头。设计并合成seqidno:36(有义)的dsgs1-t接头以在两端包含agei位点。用agei酶切dsgs1-t接头,并使用pcr纯化试剂盒(qiagen,cat#:28104)进行纯化。将经纯化的接头连接到用cip/agei处理的pcdna3.1-hygro-fvii-tf载体中。实施例5:构建包含螺旋接头的fvii-tf表达载体(pcdna3.1-hygro-fvii-螺旋-tf)通过美国公开(laid-openpublication)no.2009/0170163中公开的方法制备螺旋接头dna。通过使用引物向所制备螺旋接头dna(螺旋接头s和螺旋接头as(seqidno:37和38))的两端添加agei位点。将引物螺旋接头s和螺旋接头as退火并用agei酶切,随后插入用agei和cip处理的pcdna3.1-hygro-fvii-tf载体中。通过dna测序确证所构建的载体。比较例1:构建fvii-白蛋白融合表达载体(fvii-alb)构建欧洲专利no.1816201中所公开的fvii-白蛋白融合蛋白质。通过使用人肝mrna(clontech)作为模板以及白蛋白基因特异性引物白蛋白-s和白蛋白-as(seqidno:39和40)的rt-pcr获得人白蛋白cdna。根据制造商的说明书通过使用accuscript高保真rt-pcr系统试剂盒(cat#600180)进行rt-pcr。首先,将10μl的逆转录反应溶液(1μl10x逆转录酶缓冲液、0.6μl寡-dt引物、1μldntp、0.4μl水、5μl人肝mrna(10ng/μl))在65℃下保持5分钟,并在室温下保持5分钟,并在42℃下与1μl100mmdtt和1μl逆转录酶反应1小时。通过使用合成cdna作为模板以及引物白蛋白-s和白蛋白-as的pcr获得人白蛋白序列。在以下条件下通过处理50μl的反应溶液(1μlcdna模板、10μl5xphusionhf缓冲液、分别1μl引物白蛋白-s和白蛋白-as、1μl10mmdntp、0.5μlphusiondna聚合酶(finnzymes,#f-530s;2单位/μl)和35.5μl水)而进行pcr:在98℃下1分钟的1个变性循环,在98℃下10秒、60℃下30秒、以及在72℃下60秒的30个扩增循环,以及在72℃下7分钟的1个最终延伸循环。将合成的seqidno:41和42的寡核苷酸退火至欧洲专利no.1816201中所公开的gs接头[ss(ggs)9gs](seqidno:45)。为了将实施例1中制备的fviicdna连接至上述接头,通过使用引物mutfvii(xhoi)-s和mutfvii(xhoi)-as(seqidno:43和44)的基于pcr的诱变,用xhoi位点替换pcdna3.1-hygro-fvii载体中的fvii终止密码子。使用xhoi/apai位点,将gs接头[ss(ggs)9gs]融合至pcdna3.1-hygro-fvii载体中fviicdna的3′端。最终,将用bamhi酶切的人白蛋白cdna插入到pcdna3.1-hygro-fvii-gs-接头载体中。通过dna测序核实所制备的pcdna3.1-hygro-fvii-gs-接头-白蛋白表达载体。表1中示出实施例2至5以及比较例1中所构建表达载体的特征。表1[表1]实验例1:测量fvii-融合蛋白质的比活性在稳定表达vkorc1(维生素k环氧化物还原酶复合物亚单位1)的cho细胞(cho(vk2))中表达实施例2至5以及比较例1中所构建的fvii-融合蛋白质。通过使用无内毒素质粒大量提取试剂盒(endo-freeplasmidmaxikit)(qiagen,#27104)纯化实施例2至5以及比较例1中所构建的表达载体。使用β-半乳糖苷酶作为转染的内对照。在6孔板中以1.5×106个细胞/孔的密度接种cho(vk2)细胞。在补充了10%fbs(lonza,#14-501f)、1xht(invitrogen,#11067-030)、4mml-谷氨酰胺(lonza,#17-605e)和200μg/ml潮霉素(invitrogen,#10687-010)的α-mem(lonza,#12-169f)中孵育细胞24小时,随后根据制造商的说明书使用lipofectamine2000(invitrogen)转染。转染之后四小时,替换为无血清培养基(optimem),并补充5μg/ml维生素k。孵育48小时之后,对培养基进行取样并贮藏于-70℃。分别使用coatest因子vii测定试剂盒(chrmogenix,#821900-63)和fviielisa试剂盒(cedarlenelab,#cl20030k)分析所表达fvii-融合蛋白质的发色活性和抗原量。根据制造商的说明书进行所述测定。将相对于who标准归一化的标准入血浆用作这两种测定中的对照fvii。通过western印迹分析评估蛋白质的表达。基于elisa结果加载等量的fvii融合蛋白质。发现所表达的fvii-融合蛋白质具有预期的大小,没有可检出的片段化(见图4)。同时,fvii-转铁蛋白融合蛋白质的比活性为0.74至1,这高于fvii-白蛋白融合蛋白质的比活性(0.52)(见表2)。包含接头的fvii-转铁蛋白融合蛋白质还保留了不低于70%的fvii活性。接头长度与比活性之间没有关系,但具有较短gs接头的fvii融合蛋白质表现出比具有较长gs接头的融合蛋白质稍微更高的比活性。尤其是fvii-gs1-tf和fvii-gs1-t-tf融合蛋白质表现出相当的比活性(见图5)。表2[表2]实施例6:根据tf融合方向表征fvii融合蛋白质在该实施例中,制备了其中人转铁蛋白(tf)连接至fvii的n末端的融合蛋白质,并与其中转铁蛋白连接至fvii的c末端的融合蛋白质相比较,以检测根据融合蛋白质中融合方向的特征的改变。详细的操作如下。<6-1>构建tf-fvii和tf-gs1-t-fvii表达载体tf连接至fvii的n末端的两种融合蛋白质设计如下:(1)(tf的前导肽)-(成熟tf)-(thr-gly)-(成熟fvii);和(2)(tf的前导肽)-(成熟tf)-(thr)-(gs1-t;seqidno:12)-(thr-gly)-(成熟fvii)。首先,为了获得包含前导肽的tf基因序列,设计正向引物(nhe-tf:seqidno:46)以包含用于克隆的目的的nhei位点,并且设计反向引物(tf-age:seqidno:47)以包含用于除去转铁蛋白终止密码子以及克隆的目的的agei位点。对于克隆除去了前导肽的成熟fvii,设计正向引物(age-fvii:seqidno:48)以包含agei位点,并且设计反向引物以包含xhoi位点。对于tf基因,cdna购自origene(cat#:sc322130),在实施例2中用作pcr模板。在以下条件下通过处理50μl的反应溶液(1μl载体模板、2μl引物nhe-tf和tf-agei(10μm)、10μl5×phusionhf缓冲液、1μl10mmdntp、0.5μlphusiondna聚合酶(finnzymes,#f-530s,2单位/μl)和33.5μl水)而进行pcr:在98℃下30秒的1个变性循环,在98℃下10秒、70℃下30秒、以及在72℃下36秒的25个扩增循环,以及在72℃下10分钟的1个最终延伸循环。如实施例4中那样通过使用pcdna3.1-hygro-fvii-gs1-t-tf载体作为模板的pcr扩增fvii。pcr条件与上述tfpcr条件相同,不同之处在于施用引物age-fvii(10μm)和vii-xho(10μm)。通过使用nhei/agei将所扩增的tf基因插入到pcdna3.1-hygro-fvii-gs1-t-tf载体中以获得pcdna3.1-hygro-tf-tf载体。用agei/xhoi酶切并连接pcdna3.1-hygro-tf-tf载体和fviipcr产物以构建包含pcdna3.1-hygro-tf-fvii融合蛋白质的表达载体。通过经由agei将插入实施例4中所合成的dsgs1-t序列而构建pcdna3.1-hygro-tf-gs1-t-fvii表达载体。通过限制酶切作图和dna测序确证所构建的表达载体。<6-2>表达融合蛋白质并表征为了表征具有连接至fviin末端的tf的融合蛋白质,将其表达载体(即,pcdna3.1-hygro-fvii-tf、pcdna3.1-hygro-fvii-gs1-tf-vii、pcdna3.1-hygro-tf-fvii和pcdna3.1-hygro-tf-gs1-t-fvii)瞬时表达于cho细胞中。使用无内毒素质粒大量提取试剂盒分离所构建的4种质粒dna。转染前一天,通过胰蛋白酶分离在t75瓶中培养的cho(dg44)细胞,并以1.5×106个细胞/孔的密度接种在6孔板中。24小时之后,根据制造商的说明书转染细胞。转染四小时之后,除去每孔中的培养基并替换为2ml补充有5μg/ml维生素k的培养基。转染之后,在37℃、5%co2的培养箱中孵育6孔板,并在48小时之后收获培养基。将所收获的上清液转移至1.5ml管中并在-70℃下贮藏,用于fvii的发色测定和elisa。用每孔2mlhbss清洗除去了培养基的板并用250μl裂解液(tropix,#abx210lm,添加1mmdtt)裂解,随后贮藏在-70℃,用于β-半乳糖苷酶测定。按照实验例1进行发色测定和fviielisa。通过在实验之前融化转染之后冷冻贮藏的培养基并通过离心获得上清液而制备用于分析的样品。在测定中使用标准人血浆(dadebehring,#orkl13,lot#503216f)作为标准物。测量结果示于表3中。对于tf连接至fviin末端的tf-fvii和tf-gs1-t-tf,未测量出活性,不同于tf连接至fviic末端的fvii-tf和fvii-gs1-t-tf。此外,在fviielisa中检出了少量tf连接至n末端的融合蛋白质。但是,在使用tf的多克隆抗体的western印迹结果中,无论融合方向如何,所述融合蛋白质表现出了相似的检测灵敏度。结果表明,当将tf连接至fvii的n末端时,即便正常地进行了氨基酸的翻译,也生成没有fvii活性的融合蛋白质。表3[表3]实施例7:根据融合中使用的限制酶识别序列缺失而表征融合蛋白质在实施例2中,使用限制酶(agei)识别序列以利于在fvii与tf之间插入多种接头。结果,一些融合蛋白质变得在接头的两端具有由上述限制酶多编码的thr和gly。在该实施例中,检测了融合蛋白质的特性是否通过存在限制酶(agei)识别序列而改变。通过使用诱变引物的基于pcr的位点定向诱变,使限制酶识别序列从包含gs1-t接头的fvii-gs1-t-tf融合蛋白质中缺失。如图6所示,设计该实验以在n末端缺失“thr”并且在c末端缺失“thr-gly”,实验中使用的引物列在表4中。表4[表4]<7-1>缺失thr-gly进行基于pcr的诱变。在以下条件下通过处理50μl的反应溶液(1μlpcdna3.1-hygro-fvii-gs1-t-tf载体、0.2μl有义引物(tgdel-s10μm)、0.2μl反义引物(tgdel-as10μm)、1μl10mmdntp、4μl5xpcr缓冲液、14μl水和0.2μlphusiondna聚合酶(finnzymes,#f-530s))而进行pcr:在98℃下30秒的1个变性循环,在98℃下10秒、58℃下30秒、以及在72℃下3分钟的18个扩增循环,以及在72℃下7分钟的1个最终延伸循环。为了除去初始的模板dna,用1μldpni(neb,#r0176s)处理经扩增的pcr产物,并在37℃下孵育1小时。使用10μl经dpni-处理的dna转化50μlhit感受态细胞(dh5α,rh617),并在lb+amp(10mg/ml)固体培养基中过夜孵育。通过dna测序分析如此获得的4个克隆,并且核实了两个克隆为突变体。<7-2>缺失thr为了缺失thr,通过使用不同引物而进行与<7-1>中类似的方法。简单地说,在相同的条件下通过使用1μl克隆的质粒dna作为模板(其中在实施例<7-1>中确证突变)和1μl有义引物(tdel-s;10pmole)以及1μl反义引物(tdel-as;10pmole)而进行基于pcr的诱变。通过对所选择的四个克隆进行dna测序,确证了三个克隆具有突变。将所获得的表达载体命名为“pcdna3.1-hygro-fvii-gs1-t-tf(m3)”。<7-3>表征具有限制酶识别序列缺失的融合蛋白质用fvii-gs1-t-tf和fvii-gs1-t-tf(m3)表达载体转染cho细胞,并以fvii-alb表达载体作为对照,获得培养基的上清液。对所获得的上清液进行fvii发色测定(chromogenix)和fviielisa(cedarlane)以核实活性/抗原比值的改变。如表5所示,fvii-gs1-t-tf和fvii-gs1-t-tf(m3)融合蛋白质的抗原量和活性几乎彼此相等,并且比值(比活性)也没有改变。此外,确证了比活性显著高于fvii-alb融合蛋白质的比活性。表5[表5]实施例8:测量融合蛋白质的半衰期为了测定根据本发明的融合蛋白质半衰期的提高,将fvii-tf、fvii-gs1-tf、fvii-gs3-tf、fvii-gs15-tf和fvii-gs1-t-tf用作实验组,并将野生型fvii和市售的fviia(novonordisk)用作对照组。<8-1>样品制备1)获得表达培养基在freestyletmcho-s细胞系(invitrogen,cat.no.r800-07)中表达野生型fvii蛋白质和五种融合了tf的fvii融合蛋白质。在具有补充有8mml-glu(gibco,l-谷氨酰胺200mm(100x),cat.no.25030-081)的freestylecho表达培养基的旋转瓶(spinnerflask)中悬浮培养cho-s细胞。在转染前24小时,以4×105个细胞/ml的密度接种所培养的细胞,并且当密度变为1×106个细胞/ml时进行转染。通过使用无内毒素大量制备试剂盒(endo-freemaxiprepkit)(qiagen,cat.no.12362)或无内毒素质粒宏量制备试剂盒(endo-freeplasmidmegaprepkit)(qiagen,12381)制备转染中使用的dna,并且参照freestylemax试剂(invitrogen,cat.no.16447-100)的转染方案进行转染。向8mloptiprosfm(invitrogen,cat.no.12309-019)中添加500μgdna并混合。向另一管中添加8mloptiprosfm(invitrogen,cat.no.12309-019)和500μlfreestylemax试剂,随后将上述两种混合物缓慢混合并在室温下贮藏10分钟。10分钟之后,用所述混合物转染freestyletmcho-s细胞。在37℃,5%co2的培养箱中孵育经转染的细胞3~5天,随后获得上清液。2)纯化表达培养基将通过旋转瓶获得的培养基过滤通过0.22μm滤器(corning)以除去剩余的细胞和碎片。通过使用切向流动式膜(tangential-flowmembrane)(satorious,30kda)的超滤将经过滤的培养基浓缩10倍。将经浓缩的培养基加至装有陶瓷羟磷灰石(ceramichydroxyapatite)(bio-rad,157-0040)树脂的xk16/20柱(gehealthcare)。用超过10个柱体积的平衡缓冲液(25mm咪唑、0.02%吐温80和150mmnacl,ph6.5)平衡该陶瓷羟磷灰石柱。加载了经浓缩培养基之后,用平衡缓冲液和清洗缓冲液-1(25mm咪唑、0.02%吐温80、100mm磷酸钠,ph6.3)和清洗缓冲液-2(25mm咪唑、0.02%吐温80、100mm磷酸钠、1mnacl,ph6.3)清洗所述柱。清洗之后,用洗脱缓冲液(25mm咪唑、0.02%吐温80、500mm磷酸钠,ph6.3)洗脱捕获至所述柱的融合蛋白质。通过fvii-发色测定、fviielisa测定和sds-page/western印迹分析经洗脱的蛋白质。<8-2>western印迹测定通过sds-page/考马斯蓝染色确证了通过两步柱而部分纯化的fvii和fvii/tf融合蛋白质具有45%或更高的纯度。通过western印迹评估经纯化蛋白质中源自fvii的片段的存在,因为经纯化的融合蛋白质中的片段化fvii可能具有比完整fvii融合蛋白质更短的半衰期,并且误导对每种fvii融合蛋白质的半衰期的测定。以0.1iu(fvii活性)/10μl制备(novonordisk,1.2mg/小瓶,60kiu)和经纯化样品,随后通过使用nupage4-12%bis-tri凝胶(invitrogen)进行sds-page。完成电泳之后,将凝胶转移至pvdf膜,并在室温下通过添加10ml封闭缓冲液(25mmtris、150mmnacl(ph7.2)、5%脱脂乳和0.1%吐温80)而封闭1小时。倾泻掉封闭液,以1∶5000和1∶500的比例添加抗fvii抗体(cat.no.f8146,sigma)或小鼠抗转铁蛋白抗体(sc52256,santacruz),并在翘板摇床(rockingshaker)中孵育1小时。用清洗溶液(25mmtris、150mmnacl,ph7.2)将所述膜清洗四次,并在10ml(pbs-t中的5%脱脂乳)溶液(其中已添加比例为1∶50,000的山羊抗小鼠igg-hrp抗体(cat.no.g21040,invitrogen)作为二抗)中孵育1小时。用清洗溶液(25mmtris、150mmnacl,ph7.2)将所述膜清洗四次之后,向其上添加2mlsuper-signalwestfemtomix(thermo)5分钟。反应完成之后,使膜显影。图7中显示western印迹的结果。如图7所示,在经纯化的蛋白质中未检出源自fvii的片段。在所探查的印迹上通过抗转铁蛋白抗体未检出片段化的转铁蛋白。<8-3>测量半衰期在大鼠中测量以下蛋白质的半衰期并彼此比较:不具有接头的融合蛋白质,具有四种接头(gs1、gs1-t、gs3和gs15)的融合蛋白质,以及表达的和在相同条件下纯化的野生型fvii,并且用市售的novoseven作为对照。根据制造商的说明书,通过人fviielisa(cedarlane,用于对因子vii进行elisa的成对抗体,#cl20030k)进行对待施用样品以及从动物实验中所采集样品中的fvii量进行定量分析。通过对来自三个不同稀释度的样品的值求平均值而测定待施用样品的浓度。施用稀释溶液(administrationdilutionsolution)(nacl3mg/ml、cacl2二水合物1.5mg/ml、甘氨酰甘氨酸1.3mg/ml、聚山梨酯800.1mg/ml和甘露醇30mg/ml,ph5.5)被用作稀释剂。fviielisa定量之后,用施用稀释溶液稀释每种蛋白质,通过尾静脉以基于实验当天所测量的大鼠体重150iu/kg向大鼠(250~300g的spraguedawley,每组三只大鼠)静脉内施用经稀释的样品。在总共11个时间点取血,即施用药物之后0分钟、5分钟、15分钟、30分钟、60分钟、1.5小时、2小时、4小时、6小时、8小时和24小时。将225μl血液与25μl3.2%柠檬酸钠混合,并在4℃下以13,000rpm离心1分钟,随后将上清液贮藏在-70℃。通过用fviielisa试剂盒(cedarlane)中使用的清洗缓冲液稀释1/50或1/100而分析大鼠血浆。通过对人fvii抗原浓度的对数相对于取样时间点进行作图而获得回归曲线。通过根据公式“半衰期=ln2/回归曲线的斜率”计算而确定每种fvii的半衰期。如表6所示,与野生型fvii相比,本发明的融合蛋白质显示出3~4倍提高的半衰期。表6[表6]类型半衰期(分钟)fvii-gs1-tf254.2±19.1fvii-gs3-tf227.4±23.5fvii-gs1-t-tf235.4±27.4fvii-gs15-tf257.0±23.9fvii-tf277.0±24.5诺其(novoseven)80.3±27.4天然型fvii59.6±2.9以下内容对应于母案申请中的原始权利要求书,现作为说明书的一部分并入此处:1.包含因子vii(fvii)和转铁蛋白的融合蛋白质,其中所述转铁蛋白与所述fvii的c末端相连接。2.项1的融合蛋白质,其中所述fvii是氨基酸序列seqidno:1所示的多肽或其功能等价物。3.项2的融合蛋白质,其中所述功能等价物具有与所述fvii的功能活性基本等价的功能活性,并且在seqidno:1所示的氨基酸序列中具有氨基酸残基的缺失、插入或替换。4.项1的融合蛋白质,其中所述转铁蛋白是氨基酸序列seqidno:2所示的多肽或其功能等价物。5.项4的融合蛋白质,其中所述功能等价物具有与所述fvii的功能活性基本等价的功能活性,并且在seqidno:2所示的氨基酸序列中具有氨基酸残基的缺失、插入或替换。6.项1的融合蛋白质,其在所述fvii的c末端与所述转铁蛋白之间还包含限制酶识别序列。7.项1的融合蛋白质,其在所述fvii与所述转铁蛋白之间还包含接头。8.项7的融合蛋白质,其中所述接头由1至100个氨基酸组成。9.项7的融合蛋白质,其中所述接头由1至75个氨基酸组成。10.项7的融合蛋白质,其中所述接头由5至25个氨基酸组成。11.项7的融合蛋白质,其中翻译自限制酶识别位点的一个或更多个氨基酸存在于所述接头的一端或两端。12.项7的融合蛋白质,其中所述接头是seqidno:3至11的任一氨基酸序列所示的肽。13.项7的融合蛋白质,其中所述接头包含蛋白酶切割位点,其能够被选自凝血酶、因子xa、因子ixa和因子viia的蛋白酶切割。14.项13的融合蛋白质,其中所述接头是由seqidno:12的氨基酸序列所示的肽。15.项1至14中任一项的融合蛋白质,其中所述融合蛋白质具有与未融合天然型fvii相比不低于0.7的fvii比活性。16.编码项1至14中任一项的融合蛋白质的dna。17.项16的dna,其中所述dna由seqidno:13至24的任一核苷酸序列所示。18.包含项16的dna的重组载体。19.包含项18的重组载体的宿主细胞。20.项19的宿主细胞,其选自:cho细胞、bhk21细胞、hek293细胞和hepg2细胞。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1