一种酶法提取的川白芷多糖及制备方法和应用与流程
本发明属于生物医药技术领域,具体涉及一种酶法提取的川白芷多糖及制备方法和应用。
背景技术:
药材白芷为伞形科植物白芷angelicadahurica(fisch.exhoffm)benth.ethook.f.或杭白芷angelicadahurica(fisch.exhoffm)benth.ethook.f.var.formosana(boiss)shanetyuan的干燥根。白芷味辛,性温,归胃、大肠、肺经。具有解表散寒,祛风止痛,宣通鼻窍,燥湿止带,消肿排脓的作用,用于感冒头痛,眉棱骨痛,鼻塞流涕,鼻鼽,鼻渊,牙痛,带下,疮疡肿痛。
白芷是卫生部公布药食两用的87种中药材之一,也是我国常用的大宗药材。按产地分为川、杭、祁、禹白芷四大商品。其中产于四川的白芷则称川白芷,是著名的川产道地药材,产量约占全国商品白芷的70%。川白芷以四川遂宁出产的形优、质坚、粉多香浓、色白细腻、横切片成“菊花芯形”的川白芷品质最佳。白芷富含多种化学成分,现代研究表明,白芷的脂溶性化学成分主要为香豆素类,含量为0.211%~1.221%,其水溶性成分有棕榈酸、豆甾醇、β-谷甾醇、β-胡萝卜苷(β-daucosterin)等。白芷还富含多种多糖。
多糖广泛存在于动、植物和微生物(真菌和细菌)中,常与蛋白质和多聚核苷酸相连,是生命活动中必不可少的生物大分子,在细胞吸附、细胞与细胞信息交流、免疫系统分子识别方面扮演着重要的作用。目前多糖已成为天然药物及保健品研发中的重要组成部分。植物多糖具有多种生物活性,如抗氧化、抗肿瘤、抗病毒、降血糖血脂、抗衰老、抗炎、抗辖射、免疫调节等。采用酶解法提取多糖在近年来得到了广泛的应用。
酶解法通常是将粉碎的试验材料悬浮于水中,根据酶作用的最适条件,调节反应液ph值和温度,然后加入5%~25%根据提取目的和实验室条件选用的酶,反应1~4h,除去残渣得滤液即为多糖提取液。酶解法已在多糖保健品的制备中采用。但考虑到经济和效率因素,多采用热水提取法与酶法相结合的办法,即先用热水提取,然后残渣再用酶法提取。
酶具有专一性,能有选择地释放产物,且温度低,无污染,不易破坏多糖的生物活性。酶属于蛋白质,它的种类,酶促反应温度、ph、时间和底物量等都会影响酶的活性,过高或过低都会造成酶的失活,从而影响提取效率。同时酶解法因为酶制剂用量大、成本高,限制了它在多糖提取工业上的大规模应用。
目前对于白芷多糖的研究尚不够深入。仅个别学者对杭白芷多糖的提取以及抗氧化和增强动物皮肤组织免疫功能进行过分析,且尚未开展过酶法提取的白芷多糖的结构和活性研究。
技术实现要素:
本发明的目的之一在于针对上述问题,提供一种酶法提取的川白芷多糖。
本发明的目的之二在于提供一种酶法提取的川白芷多糖的制备方法。
本发明的目的之三在于提供一种酶法提取的川白芷多糖的应用。
为实现上述目的,本发明采用的技术方案如下:
本发明所述的一种酶法提取的川白芷多糖,该川白芷多糖同时存在α构型和β构型多糖链接方式,其表观分子量为3.93kda;由川白芷药材经酶法提取、醇沉除杂、去蛋白去盐后,再经deae-琼脂糖凝胶ff柱和琼脂糖凝胶6ff凝胶柱层析分离而得。
本发明所述的川白芷多糖的制备方法,具体包括以下步骤:
a:脱脂脱色素:将川白芷药材洗净烘干粉碎,得粉末;将所述粉末先加入石油醚脱脂后,再加入高浓度乙醇溶液脱除部分色素及低聚糖;
b:提取:将脱脂脱色素后的粉末加入含酶的缓冲液提取后,灭活酶,离心,收集上清液,浓缩,得浓缩液;
c:醇沉除杂:将所述浓缩液醇沉除去水溶性杂质后得到川白芷粗多糖;
d:去蛋白:将所述川白芷粗多糖采用木瓜蛋白酶+sevag结合法去除蛋白;
e:透析去盐:将所述去除蛋白后的川白芷粗多糖采用透析法去除使无机盐和小分子物质,得去盐川白芷多糖;
f:将所述去盐川白芷多糖进行deae-琼脂糖凝胶ff柱层析,收集并合并第一个洗脱峰的收集液,浓缩,透析,冻干;
g:将步骤f冻干后的川白芷多糖进行琼脂糖凝胶6ff凝胶柱层析,收集并合并主峰的洗脱液,浓缩,透析,冻干,即得。
进一步地,所述步骤a中,将川白芷药材洗净,于40-50℃烘干,粉碎,过40目筛,得川白芷粉末;取所述川白芷粉末加入石油醚加热回流脱脂4h,过滤,滤渣于40-50℃烘干后,再加入80%乙醇加热回流提取4h,过滤,滤渣挥干乙醇后粉碎,于40-50℃烘干,备用;所述石油醚的用量按每克川白芷粉末计为2-4ml,所述80%乙醇的用量按每克川白芷粉末计为2ml。
进一步地,所述步骤b中缓冲液的加入量以每克脱脂脱色素后的粉末计为2-6ml,所述缓冲液为ph4.5柠檬酸-柠檬酸钠缓冲液,所述酶为纤维素酶,其含量为0.45%;所述提取条件为40-60℃水浴提取120-240min,沸水浴5-15min使酶灭活,提取次数为1-4次。
进一步地,所述步骤d中,向浓缩液中加入无水乙醇至混合溶液乙醇终浓度为50-70%,于0-6℃静置12-24h,离心,取离心沉淀加水溶解后,再次离心,取再次离心后的上清液,向所述上清液加入无水乙醇至混合溶液乙醇终浓度为50-70%,于0-6℃静置12-24h,第三次离心,取第三次离心后的沉淀,得川白芷粗多糖,备用。
进一步地,所述步骤d中去蛋白所用sevag试剂为氯仿:正丁醇=4:1,所述去蛋白的次数为10次。
进一步地,所述步骤e中,采用截留分子量为8000-14000da的透析袋,室温蒸馏水透析24h后,换干净蒸馏水再透析8h,重复2次。
进一步地,所述步骤f中,将川白芷多糖加水溶解后,上样于deae-琼脂糖凝胶ff柱,先以1.5倍柱体积的蒸馏水洗脱后,再以水和2mol/l的nacl溶液混合洗脱,实现0~1.5mol/lnacl溶液的梯度洗脱,分管收集,收集液用苯酚-硫酸法检测多糖含量,绘制苯酚-硫酸反应曲线图,并绘制反应曲线,根据所述反应曲线合并第一洗脱峰的收集液,浓缩,透析,冻干。
进一步地,将步骤f冻干后的川白芷多糖加水溶解后,上样于琼脂糖凝胶6ff凝胶柱,洗脱液为0.05mol/lnacl溶液,分管收集,收集液用苯酚-硫酸法检测多糖含量,绘制苯酚-硫酸反应曲线图,采用考马斯亮蓝g250法测定蛋白含量,并绘制反应曲线,根据所述反应曲线收集主峰的洗脱液,浓缩,透析,冻干。
本发明所述的川白芷多糖在制备具有美白作用的日化用品、食品、保健品或药品中的用途。
本发明所用川白芷,其原植物为杭白芷[angelicadahurica(fisch.exhoffm.)benth.ethook.f.var.formosana(boiss.)shanetyuan],产地为四川省遂宁市。
与现有技术相比,本发明具备的有益效果为:
本发明提供了一种酶法提取的川白芷多糖,该川白芷多糖具有良好的美白活性,对酪氨酸酶活性具有抑制作用,且随着浓度的增加,酪氨酸酶抑制率逐渐升高,呈现明显的量效关系。
本发明采用0.45%纤维素酶,与传统酶解法中5%~25%的酶用量相比,极大地降低了酶的用量;采用40-60℃水浴直接加热提取,避免了传统酶解法需要先用热水提取,然后残渣再用酶法提取的缺点,减少了反应步骤。本发明方法提取川白芷多糖,酶制剂用量少、工艺简单,生产成本低。本发明所得川白芷多糖经醇沉、去蛋白去盐后,再经两次柱层析,得到良好的纯化效果,含糖量显著提高,淀粉几乎除尽。
本发明建立了一条完整可行的川白芷多糖提取、分离纯化、理化性质、结构和生物活性研究技术路线。
附图说明
附图1为本发明川白芷多糖的deae-琼脂糖凝胶ff色谱柱蒸馏水洗脱曲线图。
附图2为本发明川白芷多糖的deae-琼脂糖凝胶ff色谱柱nacl梯度洗脱曲线图。
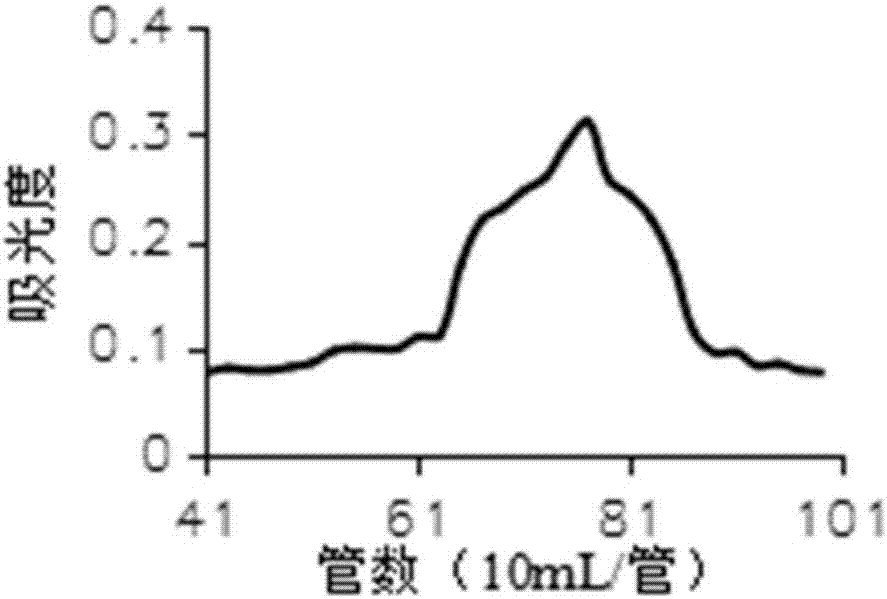
附图3为本发明川白芷多糖的琼脂糖凝胶6ff色谱柱洗脱曲线图。
附图4为本发明川白芷多糖的刚果红反应曲线图。
附图5为本发明川白芷多糖的傅里叶变换红外光谱图。
附图6为本发明川白芷多糖的1h和13cnmr图谱,其中a为1hnmr图谱,b为13cnmr图谱。
附图7为本发明川白芷多糖、实施例11制得的川白芷多糖、实施例12制得的川白芷多糖的酪氨酸酶抑制活性图。
具体实施方式
下面结合附图说明和实施例对本发明作进一步说明,本发明的方式包括但不仅限于以下实施例。
实施例1
川白芷多糖的制备
川白芷多糖的制备方法,包括以下步骤:
a:脱脂脱色
取川白芷根洗净,45℃烘干,粉碎过40目筛。称取白芷粉样,加入石油醚回流脱脂4h,45℃烘干后,再加入80%乙醇80℃回流提取4h,以除去部分色素及低聚糖,挥干乙醇后粉碎,45℃烘干备用。石油醚的用量按每克川白芷粉末计为3ml,80%乙醇的用量按每克川白芷粉末计为2ml。
b:提取
称取脱脂脱色素后的粉末500g,加入4倍量含0.45%纤维素酶、ph值为4.5的柠檬酸-柠檬酸钠缓冲液,50℃水浴提取160min,沸水浴10min使酶灭活,4000r/min离心5min收集上清液,重复提取2次,合并提取液并浓缩,得浓缩液。
c:醇沉除杂
向所述浓缩液中缓慢加入无水乙醇至混合溶液乙醇终浓度为60%,置于4℃静置12h,离心得川白芷多糖沉淀,弃上清液。川白芷多糖沉淀用水复溶,4000r/min离心10min除杂质,得上清液加乙醇浓度至60%,4℃静置12h后,第三次离心,取第三次离心后的沉淀,备用;
d:去蛋白
按照lg川白芷多糖沉淀加入200ml水的比例溶解样品,在50℃水浴加热并间歇性搅拌至其完全溶解,4000r/min离心5min除去杂质,川白芷多糖溶液ph调节到6.5。木瓜蛋白酶用ph6.5的pbs配成50mg/ml的酶溶液,按照lg川白芷多糖沉淀加20mg木瓜蛋白酶的比例加到上述的糖溶液中,然后在50℃水浴加热3h,再在100℃下灭酶15min,冷却至室温,4000r/min离心10min除去变性蛋白和酶。
在经过木瓜蛋白酶解后的混合溶液中加入等体积的sevag试剂(氯仿:正丁醇=4:1),剧烈振摇15min,5000r/min离心10min,离心后液体三分层,小心吸取上层清液并重复去蛋白10次后40℃旋转蒸发真空浓缩得到去蛋白川粗多糖。
e:透析去盐
8000-14000da透析袋处理液为0.01mol/lnahco3和lmmol/ledta的水溶液。将长度为25cm左右透析袋放入处理液加热煮沸半个h。蒸馏水清洗透析袋,置于50%乙醇中,4℃保存备用。将去蛋白川粗多糖溶液适量装于透析袋中,透析袋两头扎好后留出1/3的空间防止其中亲水性物质过度吸水使袋破裂。室温下用蒸馏水透析24h后,换干净蒸馏水再透析8h,重复2次。透析后浓缩、冻干得到川白芷粗多糖。
f:deae-琼脂糖凝胶ff柱层析
首先,将500ml的deae-琼脂糖凝胶ff装入2.5×100cm的中压层析柱中。柱子装好后用3倍柱体积的去蒸馏水,1ml/min流速进行平衡。
其次,将川白芷粗多糖配制成50mg/ml的水溶液样品,所有备用洗脱液要超声脱气,上样量5ml,流速lml/min,先以1.5倍柱体积的去蒸馏水洗脱,再以水和2mol/l的nacl混合洗脱,实现0~1.5mol/lnacl溶液的梯度洗脱,每10ml收集一管。每隔一管取样100μl用苯酚-硫酸法(100μl样品溶液+200μl5%苯酚溶液+1ml浓硫酸)检测多糖含量,绘制苯酚-硫酸反应曲线图,并绘制反应曲线。收集并合并第一个洗脱峰的收集液,浓缩,透析,冻干。川白芷多糖deae-琼脂糖凝胶ff色谱柱洗脱曲线如附图1和附图2所示。
g:琼脂糖凝胶6ff凝胶柱层析
将步骤f冻干后的川白芷多糖过琼脂糖凝胶6ff凝胶柱进行过滤层析,配制成30mg/ml的多糖水溶液,上样量10ml,洗脱液为0.05mol/lnacl溶液,流速为0.5ml/min,每管收集5ml。每管取样100μl用苯酚硫酸法检测多糖,绘制苯酚-硫酸反应曲线图,采用考马斯亮蓝g250法测定蛋白含量,并绘制反应曲线。收集主峰的洗脱液,浓缩,透析,冻干,即得。川白芷多糖初步分离组分的琼脂糖凝胶7ff色谱柱洗脱曲线如附图3所示。
实施例2
川白芷多糖的制备
川白芷多糖的制备方法,包括以下步骤:
步骤a:与实施例1的步骤a相比,烘干温度均为50℃,石油醚的用量按每克川白芷粉末计为2ml,其余条件均相同。
步骤b:称取脱脂脱色素后的粉末500g,加入2倍量含0.45%纤维素酶、ph值为4.5的柠檬酸-柠檬酸钠缓冲液,40℃水浴提取240min,沸水浴15min使酶灭活,4000r/min离心5min收集上清液,重复提取4次,合并提取液并浓缩,得浓缩液。
步骤c至步骤g同实施例1,即得。
实施例3
川白芷多糖的制备
川白芷多糖的制备方法,包括以下步骤:
步骤a:与实施例1的步骤a相比,烘干温度均为40℃,石油醚的用量按每克川白芷粉末计为4ml,其余条件均相同。
步骤b:称取脱脂脱色素后的粉末500g,加入6倍量含0.45%纤维素酶、ph值为4.5的柠檬酸-柠檬酸钠缓冲液,60℃水浴提取120min,沸水浴5min使酶灭活,4000r/min离心5min收集上清液,提取1次,合并提取液并浓缩,得浓缩液。
步骤c至步骤g同实施例1,即得。
实施例4
对实施例1得到的川白芷多糖及实施例1中步骤b所得浓缩液进行总糖检测,检测方法如下:
1mg/ml葡萄糖母液的配置:葡萄糖在烘箱中100℃烘干至恒重,称0.1033g葡萄糖于100ml容量瓶中,加蒸馏水溶解定容,备用。
葡萄糖标准曲线绘制:分别取适量1mg/ml葡萄糖母液,稀释至葡萄糖浓度为0.1、0.2、0.4、0.6、0.8、1mg/ml,吸取100μl葡萄糖溶液,然后加入5%苯酚200μl及1ml浓硫酸,摇匀,室温放置20min后于490nm下测吸光度。以加入葡萄糖浓度为横坐标,490nm下吸光度为纵坐标绘制葡萄糖标准曲线。
取实施例1中步骤b所得浓缩液干燥后,得酶提粗多糖样品;配制成1mg/ml的酶提粗多糖样品溶液,取100μl检测,做三组平行试验取平均值。
取实施例1得到的川白芷多糖,配制1mg/ml的不同样品溶液,取100μl检测,做三组平行试验取平均值。
以葡萄糖为标准品绘制标准曲线,苯酚-硫酸法检测总糖的含量,回归方程为y=2.6803x+0.1572,r2=0.994。根据样品测定的吸光度值对照标准曲线回归方程计算白芷多糖总糖含量,得到实施例1所得的川白芷多糖中总糖的含量为55.20±0.06%,实施例1步骤b所得的酶提粗多糖样品中总糖的含量为6.62±0.05%。
实施例5
对实施例1得到的川白芷多糖进行蛋白质分析,实验方法如下:
考马斯亮蓝g250是一种蛋白质染料,与蛋白质结合使之染色在595nm处有最大吸收,常用来定性和定量测定蛋白质,该方法操作简单、反应迅速适合大量样品的测定,在0-100mg/l蛋白质浓度范围内,吸光值与蛋白质含量呈良好的线性关系。
1mg/ml样品的水溶液,可见光-紫外光分光光度计进行400-200nm紫外扫描,以蒸馏水为空白对照。
蛋白质标准曲线绘制:称100mg考马斯亮蓝g250溶于50ml95%乙醇,加入100ml85%磷酸,蒸馏水定容至1l,滤纸过滤备用。配制0.1mg/ml牛血清白蛋白标准溶液,分别取0ml、0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,用蒸馏水补充至1.0ml,分别加入考马斯亮蓝g250溶液各5ml,混匀。放置2min后在595nm下测吸光度。
配制成lmg/ml的不同样品溶液,取lml检测,做三组平行试验取平均值。
以牛血清白蛋白为标准品绘制标准曲线,线性回归方程为:y=0.0025x+0.006,r2=0.9907。根据样品测定的吸光度值对照标准曲线回归方程计算白芷多糖蛋白质含量,得到实施例1所得的川白芷多糖中蛋白质含量为0.88%。由于测量存在一定误差,该值并不能代表样品蛋白质含量,只能说明样品中不含蛋白质或可能含有结合蛋白。
实施例6
对实施例1得到的川白芷多糖进行糖醛酸定性分析,实验方法如下:
咔唑试液的制备:精密称取0.125g咔唑,加无水乙醇溶解定容至100ml的量瓶中,备用。
配制0.4mg/ml葡萄糖醛酸标准溶液,1mg/ml的样品溶液。
取50μl样品溶液,加入50μl咔唑试液,冰浴加入300μl浓硫酸,观察颜色变化。
结果表明:实施例1得到的川白芷多糖醛糖酸检测与400μg/mld-葡萄糖醛酸显色一致,均呈蓝绿色阳性显色,说明其含有一定的醛糖酸,是酸性杂多糖。
实施例7
对实施例1最终得到的川白芷多糖及其步骤b浓缩液干燥后所得的酶提粗多糖进行碘-碘化钾反应检测,方法如下:
碘试剂为含0.02%碘的0.2%ki溶液,川白芷多糖及水提粗多糖的样品浓度均为lmg/ml。分别取50μl各样品溶液,加入200μl碘试剂,观察颜色变化后用酶标仪(thermoscientificmultiskango)进行200—800nm扫描。
结果显示,川白芷多糖与碘试剂反应呈阴性,酶提粗多糖与碘试剂反应后呈阳性显色反应,说明通过两次过柱后酶粗多糖中淀粉已几乎除尽。
将川白芷多糖样品溶液与碘试剂(含0.02%碘的0.2%ki溶液)混匀,测定300-700nm范围内的吸收光谱,在565nm附近有最大吸收,说明其有较少的分支和较短的侧链。
实施例8
对实施例1得到的川白芷多糖进行刚果红试验,具体实施方法如下:
配置1mg/ml川白芷多糖溶液,80μmol/l刚果红溶液,1mol/l的naoh溶液。
吸取0.5ml川白芷多糖溶液,加入80μmol/l刚果红溶液0.5ml,混匀。然后加入适量1mol/l的naoh溶液,使反应溶液中naoh的终浓度分别为0.0mol/l、0.1mol/l、0.2mol/l、0.3mol/l和0.4mol/l混匀室温静置后,200-800nm区间进行光谱扫描,测量不同浓度下溶液的最大吸收波长。另取空白对照,即不加川白芷多糖样品,以相同方法测量不同浓度下溶液的最大吸收波长。
比较样品溶液与空白对照溶液的最大吸收波长变化情况。
结果如附图4所示,随着naoh浓度从低到高,川白芷多糖的最大吸收波长呈先增加后减少的趋势,说明其可能具有三股螺旋结构。
实施例9
对实施例1得到的川白芷多糖进行多糖分子量分析,具体实验方法如下:
采用高效液相色谱-蒸发光散射检测法(hplc-elsd)对川白芷多糖分子量进行测定。hplc色谱条件如下:色谱柱:tsk-gelg5000pwxl凝胶柱(7.8mm×30cm);流动相:水;流速:0.5ml/min;进样量:20μl;检测器:alltech2000型elsd检测器,漂移管温度:115℃,气体流量:3.2(slpm)。
标准曲线的制备:取平均分子量分别为t10、t40、t70、、t500、t2000道尔顿的标准葡聚糖对照品,以水溶解,配制为2mg/ml的标准溶液。照上述hplc色谱条件检测,以标准葡聚糖的保留时间和分子量分别取自然对数绘制。
用高纯水将多糖样品制成2mg/ml的溶液,在相同色谱条件下进样分析,记录保留时间,带入回归方程算得平均分子量。
标准多糖参照品:blue2000(mw:2000kda),t-700(mw:700kda),t-500(mw:500kda),t-70(mw:70kda)t-40(mw:40kda),t-10(mw:10kda)经hplc‐elsd分析后。以保留时间对分子量对数作图,得到回归方程y=-1.99x+21.104,r2=0.9991。由回归方程可以计算得到川白芷多糖的表观分子质量。
结果表明,川白芷多糖的表观分子质量为3.93kda。
实施例10
对实施例1得到的川白芷多糖进行红外光谱和核磁共振分析
将干燥的川白芷多糖样品3.0mg与干燥的kbr在玛瑙研钵中研磨均匀后压片,在4000-400cm-1,范围内进行扫描。取20mg川白芷多糖溶于0.5ml重水中,核磁共振仪进行600mhznmr分析。
如附图5红外图谱显示,1022.20cm-1、1085.85cm-1、1095.49cm-1三处有吸收峰,表明川白芷多糖的糖环结构为呋喃糖型。在846.692cm-1处有吸收峰说明其存在α构型的多糖链接方式。在891±7cm-1的吸收峰说明存在β构型多糖链接方式。
在1647.095cm-1附近的吸收峰表明在川白芷多糖各组分中存在乙酰氨基(-nhcoch3)的c=o键伸缩振动,说明川白芷多糖可能是一种氨基多糖。在1733.886cm-1和1261.359cm-1为酰基或o-乙酰基(o-ac)的特征吸收峰。
综上,川白芷多糖为同时存在α构型和β构型多糖链接方式,具有酰基或o-乙酰基(o-ac)的一种呋喃糖型氨基多糖片段。
如附图6核磁共振图谱显示,川白芷多糖在1hnmr信号指纹区δ4.4-5.5ppm,有信号δ4.91、δ5.18和δ5.34ppm,可见其有至少3种单糖残基,h信号δ4.91表示多糖为存在β构型链接糖残基,而h信号δ5.18、δ5.34表明酶峰1也存在α构型链接糖残基并且以α构型为主。在13cnmr中,在δ90-112ppm区域,有δ99.68、95.83、91.38ppm共振信号表示川白芷多糖存在α构型链接糖残基,这与1hnmr分析结果一致。在1hnmr,δ4.68ppm的共振峰为doh吸收峰,而在δ3.5-4.4ppm之间的信号主要是由于糖残基上c2-c6上的h信号位移峰[87],在δ3.3-3.5ppm之间,有h信号,表示存在meo。在13cnmr中,在δ181.5ppm的共振峰表示存在糖醛酸,δ70-75ppm的共振区为未发生取代多糖部位(c2、c3、c4)的碳信号,而在δ76-85ppm的c信号表示c2、c3、c4上发生取代,而在δ67ppm附近有c信号,表示c6位也发生了取代,在δ60ppm附近的c信号表示存在meo基团。
综上,川白芷多糖可能为多种糖残基组成并且同时存在α构型和β构型链接糖残基的酸性杂多糖,还有meo、糖醛酸等基团,在c2、c3、c4、c6均可能存在取代。
实施例11
对比例
采用sb25-12dtd超声清洗机(宁波新芝生物科技股份有限公司)超声提取制备川白芷多糖,包括以下步骤:
步骤a:同实施例1中的步骤a。
步骤b:称取步骤a所得的白芷粉样100g,加入3倍量去蒸馏水400w超声1h,然后4000r/min离心5min,收集上清液,重复提取一次,合并提取液并浓缩。
步骤c至步骤e同实施例1。
步骤f中,收集并合并第三个洗脱峰的收集液,其余条件同实施例1。
步骤g同实施例1,得超声提取川白芷多糖。
实施例12
对比例
采用热水提取法制备川白芷多糖,包括以下步骤:
步骤a:同实施例1中的步骤a。
步骤b:将脱脂脱色素后的粉末加水于81℃浸提3次,每次浸提1h,所述水与川白芷粉末的用量比为68ml/g,合并提取液,过滤,滤液浓缩,得浓缩液。
步骤c至步骤e同实施例1。
步骤f中,收集并合并第二个洗脱峰的收集液,其余条件同实施例1。
步骤g同实施例1,得热提川白芷多糖。
实施例13
对实施例1得到的川白芷多糖、实施例11得到的超声提取川白芷多糖,实施例12得到的热提川白芷多糖进行美白活性分析,具体实验方法如下:
采用酪氨酸酶多巴速率氧化法,以酪氨酸酶抑制率为指标进行评价,具体操作如下。
试剂配制
(1)pbs缓冲液配制
0.2mol/l的nah2po4:称取nah2po4·h2o3.12g加蒸馏水定容至100ml溶解。
0.2mol/l的na2hpo4:称取na2hpo4·2h2o3.56g加蒸馏水定容至100ml溶解。
0.1mol/l的pbs,49ml0.2mol/l的na2hpo4,51ml0.2mol/l的na2hpo4,加100ml蒸馏水稀释。
(2)多巴配制
取0.075g左旋多巴,用pbs缓冲液定容到100ml容量瓶。
(3)酪氨酸酶配制
取0.5mg的酪氨酸酶,用pbs缓冲液溶解,定容值2ml。酶活力单位在250u/ml左右。
(4)样品处理
将待测样品配置成1mg/ml、0.5mg/ml、0.25mg/ml,0.125mg/ml,0.0625mg/ml,0.03125mg/ml、0.01625mg/ml,相应浓度曲酸做阳性对照,反应体系如下表。
酪氨酸酶抑制反应体系表
按照顺序加入多巴、pbs、多糖/曲酸,加入酶液后迅速放入37℃的酶标仪孵育5min测值,每个样品做三个重复。
a:处理组样品在475nm处的吸光度;
a0:空白组样品在475nm处的吸光度;
b:对照组样品在475nm处的吸光度;
b0:总空白组样品在475nm处的吸光度。
如附图7所示,结果表明,本发明川白芷多糖、热提川白芷多糖和超声提取川白芷多糖对酪氨酸酶活性均具有抑制作用;且随着浓度的增加,酪氨酸酶抑制率逐渐升高,呈现明显的量效关系。本发明川白芷对多糖酪氨酸酶抑制活性明显大于热提川白芷多糖和超声提取川白芷多糖。
上述实施例仅为本发明的优选实施方式之一,不应当用于限制本发明的保护范围,但凡在本发明的主体设计思想和精神上作出的毫无实质意义的改动或润色,其所解决的技术问题仍然与本发明一致的,均应当包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!