大豆耐盐基因GmCHS5及其应用的制作方法
本发明属于基因工程技术领域,具体涉及大豆耐盐基因gmchs5及其应用。
背景技术:
土壤盐渍化是一个全球性的生态问题,全世界接近20%的灌溉土壤受到盐渍化的威胁,且耕地盐渍化状况日趋严峻。其中,我国约有3460万公顷的土地遭受盐渍化危害,约占全国可耕地面积的25%。土壤中高浓度的盐分导致植物生长减缓甚至死亡,是限制作物产量的主要非生物逆境因子之一。
如何减轻盐胁迫对农业生产的影响,从而使大面积的盐碱地、荒漠化土地和丰富的盐水资源可以为农业生产所利用,这是未来农业发展所面临的一个重要课题。土壤中盐胁迫涉及多种基因和大分子的协调作用,主要通过na+对植物造成损伤。na+进入细胞后引起过氧化物的积累,过量的活性氧化物会损害蛋白质、核酸以及其它大分子的结构与功能,甚至导致细胞死亡
目前,研究耐盐胁迫的主要方法是耐盐植物盐胁迫下相关基因的表达产物蛋白的研究日益受到重视,通过对基因敲除突变体或转基因重组体与野生型个体间蛋白图谱的差异进行对比分析,从而实现对新基因的功能分析。目前,公开的耐盐基因很多,例如,耐盐基因ccsos1具有菊花耐盐功能(cn102559702a)、ospex11基因具有提高水稻耐盐性中的应用(cn105838722a),耐盐基因taopr具有提高小麦耐盐性中的应用(cn102121008a)、耐盐基因tasod2具有提高小麦耐盐性中的应用(cn104328128a)。但是目前大豆耐盐基因报道较少,其中较为常见的为大豆耐盐基因gmcipk2(cn104726476a)和大豆耐盐基因gmcbl3(cn104726479a),但是这两种基因具体是在提高拟南芥的耐盐性方面有较好的生物学功能。
技术实现要素:
有鉴于此,本发明的目的在于提供提供大豆耐盐基因gmchs5及其应用,使所述基因或重组载体能够有效提高大豆的耐盐性。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种大豆耐盐蛋白gmchs5,具有如序列表中seqidno.1所示的氨基酸序列。
本发明提供了所述的大豆耐盐蛋白gmchs5的编码基因,具有如序列表中seqidno.2所示的核苷酸序列。
本发明提供了一种含有所述大豆耐盐蛋白gmchs5的编码基因的表达载体pdl28-ha-gmchs5-gfp。
本发明提供了所述的编码基因或所述的表达载体pdl28-ha-gmchs5-gfp在培育抗盐植物中的应用。
优选的,所述抗盐植物包括大豆。
优选的,所述大豆包括中黄35品种。
本发明还提供了一种提高大豆耐盐性的方法,其特征在于,包括以下步骤:
1)克隆大豆耐盐蛋白gmchs5的编码基因,得到大豆耐盐蛋白gmchs5的编码基因;
2)将所述大豆耐盐蛋白gmchs5的编码基因构建重组载体,将得到的重组载体转化入宿主菌中,筛选阳性菌,提取阳性质粒;
3)将所述步骤2)得到的阳性质粒构建真核表达载体;
4)将所述步骤3)得到的真核表达载体转化入大豆宿主。
优选的,所述步骤1)中克隆用的引物对具有序列表中seqidno.3和seqidno.4所示的核苷酸序列。
优选的,所述步骤4)中转化的方法为农杆菌转化。
本发明提供了一种大豆耐盐蛋白gmchs5,具有如序列表中seqidno.1所示的氨基酸序列。本发明提供了所述的大豆耐盐蛋白gmchs5的编码基因,具有如序列表中seqidno.2所示的核苷酸序列。本发明提供了一种含有权利要求2所述大豆耐盐蛋白gmchs5的编码基因的表达载体pdl28-ha-gmchs5-gfp。本发明利用植物基因工程技术,通过根瘤农杆菌介导的方法将基因转入大豆中,gmchs5过量表达的转基因大豆根耐盐能力显著提升。经过200mm的高浓度盐水处理后,转基因大豆根的鲜重是野生型盐处理组的1.20倍,是基因沉默组的2.08倍。
附图说明
图1为本发明构建的表达载体图谱;
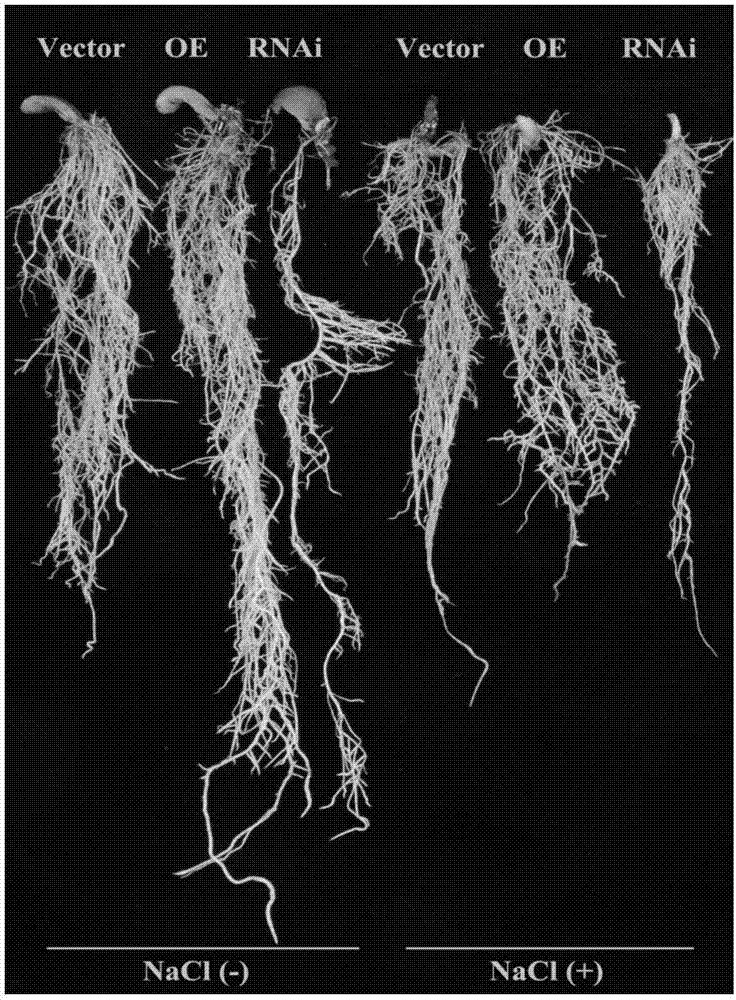
图2为实施例4中各实验组盐胁迫表型结果。
具体实施方式
本发明提供了一种大豆耐盐蛋白gmchs5,具有如序列表中seqidno.1所示的氨基酸序列。
本发明中,所述一种大豆耐盐蛋白gmchs5的制备方法没有特殊限制,采用本领域技术人员所熟知的蛋白合成方法即可。
本发明提供了所述大豆耐盐蛋白gmchs5的编码基因,具有如序列表中seqidno.2所示的核苷酸序列。
本发明中,所述大豆耐盐蛋白gmchs5的编码基因的合成方法没有特限制,采用本领域技术人员所熟知的合成方法即可。
本发明中,所述大豆耐盐蛋白gmchs5的编码基因的获得方法,优选采用根据大豆的cdna为模板扩增得到。所述扩增用引物优选为oechs5-f和oechs5-r。所述oechs5-f优选为具有序列表中seqidno.3所示的核苷酸序列的引物。所述oechs5-r优选为具有序列表中seqidno.4所示的核苷酸序列的引物。
本发明提供了一种含有所述大豆耐盐蛋白gmchs5的编码基因的表达载体pdl28-ha-gmchs5-gfp。
本发明中,含有所述大豆耐盐蛋白gmchs5的编码基因的表达载体pdl28-ha-gmchs5-gfp构建方法没有特殊限制,采用本领域技术人员所熟知的构建方法即可。
本发明提供了所述的编码基因或所述的表达载体pdl28-ha-gmchs5-gfp在培育抗盐植物中的应用。
本发明中,所述抗盐植物优选包括大豆。所述大豆优选包括中黄35品种。
本发明还提供了一种提高大豆耐盐性的方法,其特征在于,包括以下步骤:
1)克隆大豆耐盐蛋白gmchs5的编码基因,得到大豆耐盐蛋白gmchs5的编码基因;
2)将所述大豆耐盐蛋白gmchs5的编码基因构建重组载体,将得到的重组载体转化入宿主菌中,筛选阳性菌,提取阳性质粒;
3)将所述步骤2)得到的阳性质粒构建真核表达载体;
4)将所述步骤3)得到的真核表达载体转化入大豆宿主。
本发明先克隆大豆耐盐蛋白gmchs5的编码基因,得到大豆耐盐蛋白gmchs5的编码基因。
本发明中,所述步骤1)中克隆用的引物对优选具有序列表中seqidno.3和seqidno.4所示的核苷酸序列。
得到大豆耐盐蛋白gmchs5的编码基因后,本发明将所述大豆耐盐蛋白gmchs5的编码基因构建重组载体,将得到的重组载体转化入宿主菌中,筛选阳性菌,提取阳性质粒。
本发明中,所述构建重组载体的方法没有特殊限制,采用本领域技术人员所熟知的构建方法即可。所述转化的方法没有特殊限制,采用本领域技术人员所熟知的转化方法即可。所述筛选阳性菌和提取阳性质粒的方法没有特殊限制,采用本领域技术人员所熟知的方案即可。
得到阳性质粒后,本发明将所述阳性质粒构建真核表达载体。
本发明中,所述构建真核表达载体的方法没有特殊限制,采用本领域技术人员所熟知的构建方法即可。
得到真核表达载体后,本发明将所述的真核表达载体转化入大豆宿主。
本发明中,所述转化的方法优选为农杆菌转化。
得到转化后的大豆宿主后,本发明优选对大豆宿主进行验证。
所述验证的方法优选采用基因敲除的方法验证。所述基因敲除的方法优选包括一下步骤:a.构建基因沉默载体;b.将所述基因沉默载体侵染有真核表达载体转化的转基因大豆根中;c.将所述基因沉默载体侵染的转基因大豆根与未被侵染的转基因大豆根同时进行盐处理,统计两者的根部鲜重;若基因沉默的盐处理组与基因沉默的对照组相比,转基因大豆根的鲜重差异明显,则判断为耐盐蛋白gmmyb173的编码基因具有耐盐特性。
本发明中,所述基因沉默载体包括沉默基因和重组质粒。
本发明中,所述沉默基因的构建方法优选包括如下步骤:
1)用软件找出gmchs5序列中的沉默片段;
2)根据沉默片段设计rnai引物,得到引物沉默片段引物对;
所述沉默片段引物对的正向引物优选为rnaigmchs5-f(seqidno.5)
c
所述沉默片段引物对的正向引物优选为rnaigmchs5-r(seqidno.6)
gc
3)利用所述rnaigmchs5-f和rnaigmchs5-r循环扩增获取rnai序列;
4)将得到沉默片段的正向序列和反向序列用pdkintron隔开,形成沉默基因。
本发明中,所述软件优选为oligoengineworkstation2。
本发明中,所述沉默片段为如序列表中seqidno.7(gmchs5-1正向链)序列所示。所述沉默片段的反向链如序列表中seqidno.8(gmchs5-1反向链)序列所示。
本发明中,所述沉默基因设计的方法具体优选包括以下内容:pdkintron是形成发夹结构的重要片段,故从pdk两段选取合适的酶切位置,将两段沉默片段连入,分别以hindⅲ/xbai(这两个酶切位点在pdkintron右边,顺序是hindⅲ到xbai,gmchs5-1正向链)和sali/ecori(这两个酶切位点在pdkintron左边,顺序是sali到ecori,gmchs5-1反向链)构入pkannibal载体中,seqidno.7和seqidno.8片段被pdkintron隔开,转录后形成发卡结构。
本发明中,所述基因沉默载体的构建方法没有特殊限制,采用本领域人员所熟知的构建方法即可。
下面结合实施例对本发明提供的大豆耐盐基因gmchs5及其应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
材料与试剂来源说明
1、质粒和菌株
1.1大肠杆菌(escherichiacoli)菌株dh5α、农杆菌k599感受态及
pdl28-ha及其改造质粒由本实验室保存。
2、植物材料
本实验用于研究的植物材料是中黄35品种大豆。
3、实验试剂
1.植物rna提取试剂盒(含dnasei),superrtcdna第一链合成试剂盒,质粒提取试剂盒、胶回收试剂盒购自北京康为世纪生物科技有限公司。
2.
3.所用限制性内切酶购自宝生物工程(大连)有限公司。
4.t4dnaligase(t4dna连接酶)购自普洛麦格(北京)生物技术有限公司。
5.所有生化试剂均购自中国国药集团化学试剂有限公司。
6.合成pcr反应引物由上海生工生物工程技术服务有限公司完成。
7.dna测序工作主要由杭州擎科梓熙生物技术有限公司完成。
4、培养基
1.lb(生工生物)
2.fm(fahraeusmedium)培养基(14g/lagar):
(分别称取0.1geach溶于1l,从中取出1ml再次溶于1l中即1000×母液)
实施例1
gmchs5的克隆
1.1大豆总rna的提取:
1.提前在研钵中倒入液氮,从大豆中剪取新鲜根样100-500mg直接放在研磨中充分研磨,将粉末移至事先准备好的1.5mlep管中,加入600ml的康为世纪公司rlc或者rl(预先加入1%体积的β-巯基乙醇)。
2.涡旋数分钟,使样本充分裂解。冰上静置5min,使样品充分裂解。
3.将上述裂解液缓慢转移至2mlshredderspin管,4℃,12000rpm离心2min;(若体积过大可分两次转入;若裂解液中样品颗粒较大,可将枪头尖剪除0.5cm)
4.准备新的1.5mlrnase-free离心管,将步骤3中滤液转入,加入0.5倍体积的无水乙醇(滤液颜色很深,可以适当多添加),迅速混匀。
5.将上述混合液装入2mlspincolumn管(若体积过大可分两次转入),弃滤液,并将spincolumn重新放回离心管中。
6.向spincolumn加入350ulbufferrw1,4℃,12000rpm离心1min,弃滤液,并将spincolumn重新放回离心管中。
7.加入80μldnasei混合液(52ulrnase-freewater,8μl10×reactionbuffer,20μldnasei),20-30℃孵育15min。
8.向spincolumn加入350ulbufferrw1,4℃,12000rpm离心1min,弃滤液,并将spincolumn重新放回离心管中。
9.向spincolumn加入500ulbufferrw2,4℃,12000rpm离心1min,弃滤液,并将spincolumn重新放回离心管中。
10.向spincolumn加入500ulbufferrw2,4℃,12000rpm离心1min,弃滤液,并将spincolumn重新放回离心管中。
11.4℃,12000rpm空转离心1min,将spincolumn置于一个新的1.5mlrnase-free离心管中,室温吹干10min。
12.向spincolumn加入30rnase-freewater,室温放置1分钟,4℃12000rpm离心1分钟,保存于-80℃。
1.2cdna反转录
1.将primermix、dntpmix、dtt、5×rtbuffer和rnase-freewater溶解并置于冰上备用,待其溶解后,将rna模板和hifiscript取出放置于冰上。
2.按照下表配制反应体系,总体积为20μl。
3.用手指轻轻拨动,短暂离心。
4.在pcr仪上42℃孵育2小时,85℃孵育5分钟。反应结束后,短暂离心,置于冰上冷却,-20℃保存。
1.3目的基因的克隆
1.扩增正向引物为oechs5-f(seqidno.3)
cgcggatccatggttagcgtagctgagatcaggc
扩增正向引物为oechs5-r(seqidno.4)
cgcggatccgatggccacactatgcaaaacaac
以1.2中反转录cdna为模板,进行pcr扩增:
2.程序设置:
pcr产物进行电泳,并且琼脂糖回收。
1.4gmchs5tvector克隆载体的构建
1.4.1回收目的基因dna片段
1.在紫外灯下将目的片段从琼脂糖凝胶中切下(条带尽量单一,体积尽量小),放入离心管中,在天平上称重并记录。
2.用无菌枪头将切下的凝胶块捣碎,加入3倍体积的de-a溶液(如凝胶重为100mg,其体积可视为100μl,依此类推),75℃孵育6~8min,间隔两分钟温和的上下颠倒几下,直至胶块完全溶解。
3.将溶解后的离心管从水浴锅中取出,冷却至室温,加入1.5倍体积de-b溶液,上下颠倒混匀。
4.吸取混合液,转入含有吸附柱的2ml收集管中,12000g离心2min,弃滤液,将吸附柱放回收集管中(若体积过大可分两次转入)。
5.向吸附管中加入500μlbufferw1,12000rpm离心1min,弃滤液,并将吸附柱重新放回离心管中。
6.向吸附管中加入700μlbufferw2,12000rpm离心1min,弃滤液,并将吸附柱重新放回离心管中。
7.向吸附管中加入700μlbufferw2,12000rpm离心1min,弃滤液,并将吸附柱重新放回离心管中。
8.12000rpm空转离心1min,将spincolumn置于一个新的1.5mlrnase-free离心管中,室温吹干10min。
9.向离心管中加入10μl洗脱液,室温放置1min,12000rpm离心1min,保存于-20℃。
1.4.2gmchs5-tvector载体连接和转化
采用
上述反应在200μl的pcr管中进行,用手指轻轻拨动,短暂离心,在pcr仪上25℃孵育30min,反应结束后置于冰上,然后准备转化dh5α感受态细胞。
1.将dh5α感受态细胞从-80℃冰箱取出置于冰上融化5min,作好标记。将上述连接体系5μl全部加入到感受态细胞,冰置30min;
2.将感受态细胞置于42℃水浴锅中孵育90s,然后置于冰上10min;
3.加入提前预热的400μllb液体培养液(不含kan抗生素),37℃、200rpm复苏培养60min;
4.准备lb+kan50mg/l固体培养基平皿,4000rpm、1min离心菌液,吸去400μl上清液,其余上清用来悬浮菌体,涂布,将培养皿倒置于37℃培养箱12~14h。待第二天克隆长出后,挑取3个克隆用引物m13-f/r(
1.4.3质粒dna的提取
1.挑取测序正确的阳性克隆dmi3-tvector,在3~5mllb+kan50mg/llb液体培养基中过夜培养;
2.室温6200g,离心3min收集菌体沉淀,向留有菌体沉淀的离心管中加入250μlbuffers1(实验前已加入rnasea),用移液枪反复吹打,充分悬浮菌体,转至新的1.5mlep管中(自备)。
3.向1.5mlep离心管中加入250μlbuffers2,温和地上下颠倒混匀4~6次,是菌体尽可能裂解,此时菌液应慢慢变清(从加入buffers2至加入buffers3时间间隔尽量不要超过5min);
4.向离心管中加入350μlbuffers3,迅速上下颠倒6~10次,使菌液充分混匀,此时应有白色絮状沉淀出现,室温12000rpm离心10min;
5.吸取上清液,转入含有吸附柱的2ml收集管中,12000g离心1min,弃滤液,将吸附柱放回收集管中(若体积过大可分两次转入)。
6.向吸附管中加入500μlbufferw1,12000rpm离心1min,弃滤液,并将吸附柱重新放回离心管中。
7.向吸附管中加入700μlbufferw2,12000rpm离心1min,弃滤液,并将吸附柱重新放回离心管中。
8.向吸附管中加入700μlbufferw2,12000rpm离心1min,弃滤液,并将吸附柱重新放回离心管中。
9.12000rpm空转离心1min,将吸附柱置于一个新的1.5mlrnase-free离心管中,室温吹干10min。
10.向离心管中加入50μl洗脱液,室温放置1min,12000rpm离心1min,保存于-20℃。
实施例2
含gmchs5基因的真核表达载体构建
2.1过表达载体构建
2.1.1载体pdl28-ha-rfp(内含cam35s启动子)经bamhi单酶切,电泳,回收10k片段,得到酶切产物;酶切体系为载体1ug,sacⅰ2ul,kpnⅰ2ul,2ul10×kbuffer,2ul0.1%bsa,双蒸水补足至20ul;酶切条件为37℃反应2h。
2.1.2克隆载体gmchs5tvector经bamhi单酶切,电泳,回收0.8k左右片段,得到酶切产物;酶切体系为克隆载体1ug,sacⅰ2ul,kpnⅰ2ul,2ul10×kbuffer,2ul0.1%bsa,双蒸水补足至20ul;酶切条件为37℃反应2h。
2.1.3将2.1.1.1中的酶切产物和2.1.1.2中的酶切产物混合后,16℃连接过夜,得到质粒pdl28-ha-gmchs5-rfp,转再化至大肠杆菌,挑选阳性克隆;连接体系为:gmchs5tvector片段7ul,pdl28-ha-rfp片段1ul,t4dna连接酶1ul,t4dna缓冲液1ul。16℃连接过夜。将连接产物转化至大肠杆菌dh5α中,转化方法如1.4.2。
2.1.4挑选2.1.3转化生长出来的克隆以oechs5-f和oechs5-r为引物进行pcr验证质粒pdl28-ha-gmchs5-rfp,电泳得到0.9k左右的单一亮带。限制性内切酶bamhi酶切验证质粒pdl28-ha-gmchs5-rfp,电泳得到0.8k和10k左右两条亮带。
2.2农杆菌k599转化
2.2.1将k599感受态细胞从-80℃冰箱取出置于冰上融化5min,作好标记。将质粒pdl28-ha-gmchs5-rfp取1ug加入到100ul感受态细胞,冰置30min。
2.2.2将感受态细胞置于液氮中孵育1min,37℃水浴锅中孵育2min,重复此操作一次,然后置于冰上10min。
2.2.3再加入37℃的200μllb液体培养液(不含kan抗生素),在37℃、200rpm的条件下复苏培养60min,得菌液。
2.2.4准备lb+kan50mg/l固体培养基平皿,将菌液在室温、4000rpm离心1min,去掉400μl上清液,其余约100ul上清用来悬浮菌体,然后全部涂布在固体培养基平皿上,将培养皿倒置于37℃培养箱12~14h,挑点,用m13f/r引物pcr循环扩增进行验证,电泳得到0.9k左右单一条带即验证正确,命名为k599-pdl28-ha-gmchs5-rfp,用4mllb(含50mg/lkan)液体培养基摇菌,在37℃、220rpm的条件下培养16h。培养后加入1ml二甲亚砜,分装,-80℃保存。
2.3基因沉默载体构建
2.3.1用软件(oligoengineworkstation2)找出gmchs5完整序列中的沉默片段,序列如gmchs5-1正向链序列所示;
2.3.2设计rnai引物,pdkintron是形成发夹结构的重要片段,故从pdk两段选取合适的酶切位置,将两段沉默片段连入,分别以hindⅲ/xbai(这两个酶切位点在pdkintron右边,顺序是hindⅲ到xbai,设计1号正向链)和sali/ecori这两个酶切位点在pdkintron左边,顺序是sali到ecori,设计2号反向链)构入pkannibal载体中,两个方向相反的片段被pdkintron隔开,转录后形成发卡结构。
设计引物:
沉默基因的正向引物为rnaigmchs5-f(seqidno.5)
cg
ecorihindiii
沉默基因的反向引物为rnaigmchs5-r(seqidno.6)
gc
salixbai
2.3.3以rnaigmchs5-f和rnaigmchs5-r循环扩增获取rnai序列,方法见1.3.2。
2.3.4构建rnaigmchs5-t载体,转化,测序,提质粒,方法见1.4;
2.3.5先插入1号正向链,用xbai、hindⅲ酶切gmchs5-t载和pkannbal载体,载体各1ug,xbai、hindⅲ酶各2ul,2ul10×mbuffer,双蒸水补足至20ul,37℃酶切2h。胶回收,再进行t4连接,验证成功后,将连接后的载体命名为pkannibal-gmchs5-1;
2.3.6再插入2号反向链,以正确的pkannibal-gmchs5-1为模板,用sali和ecori酶切rnaigmchs5gmchs5-t载和pkannibal-gmchs5-1载体,载体各1ug,sali和ecori酶各2ul,2ul10×hbuffer,双蒸水补足至20ul,37℃酶切2h。胶回收,再t4连接,验证成功后,将连接后的载体命名为pkannibal-rnai-gmchs5;
2.3.7用限制酶sali、spei将pkannibal-rnai-gmchs5和表达载体pdl28-ha-rfp酶切,所述酶切的体系优选包括载体1ug,sali2ul,spei2ul,2ulhbuffer,双蒸水补足至20ul;所述酶切的条件优选为37℃酶切2h。胶回收,再t4连接,验证成功后,将连接后的载体命名pdl28-ha-rnaigmchs5-rfp;
2.3.8按照2.1.2转化质粒pdl28-ha-rnaigmchs5-rfp,涂板,挑点,验证正确,如2.1.3。命名k599-pdl28-ha-rnaigmchs5-rfp,用lb+kan(终浓度50ug/ml)液体培养基摇菌,-80℃保存。
实施例3
大豆毛状根转化
1.划线活化农杆菌和萌发大豆种子。在含kan的lb板上划线活化农杆菌k599;按漂白水:无菌水=1:2配方配置混合液,加入到含50颗左右完整饱满的大豆的50ml离心管中(完全浸没),侧放处理5min,用无菌水清洗5次,其中第4次清洗需要侧放5min。洗涤结束后,将大豆平铺于加入了滤纸与适量无菌水的方皿中,上下两层每层各放置15-20颗大豆,再在培养箱中25~28℃,暗培养48h;
2.大豆侵染。配制fm培养基→高温高压灭菌→冷却倒板→切根→沿子叶方向将子叶节对半切→子叶节部分划伤处理→农杆菌侵染外植体伤口部分→将外植体置于fm培养基上(每板10-12颗)→弱光共培养8d左右;将大豆转移到生根培养基,fm+1.5%蔗糖+cef;
3.阳性根筛选。体式荧光显微镜观察阳性根,切除不带荧光的非阳性根,阳性苗炼苗过夜;
4.大豆种植。每个品种都需要2个处理样(加盐、不加盐),每个处理样至少有3个重复;
5.盐胁迫处理。大豆加盐组浇100ml(200mm)的盐水,一共浇两次,每次间隔3~5d;
6.收样。盐处理结束,收集各实验组大豆根,共有野生型(加盐)、野生型(不加盐)、过表达(加盐)、过表达(不加盐)、基因敲低(加盐)、基因敲低(不加盐)6组。荧光显微镜观察切下阳性根收集,液氮处理,-80度保存。经统计,6组大豆根部鲜重结果如表1所示。
表16组大豆根部鲜重结果一览表
注:ev表示野生型,oe表示过量表达,kd表示基因敲除,control表示对照组(不加盐处理),nacl处理组表示实验组。
从表1中结果中可以得出,经过高浓度盐水处理的大豆各实验组均受到不同程度的盐胁迫,生物量不同程度降低;基因gmchs5的过量表达,实验组的根样鲜重是野生型加盐组的1.20倍,是基因敲除组的2.08倍。由此结果可以看出,基因gmchs5对于大豆的耐盐是具有积极的作用。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
sequencelisting
<110>杭州师范大学
<120>大豆耐盐基因gmchs5及其应用
<130>2017
<160>8
<170>patentinversion3.3
<210>1
<211>389
<212>prt
<213>人工序列
<400>1
metvalservalalagluileargglnalaglnargalagluglypro
151015
alathrileleualaileglythralaasnproproasnargvalasp
202530
glnserthrtyrproasptyrtyrpheargilethrasnserasphis
354045
metthrgluleulysglulyspheglnargmetcysasplyssermet
505560
ilelysthrargtyrmettyrleuasnglugluileleulysgluasn
65707580
proasnmetcysalatyrmetalaproserleuaspalaargglnasp
859095
metvalvalvalgluvalprolysleuglylysglualaalavallys
100105110
alailelysglutrpglyglnprolysserlysilethrhisleuile
115120125
phecysthrthrserglyvalaspmetproglyalaasptyrglnleu
130135140
thrlysglnleuglyleuargprotyrvallysargtyrmetmettyr
145150155160
glnglnglycysphealaglyglythrvalleuargleualalysasp
165170175
leualagluasnasnlysglyalaargvalleuvalvalcysserglu
180185190
ilethralavalthrpheargglyproseraspthrhisleuaspser
195200205
leuvalglyglnalaleupheglyaspglyalaalaalavalileval
210215220
glyseraspproileproglnvalglulysproleutyrgluleuval
225230235240
trpthralaglnthrilealaproaspsergluglyalaileaspgly
245250255
hisleuarggluvalglyleuthrphehisleuleulysaspvalpro
260265270
glyilevalserlysasnileasplysalaleupheglualapheasn
275280285
proleuasnileserasptyrasnserilephetrpilealahispro
290295300
glyglyproalaileleuaspglnvalgluglnlysleuglyleulys
305310315320
proglulysmetlysalathrargaspvalleuserglutyrglyasn
325330335
metserseralacysvalleupheileleuaspglumetargarglys
340345350
seralagluasnglyhislysthrthrglygluglyleuglutrpgly
355360365
valleupheglypheglyproglyleuthrilegluthrvalvalleu
370375380
hisservalalaile
385
<210>2
<211>1170
<212>dna
<213>人工序列
<400>2
atggttagcgtagctgagatcaggcaggcacaaagggcagaaggcccagcaaccatcctt60
gccattggaactgcaaacccaccaaaccgtgttgatcagagcacctatcctgattactac120
ttcagaatcaccaacagtgaccacatgaccgagctcaaagagaaatttcagcgcatgtgt180
gacaagtctatgatcaagacgagatatatgtacctaaacgaagagatcttgaaagagaat240
ccaaacatgtgtgcttacatggcaccttctttggatgctaggcaagacatggtggtggta300
gaggtaccaaagctagggaaagaggctgcagtaaaggccataaaggagtggggccagcca360
aagtcaaagattacccacttgatcttctgcaccactagcggtgtggacatgcctggtgct420
gattaccaactcaccaaacaattgggccttcgcccttatgtgaagaggtacatgatgtac480
caacaaggttgctttgcaggtggcacggttcttcgtttggccaaggatttggctgagaac540
aacaagggtgcacgtgtgcttgttgtctgctctgagatcactgcagtcacattccgtggg600
ccaagtgacactcaccttgatagtcttgtgggccaagcattgtttggagatggagctgct660
gcagtcattgttggttctgacccaattccacaagttgagaagcctttgtatgagcttgtt720
tggactgcacaaacaattgctccagacagtgaaggtgctattgatggacaccttcgtgaa780
gttggactcacatttcacctcctcaaggatgttcccgggattgtctcaaagaacattgat840
aaggcactttttgaggctttcaacccattgaacatctctgattacaactccatcttttgg900
attgcacaccctggtgggcctgcgattttagaccaagttgagcaaaagttgggtctcaaa960
cctgagaagatgaaggccactagagatgtgcttagtgaatatgggaacatgtcaagtgct1020
tgtgttcttttcatcttggatgagatgaggaggaaatctgctgaaaatggacataaaacc1080
acaggtgaaggacttgaatggggtgtgttgttcggttttggacctggacttaccattgaa1140
actgttgttttgcatagtgtggccatctga1170
<210>3
<211>34
<212>dna
<213>人工序列
<400>3
cgcggatccatggttagcgtagctgagatcaggc34
<210>4
<211>33
<212>dna
<213>人工序列
<400>4
cgcggatccgatggccacactatgcaaaacaac33
<210>5
<211>36
<212>dna
<213>人工序列
<400>5
cggaattcaagcttaatcaccaacagtgaccacatg36
<210>6
<211>33
<212>dna
<213>人工序列
<400>6
gcgtcgactctagaggccaaacgaagaaccgtg33
<210>7
<211>397
<212>dna
<213>人工序列
<400>7
aatcaccaacagtgaccacatgaccgagctcaaagagaaatttcagcgcatgtgtgacaa60
gtctatgatcaagacgagatatatgtacctaaacgaagagatcttgaaagagaatccaaa120
catgtgtgcttacatggcaccttctttggatgctaggcaagacatggtggtggtagaggt180
accaaagctagggaaagaggctgcagtaaaggccataaaggagtggggccagccaaagtc240
aaagattacccacttgatcttctgcaccactagcggtgtggacatgcctggtgctgatta300
ccaactcaccaaacaattgggccttcgcccttatgtgaagaggtacatgatgtaccaaca360
aggttgctttgcaggtggcacggttcttcgtttggcc397
<210>8
<211>403
<212>dna
<213>人工序列
<400>8
ggccaaacgaagaaccgtgccacctgcaaagcaaccttgttggtacatcatgtacctctt60
cacataagggcgaaggcccaattgtttggtgagttggtaatcagcaccaggcatgtccac120
accgctagtggtgcagaagatcaagtgggtaatctttgactttggctggccccactcctt180
tatggcctttactgcagcctctttccctagctttggtacctctaccaccaccatgtcttg240
cctagcatccaaagaaggtgccatgtaagcacacatgtttggattctctttcaagatctc300
ttcgtttaggtacatatatctcgtcttgatcatagacttgtcacacatgcgctgaaattt360
ctctttgagctcggtcatgtggtcactgttggtgattgaattc403
- 还没有人留言评论。精彩留言会获得点赞!