培育抗大豆胞囊线虫转基因植物的方法与流程
本发明涉及植物基因工程技术领域,尤其涉及一种培育抗大豆胞囊线虫转基因植物的方法。
背景技术:
大豆胞囊线虫病(soybeancystnematode,scn)是由大豆胞囊线虫(heteroderaglycineslchinohe)引起的一种世界性大豆病害,在世界各大豆主要生产国如美国、巴西、阿根廷、中国等均广泛分布。据统计,scn每年可造成大豆减产7%~10%,严重地块可达15-50%,甚至绝产,每年造成约10-15亿美元的经济损失。scn在我国东北和黄淮海大豆主产区均有发生,其中又尤以黑龙江省、吉林省中西部及内蒙古的盐碱地、白浆土和沙壤土地发生较为严重,每年危害面积达2.0×106hm2以上。除危害大豆外,scn还危害小豆,赤小豆、饭豆、菜豆等170多种作物。
大豆胞囊线虫是一种定居型内寄生线虫,主要通过2龄幼虫侵染大豆根部维管束,危害大豆生长发育,造成植株发育不良,根系减少,根上附有白色的球状物,茎和叶变淡黄色,豆荚和种子萎缩瘪小,甚至不结荚。大豆胞囊线虫是严格的专性寄生物,存在明显的寄主分化现象,不同生理小种致病性存在明显差异。按照riggs等提出的分类标准,scn存在16个生理小种。目前在我国已发现1、2、3、4、5、7、14号小种,其中东北大豆产区主要是3号小种,另外还有1号、4号和14号;黄淮海大豆产区主要以1号和4号小种为主。
scn作为一类土传病害,其分布与大豆种植区域高度重叠,是一种极难防治的土传病害。由于目前尚无有效可使用的化学杀线虫剂,生产上主要通过种植抗性品种和田间轮作控制scn的发生与危害。由于胞囊在土壤中存活时间较长(9-10年),采用轮作等方式虽然可以减少大豆产量损失,但并不能完全控制scn的危害,实际应用效果有限。目前生产上利用的抗线虫大豆品种主要来源于peking、pi88788等少数几个抗源品种,其中,具有peking或pi88788血缘的品种占所有抗线虫大豆品种的97%以上,抗性遗传基础极为狭窄。同时,由于大豆胞囊线虫生理小种较为复杂,易造成线虫产生抗性,目前已报道多个胞囊线虫生理小种对pi88788血缘的抗线虫品种产生了抗性。因此,如何进一步开发新型抗线虫资源,拓宽抗性资源遗传基础,加快抗线虫大豆品种的选育是我国抗病大豆育种中面临的主要问题之一。
rna沉默(rnainterference,rnai)是植物抵抗外来核酸(病毒、转座子等)入侵以保持自身基因组完整性的一种防御机制。双链rna(double-strandedrna,dsrna)导入细胞后被切割成21-23个核苷酸长度的短核苷酸双链,在其它作用因子的参与下能够特异地与其同源的mrna结合并导致该同源mrna降解,进而导致内源基因发生沉默。rnai作为一种简单有效的基因敲除的工具,已广泛应用于秀丽隐杆线虫(c.elegans)等多种线虫基因功能研究及转基因研究。目前,国际上已完成基因组测序工作的线虫有10种(nematode.net),其中2种为植物寄生线虫,包括北方根结线虫(meloidogynehapla)和南方根结线虫(m.incognita)。截至2011年,nematode.net数据库中已经有超过一千万个ests序列和二十多万个来自50多种线虫的基因组序列。尽管目前h.glycines基因组测序工作尚未完成,但利用已有的ests数据库信息,结合模式生物c.elegans生物学数据信息,筛选与大豆胞囊线虫寄生、发育及繁殖等相关的关键基因,开展大豆胞囊线虫基因功能验证和转基因研究提供了极大的便利。
目前已发现h.glycines和c.elegans基因组中共有8334个基因存在保守性,其中1508个基因在c.elegans中为致死基因。通过dsrna溶液浸泡或毛状根表达系统,进一步研究这些致死基因在h.glycines中的功能时发现,抑制这些基因的表达可以显著降低胞囊线虫的胞囊或雌虫数量。urwin等(2002)利用半胱氨酸蛋白酶编码基因(hgcp-i)dsrna溶液浸泡线虫后,寄生胞囊线虫雌虫和胞囊数都显著降低。klink等(2009)利用毛状根表达系统研究了核糖体蛋白3a(hg-rps-3a)、核糖体蛋白4(hg-rps-4)、剪接体蛋白(hg-spk-1)及突触小泡蛋白(hg-snb-1)的抑制表达对h.glycines的影响。结果表明,抑制上述4个基因的表达,均可以显著降低毛状根中胞囊线虫的雌虫数量。li等(2010)利用毛状根表达系统对h.glycines的3个靶基因(y25、prp-17、cpn-1)进行rnai研究表明,与对照相比,转基因毛状根中的胞囊数量降低79-95%。此外,huang等(2006年)将线虫寄生必需的分泌肽-16d10基因反向重复序列导入拟南芥,转基因植物对4种根结线虫均产生较高的抗性。steeves等(2006)将h.glycines精子蛋白基因msp的一段反向重复序列构建到rnai表达载体上并转化大豆。接种鉴定结果表明,转基因大豆植株根部胞囊数比对照降低68%。可见,rnai介导h.glycines基因沉默技术不仅可以为胞囊线虫抗性育种研究提供了更为广泛的抗源基因。同时,通过选择高度保守的rnai靶序列,也为兼抗多个胞囊线虫生理小种提供了一条更为有效的技术途径。
目前,有关rnai介导h.glycines基因沉默的研究主要集中在基因功能验证方面,尚未见到利用胞囊线虫hg-snb1基因片段培育抗大豆胞囊线虫转基因植物的技术资料。基于上述理解,本研究通过rnai介导胞囊线虫hg-snb1基因沉默,获得了对scn抗性显著提高的转基因大豆。在此研究基础上,提出本发明。
技术实现要素:
针对上述技术中存在的不足之处,本发明提供一种培育抗大豆胞囊线虫转基因植物的方法,本发明对于培育抗大豆胞囊线虫植物(特别是大豆)具有重要的应用价值。
为实现上述目的,本发明提供一种培育抗大豆胞囊线虫转基因植物的方法,该方法为将dna片段与根特异性启动子组合导入目标植物,使所述dna片段主要在目标植物根中特异表达,获得对大豆胞囊线虫抗性显著提高的转基因植物。
其中,所述dna片段与根特异性启动子组合形成培育抗大豆胞囊线虫转基因植物的方法,所述根特异性启动子可以驱动下游基因或dna片段在植物根中的特异性表达。
其中,该载体还包括含有dna片段的质粒,dna片段包括dna片段1,dna片段2,以及位于所述dna片段1和dna片段2之间的间隔序列;且所述dna片段1和dna片段2反向互补。
其中,所述dna片段从上游至下游依次包括如下元件:根特异性启动子、dna片段1、间隔序列、dna片段2和终止子。
其中,所述在目标植物中表达dna片段的实现方式有:将上述任一所述质粒导入目标植物。
其中,在转基因植物或细胞中含有所述dna片段,其主要在植物根细胞中特异表达,并直接或间接调控寄生大豆胞囊线虫内源基因的表达。
其中,所述目标植物和转基因植物为双子叶植物或单子叶植物,所述双子叶植物为大豆。
本发明的有益效果是:与现有技术相比,本发明提供的培育抗大豆胞囊线虫转基因植物的方法,本发明将dna片段与根特异性启动子组合导入目标植物,使所述dna片段主要在目标植物根中特异表达,获得对大豆胞囊线虫抗性显著提高的转基因植物。本发明对于培育抗大豆胞囊线虫植物(特别是大豆)具有重要的应用价值。
附图说明
图1为重组质粒pcambia3301-cpn1rnai的结构示意图
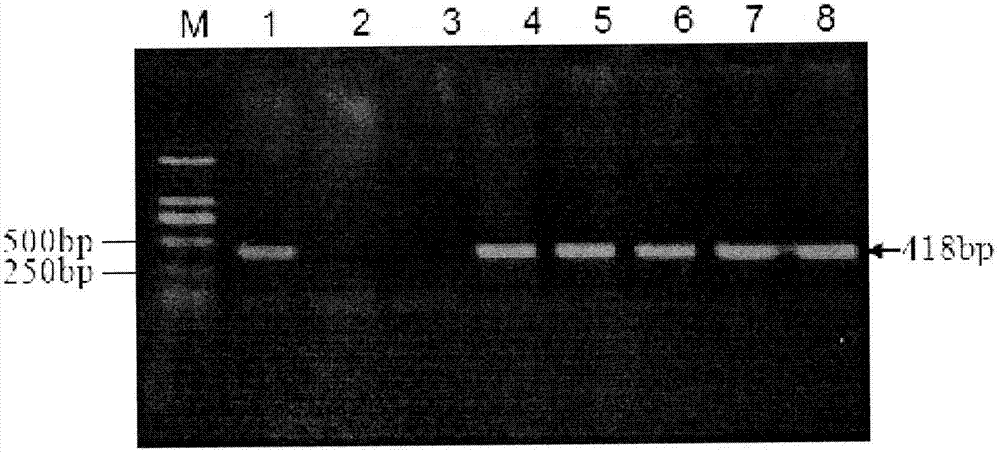
图2为t1代pat/bar试纸条阳性植株pcr鉴定结果.m:dna标准分子量,1:质粒pcambia3301-cpn1rnai,2:非转化植株,3:ddh20对照,4~8为pat/bar试纸条阳性植株c845、c815、c810、c842、c847图
图3为t1代转基因大豆southern杂交鉴定结果.m:dna标准分子量,1:质粒pcambia3301-cpn1rnai,2:非转化植株,3~7为t1代转基因植株c810、c815、c845、c842、c847.植物基因组dna(50μg)用ecori酶切后,用dig标记cpn1基因探针进行检测图;
图4为t3代转基因大豆植株接种大豆胞囊线虫3号生理小种鉴定图;
图5为dna片段的seq-1序列图;
图6为根特异性启动子的seq-2序列图;
图7为质粒中dna片段的seq-3序列图。
具体实施方式
为了更清楚地表述本发明,下面结合附图对本发明作进一步地描述。
本发明的培育抗大豆胞囊线虫转基因植物的方法,该方法为将dna片段与根特异性启动子组合导入目标植物,使所述dna片段主要在目标植物根中特异表达,获得对大豆胞囊线虫抗性显著提高的转基因植物。
在本实施例中,所述dna片段与根特异性启动子组合形成培育抗大豆胞囊线虫转基因植物的方法,所述根特异性启动子可以驱动下游基因或dna片段在植物根中的特异性表达。所述dna片段如图5中的seq-1所示,所述根特异性启动子序列如图6中的seq-2所示。
在本实施例中,该载体还包括含有dna片段的质粒,dna片段包括dna片段1,dna片段2,以及位于所述dna片段1和dna片段2之间的间隔序列;且所述dna片段1和dna片段2反向互补。dna片段如图7中的seq-3所示。
在本实施例中,所述dna片段从上游至下游依次包括如下元件:根特异性启动子、dna片段1、间隔序列、dna片段2和终止子。
在本实施例中,所述dna片段可以通过反转录获得cdna片段,或是通过人工合成获得;所述根特异性启动子可以通过pcr扩增获得,或是通过人工合成获得。
在本实施例中,所述在目标植物中表达dna片段的实现方式有:将上述任一所述质粒导入目标植物。
在本实施例中,在转基因植物或细胞中含有所述dna片段,其主要在植物根细胞中特异表达,并直接或间接调控寄生大豆胞囊线虫内源基因的表达。
在本实施例中,所述目标植物和转基因植物为双子叶植物或单子叶植物,所述双子叶植物为大豆。更具体可为栽培大豆,所述质粒可通过农杆菌介导法、基因枪法、花粉管介导法等导入目标植物。
本发明中dna片段与根特异性启动子的组合或是上述任一质粒在培育抗大豆胞囊线虫转基因植物的应用。另外,dna片段与根特异性启动子的组合或是上述任一质粒在制备大豆胞囊线虫生防制剂中的应用。大豆胞囊线虫具体可为大豆胞囊线虫3号生理小种,尽管本发明仅利用大豆胞囊线虫3生理号小种进行了接种鉴定,但所述dna片段为保守序列,具有广谱性,所以对其它大豆胞囊线虫生理小种也具有相同的抑制效果。
相较于现有技术,本发明提供的培育抗大豆胞囊线虫转基因植物的方法,具有如下优势:本发明将dna片段与根特异性启动子组合导入目标植物,使所述dna片段主要在目标植物根中特异表达,获得对大豆胞囊线虫抗性显著提高的转基因植物。本发明对于培育抗大豆胞囊线虫植物(特别是大豆)具有重要的应用价值。
以下实施例进一步说明本发明的内容,但不应理解为对本发明的限制。下述实施例中的实验方法,如无特殊说明,均为本领域技术人员所熟知的常规方法。实施例中所用的试验材料,如无特殊说明,均可通过商业途径获得。
质粒phannibal-attb1:公众可以从吉林省农业科学院农业生物技术研究所获得。
质粒pcambia3301:公众可以从商业途径获得。
栽培大豆品种williams82:公众可以从国家作物种质资源数据库(http://www.cgris.net/query/croplist.php)获得。
大豆胞囊线虫3号生理小种:为我国东北大豆产区scn主要生理小种,公众可以从吉林省农业科学院植物保护研究所获得。参考文献:段玉玺周博陈立杰吴海燕.抗大豆胞囊线虫3号生理小种(scn3)核心种质代表性分析.大豆科学,2008,3:366-372.
实施例中所用到的各种培养基配方见表1。ms合成盐购自sigma公司,货号为m5524。b5合成盐购自sigma公司,货号为g5768。
表1.培养基配方
实施例1.大豆胞囊线虫hg-cpn-1基因片段及大豆根特异性启动子的发现
利用线虫基因组数据库(nematode.net)及大豆胞囊线虫ests数据库信息,发现大豆胞囊线虫基因hg-cpn-1存在保守序列。在该保守区内最终确定了序列长度为418bp的片段,如seq-1所示。预期通过seq-1所示片段编码的rna抑制胞囊线虫。
利用phytozome数据库(https://phytozome.jgi.doe.gov/pz/portal.html)、大豆公共数据库(http://soybase.org/),根据大豆根特异表达基因glyma.11g236700.1上游序列设计特异性引物gmp-f1/gmp-r1。以大豆栽培品种williams82基因组dna为模板,利用kodfx高保真酶(日本toyobo公司)进行pcr扩增,pcr反应条件为:95℃5min;(94℃30s,59℃45s,72℃2min),30个循环;72℃10min。用1.0%琼脂糖凝胶电泳回收纯化pcr扩增产物并测序。扩增产物长度为1411bp,如seq-2所示。
gmp-f1:5’-tgaatcatatcttatcaactagttag-3’
gmp-r1:5’-cccaatggaagacatgttattctctgcaaaac-3’。
实施例2.rnai重组质粒的构建
(1)以seq-2所示的双链dna分子为模板,采用gmp-f2-attb5r和gmp-r2组成引物对,用kodfx高保真酶进行pcr扩增,得到pcr扩增产物。
gmp-f2-attb5r:5’-cgagctcggggacaactttgtatacaaaagttgttgaatcaaagcttatcaactagttag-3’
gmp-r2:5’-ccgctcgagcccaatggaagacatgttattctctgcaaaac-3’
gmp-f2-attb5r和gmp-r2引物对中,下划线标注酶切识别序列,其中,“gagctc”为限制性内切酶saci的酶切识别序列。“ctcgag”为限制性内切酶xhoi的酶切识别序列,“ggggacaactttgtatacaaaagttgtt”为重组位点attb5r。
pcr反应条件为:95℃2min;(94℃30s,56℃45s,72℃2min),共35个循环;72℃10min。用1.0%琼脂糖凝胶电泳回收纯化pcr扩增产物,产物大小约为1.42kb。
(2)用限制性内切酶saci和xhoi双酶切步骤(1)的pcr扩增产物,并回收酶切产物。
(3)用限制性内切酶saci和xhoi双酶切质粒phannibal-attb1,回收载体骨架序列。
(4)将步骤(2)的酶切产物和步骤(3)的载体骨架序列连接,得到重组质粒phannibal-attb5r-gmp-attb1。
(5)合成序列表的seq-1所示的双链dna分子。
(6)以步骤(5)合成的双链dna分子为模板,采用hg-cpn1-f1和hg-cpn1-r1组成的引物对
hg-cpn1-f1:5’-ccgggatccctcgagaaggcatcgatcaggctgtg-3’
hg-cpn1-r1:5’-ggttcgaaggtacctcgctttttgtatcctccgc-3’
hg-cpn1-f1:与hg-cpn1-r1:中,下划线标注酶切识别序列,其中“ggatcc”为限制性内切酶bamhi的酶切识别序列,“ctcgag”为限制性内切酶xhoi的酶切识别序列,“ttcgaa”为限制性内切酶bstbi的酶切识别序列,“ggtacc”为限制性内切酶kpni的酶切识别序列。
pcr反应条件为:95℃2min;(94℃30s,56℃45s,72℃1min),共30个循环;72℃5min。用1.0%琼脂糖凝胶电泳回收纯化pcr扩增产物,产物大小约为418bp。
(7)用限制性内切酶xhoi和kpni双酶切步骤(6)的pcr扩增产物,并回收酶切产物。
(8)用限制性内切酶xhoi和kpni双酶切质粒phannibal-attb5r-gmp-attb1,回收载体骨架序列(约6.0kb)。
(9)将步骤(7)的酶切产物和步骤(8)的载体骨架序列连接,得到重组质粒phannibal-gmp-revcpn1。
(10)用限制性内切酶bamhi和bstbi双酶切步骤(6)的pcr扩增产物,并回收酶切产物。
(11)用限制性内切酶bamhi和bstbi双酶切重组质粒phannibal-gmp-revcpn1,回收载体骨架序列(约6.3kb)。
(12)将步骤(10)的酶切产物和步骤(11)的载体骨架序列连接,得到重组质粒phannibal-gmp-cpn1rnai。
(13)用限制性内切酶saci和bamhi双酶切质粒phannibal-gmp-cpn1rnai,回收约2.88kb酶切小片段(该片段自上游至下游依次包括大豆根特异启动子gmp,正义rna片段的编码序列,间隔内含子序列,反义rna片段的编码序列。酶切dna片段两端均为粘末端。
(14)用限制性内切酶saci和bamhi双酶切质粒pcambia3301,回收载体骨架序列(约9.5kb),dna酶切片段两端均为粘末端。
(15)将步骤(13)的酶切小片段和步骤(14)的载体骨架序列连接,得到rnai重组质粒pcambia3301-cpn1rnai。载体质粒示意图见图1。
实施例3.抗大豆胞囊线虫转基因植物的获得与鉴定
1.农杆菌介导大豆遗传转化
(1)采用冻融法将重组质粒pcambia3301-cpn1rnai导入农杆菌eha105,得到重组农杆菌。
(2)用附表1中的ccm液体培养基重悬农杆菌,得到od600nm=0.5-0.8的菌悬液备用。
(3)选取栽培大豆williams82种子,在含有氯气的密闭容器中灭菌12-16个小时。
(4)取步骤(3)中灭菌后的种子,种脐朝下置于gm培养基平板上,23℃暗培养24小时。
(5)用无菌解剖刀沿种脐切开,并去掉腋芽,在腋芽上方子叶节位置轻微划伤,然后置于步骤(2)得到的菌悬液中浸泡30-40分钟。
(6)完成步骤(5)后,将种子移至铺有无菌滤纸的ccm培养基平板上,23℃暗培养4-5天。
(7)完成步骤(6)后,将种子转移至sim培养基上,25℃,16h(光)/8h(暗)条件下培养,每2-3周继代1次。
(8)将诱导出丛生芽的外植体上的子叶切去,置于sem培养基平板上,25℃,16h(光)/8h(暗)条件下培养,每2-3周继代1次,继代时切去褐化的愈伤组织。
(9)当再生芽生长至4-8cm时,将再生芽切下,然后转移至rm培养基平板上,25℃,16h(光)/8h(暗)条件下培养。
(10)待抗性芽长至3-5cm,且长出健壮的根时,移至炼苗室驯化3-5天,然后转移至温室中生长,得到t0代植株。
(11)对移栽至温室的t0代植株进行pat/bar试纸条(libertylinkstrips,envirologix,usa)检测。试纸条检测阳性的植株即初步鉴定为转基因植株。
2.转基因大豆分子鉴定
(1)pat/bar试纸条试纸条检测的阳性t0代植株自交后即获得t1代株系。采用500mg/l除草剂-草丁膦进行喷施,7天后非转基因苗出现叶片黄化或枯死现象,而转基因植株则表现正常。
(2)剪取t1代pat/bar试纸条阳性植株叶片进行pcr检测。提植株叶片的基因组dna,采用hg-cpn1-f2和hg-cpn1-r2组成的引物进行pcr扩增,扩增片段大小为418bp。部分幼苗pcr鉴定结果见图2。
hg-cpn1-f2:5’-aaggcatcgatcaggctgtggcag-3’
hg-cpn1-r2:5’-tcgctttttgtatcctccgccac-3’
(3)剪取t1代转基因植株叶片进行southern杂交检测。采用高盐ctab法提取高纯度大豆基因组dna。ecori酶切总dna(~50μg)后,用0.8%琼脂糖凝胶分离酶切片段。采用高盐转移法,将酶切片段转移至带正电荷的hyboodtm-n+尼龙膜(amersham,usa)上。以重组质粒pcambia3301-cpn1rnai为模板,采用hg-cpn1-f2和hg-cpn1-r2引物对扩增cpn1基因片段,扩增片段大小为418bp。利用dig随机引物标记试剂盒(roche,usa)标记探针。
采用roche公司的dighighprimednalabelinganddetectionstarterkitii试剂盒进行分子杂交。杂交温度为42℃,洗膜条件为2×ssc(含0.1%sds),37℃条件下洗膜2次,每次5min;0.5×ssc(含0.1%sds),66℃条件下洗膜2次,每次15min。然后利用bcip/nbt在杂交膜上化学显色。部分转基因大豆southern杂交检测结果见图3。
(4)收获southern杂交阳性的t1代植株自交后得到的种子,播种后长出t2代幼苗,继续采用500mg/l除草剂-草丁膦和pcr进行跟踪检测,直至获得纯合的转基因株系。
4.转基因大豆抗胞囊线虫鉴定
采用病土接种法(病土采自吉林省洮南地区),对t3代转基因大豆株系c845、c815、c810、c842和c847进行抗性鉴定,受体对照品种为栽培大豆品种williams82。采用golden(1970)确立的一套鉴别寄主,riggs(1988)给定的鉴定模式,确定接种土样中胞囊线虫为3号小种。抗性分级参照shannon1992年提出鉴定大豆抗病性的ip标准进行分级,高抗0~9%(hr),中抗10%~30%(mr),中感31%~60%(ms),高感大于60%(vs)。ip指数=(供试品种的平均胞囊量/感病对照品种的平均胞囊量)×100。抗性鉴定试验重复3次,每个重复鉴定植株15株。
鉴定结果表明,与受体对照品种相比,转基因大豆c845、c815、c810、c842和c847平均胞囊数量均显著降低,表现为中抗(mr)。对照非转基因大豆williams82表现为中感(平均胞囊数量为)。表明将重组rnai表达载体导入植物,可以显著提高植物对大豆胞囊线虫的抗性。抗性鉴定结果见表2和图4。
表2.t3代转基因大豆抗scn鉴定结果
以上公开的仅为本发明的几个具体实施例,但是本发明并非局限于此,任何本领域的技术人员能思之的变化都应落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!