一种放射性同位素碳-14标记吡虫啉的合成方法与流程
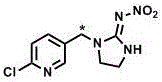
本发明属于放射化学合成领域,具体涉及到一种放射性同位素碳-14标记吡虫啉(1-(6-氯-3-吡啶[14c]甲基)-n-硝基咪唑-2-亚胺)的合成方法。
背景技术:
吡虫啉(imidacloprid;1-(6-氯-3-吡啶甲基)-n-硝基咪唑-2-亚胺)是由德国拜耳公司于1991年开发上市的高效烟碱类内吸性杀虫剂,其作用于害虫的烟酸乙酰胆碱酯酶受体,具有高效、广谱、低毒、安全和高选择性的特点。通过120多个国家数百种作物登记相关试验要求并已应用于农业生产25年。随着吡虫啉的用量持续增长,其可能引发的食品安全、环境污染环境和人体健康等问题愈发引起国内外研究者的重视。当前,在国际农药的代谢途径、作用机理、环境行为与归宿等研究,大多借助了放射性同位素示踪技术,而放射性同位素碳-14标记吡虫啉是开展上述示踪研究所必需的示踪剂。
在国际上,碳-14标记吡虫啉的合成主要由德国拜耳公司完成,其标记位点为吡虫啉分子中亚甲基和咪唑啉环2位碳原子,但因保密原因其标记合成方法尚未公开报道。综合考虑吡虫啉的分子结构特点和下游放射性同位素示踪试验要求等因素,本发明选取亚甲基作为标记位点,提供了一种放射性同位素碳-14标记吡虫啉的合成方法。
技术实现要素:
本发明提供一种高比活度、高放化纯度的放射性同位素碳-14标记吡虫啉(6-氯-3-吡啶[14c]甲基)-n-硝基咪唑-2-亚胺,下文简称亚甲基碳-14标记吡虫啉;结构如图1所示,星号代表碳-14标记位置)的合成方法。该标记物选取吡虫啉分子中亚甲基作为标记位置,以碳-14标记碳酸钡为放射性同位素原料,经格氏、还原、溴化和亲核取代四步反应和制备型高效液相色谱纯化制备了亚甲基碳-14标记吡虫啉。本发明制备的放射性同位素碳-14标记吡虫啉比活度和放化纯度高,可作为放射性示踪剂,用于吡虫啉在植物和动物的代谢残留研究及其土壤、水体中环境行为研究;同时,本发明也建立了一些含碳-14合成砌块的制备方法。
本发明中亚甲基碳-14标记吡虫啉合成实现的技术方案(如图2所示,图中用星号代表碳-14标记位置)如下:
以[14c]碳酸钡为放射性同位素原料,经有机金属试剂(2)吸收[14c]二氧化碳而转化6-氯-吡啶-3-[14c]甲酸(3);通过还原试剂还原,得到6-氯-吡啶-3-[14c]甲醇(4);再与溴代试剂反应得到2-氯-5-溴[14c]甲基吡啶(5);最后在缚酸剂(碳酸钾、碳酸铯、碳酸钠、碳酸氢钠等)作用下,与n-硝基亚氨基咪唑烷缩合得到粗产品,粗产品经经制备型hplc纯化得到本发明的目标产物亚甲基碳-14标记吡虫啉(6,6-氯-3-吡啶[14c]甲基)-n-硝基咪唑-2-亚胺);标记物的其比活度范围为1.0~58.0mci/mmol;化学纯度和放化纯度均大于98%。
本发明为一种亚甲基碳-14标记吡虫啉的合成方法,其特征在于以下步骤:
在氩气保护下,利用2-氯-5-卤吡啶(1,卤素可以是氯、溴、碘)转化的金属有机试剂吸收[14c]二氧化碳,其中有机金属试剂优选格氏试剂、有机锂试剂、有机铜试剂和有机锌试剂;反应温度为-10~60℃;酸优选浓硫酸、磷酸、高氯酸等无机酸;[14c]二氧化碳用量为2-氯-5-卤吡啶转化的金属有机试剂的0.3~2.5倍量;反应时间为30min~24h;反应结束后将反应淬灭,并将反应混合物碱化,用二氯甲烷萃取除杂;所得水相经酸化,乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩得6-氯-吡啶-3-[14c]甲酸(3)。
6-氯-吡啶-3-[14c]甲酸(3)经还原试剂还原得到6-氯-吡啶-3-[14c]甲醇(4),其中还原试剂优选氢化铝锂、硼烷、红铝、硼氢化钠-路易斯酸等;溶剂优选二氯甲烷、四氢呋喃等;反应温度为-10~60℃;反应结束后用水将反应淬灭,分液,乙酸乙酯萃取,合并有机相,无水硫酸钠干燥,过滤浓缩后得无色透明液体6-氯-吡啶-3-[14c]甲醇(4)。
在氩气保护下,6-氯-吡啶-3-[14c]甲醇(4),在溴化试剂作用下溴代得到2-氯-5-溴[14c]甲基吡啶(5),其中溴代试剂优选三溴化磷、二溴亚砜、五溴化磷、n-溴代丁二酰亚胺等;反应温度为-10~50℃;反应时间为1h~12h;反应结束,用碱将反应淬灭,碱优选碳酸钠、碳酸钾、氢氧化钠等;二氯甲烷萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,抽滤,减压浓缩得浅黄色液体2-氯-5-溴[14c]甲基吡啶(5)。
氩气保护下,2-氯-5-溴[14c]甲基吡啶(5)与n-硝基亚氨基咪唑烷在碱性条件下缩合得到本发明的目标产物亚甲基碳-14标记吡虫啉(6),其中缚酸剂优选碳酸钠、碳酸钾、碳酸铯、三乙胺、二异丙基乙基胺等;反应温度为0~100℃;反应时间为1~10h。
将粗产物用制备型高效液相色谱纯化得到纯品(比活度:1.0~58.0mci/mmol),所用色谱条件如下:色谱条件:xbridgeprepc18柱(10μm,150×19mm;watersco.,ma,usa),梯度洗脱(min/%a)控制:0/15,5/15,10/30,25/30,30/15,35/15.a为乙腈,b为水.流速12ml/min,进样量600ul,波长270nm。收集保留时间为11.0~15.0min组分。浓缩脱溶获得目标物[14c]亚甲基标记吡虫啉(6,1-(6-氯-3-吡啶[14c]甲基)-n-硝基咪唑-2-亚胺)。
本发明具有如下优点:
(1)本发明所合成并制备得到的碳-14标记吡虫啉标记位点确定,标记核素不易从标记物分子骨架脱落,能够满足生物体和环境中示踪实验的要求。
本发明以[14c]碳酸钡为放射性同位素原料,合成技术路线简便易行,总反应收率高,且反应条件温和,实验操作简便、安全,标记合成成本较低,适合比活度在1.0~58.0mci/mmol范围之内吡虫啉的制备。
附图说明
图1.本发明中放射性同位素碳-14标记吡虫啉的结构图(星号代表标记位置)
图2.本发明中放射性同位素碳-14标记吡虫啉的合成技术路线(星号代表标记位置)
图3:本发明中放射性同位素碳-14标记吡虫啉的放射性色谱图
图4:文献中本发明中放射性同位素碳-14标记吡虫啉的高效液相色谱图
图5:文献中本发明中放射性同位素碳-14标记吡虫啉的质谱图
具体实施方式
以下列举实施实例对本发明进行说明。实施实例只用于对本发明进一步说明,不代表本发明的保护范围,其他人根据本发明做出的非本质的修改和调整,依然属于本发明的保护范围。
在室温和氩气保护下,将无水氯化锂(420mg)加入反应瓶中,热风枪加热,重复抽真空—充氩气操作5次以彻底干燥氯化锂。加入镁屑(480mg),氩气置换3次,加入无水thf(10ml),搅拌至氯化锂完全溶解。再加入二异丁基氢化铝(1m,160μl),室温搅拌5min。降温至0℃,缓慢滴加2-氯-5-溴吡啶(1.540g)无水thf溶液(10ml),继续搅拌至2-氯-5-溴吡啶彻底消耗,反应溶液由无色变为茶色悬浊液。
利用自制的集成式微量放射性二氧化碳反应系统进行格氏反应制备标记物6-氯-吡啶-3-[14c]甲酸(3),具体方法见文献。投料量:[14c]碳酸钡77.8mci,浓硫酸用量为10ml。搅拌2h。反应降温至-10℃,加入饱和氯化铵淬灭反应,浓缩除去四氢呋喃,剩余水相用稀盐酸调ph至2~3,二氯甲烷萃取三次,合并有机相,再用naoh溶液洗涤有机相,合并碱水相,用稀盐酸调节ph2~3,二氯甲烷萃取三次,饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩得淡黄色固体6-氯-吡啶-3-[14c]甲酸(3,74.6mci)。
在氩气保护下,将6-氯-吡啶-3-[14c]甲酸(3,74.0mci)加入干燥反应管;加热,用氩气置换反应管内气体5次,冷至室温;用注射器加入无水thf(5ml),用冰-盐浴冷却至0~5℃,缓慢逐滴加入硼烷-四氢呋喃溶液(1m,3.7ml)。滴毕,继续搅拌1.5h;自然升至室温,搅拌1.5h后反应结束。向反应体系中缓慢滴加水直至无气泡生成以淬灭反应。减压浓缩除去thf,所得混合液用二氯甲烷萃取,合并有机层,用饱和食盐水洗涤,无水硫酸钠干燥,抽滤,减压浓缩得浅黄色液体6-氯-吡啶-3-[14c]甲醇(4,66.9mci)。
在氩气保护下,将6-氯-3-吡啶[14c]甲醇(4,65.8mci)加入干燥反应管,加热,用氩气置换至反应管内气体5次;用注射器加入无水二氯甲烷(8ml)搅拌溶解,用冰-盐浴冷至0~5℃,逐滴加入pbr3(246mg)。滴加完毕,保持此温度搅拌1h;自然升至室温,搅拌1h后反应结束。向反应体系缓慢滴加5%na2co3溶液直至无co2气泡生成以淬灭反应,用二氯甲烷萃取。合并有机层,用饱和食盐水洗涤,无水硫酸钠干燥,抽滤,减压浓缩得浅黄色液体2-氯-5-溴[14c]甲基吡啶(5,56.4mci)。
在氩气保护下,将2-氯-5-溴[14c]甲基吡啶(5,55.4mci)、n-硝基亚氨基咪唑烷(149mg)和无水cs2co3(391mg)加入干燥schlenk反应管;用氩气置换5次,注入干燥乙腈(8ml),内温升至80℃,剧烈搅拌50min后反应结束。将反应液冷却至室温,用硅藻土层滤除残余cs2co3,滤液减压浓缩得淡黄色固体粗产品。粗品经制备型rp-hplc纯化得亚甲基碳-14标记吡虫啉(6,1-(6-氯-3-吡啶[14c]甲基)-n-硝基咪唑-2-亚胺,36.7mci)纯品。色谱条件:xbridgeprepc18柱(10μm,150×19mm;watersco.,ma,usa),流速12ml/min,波长270nm,进样量600ul;梯度洗脱(min/%a)控制:0/15,5/15,10/30,25/30,30/15,35/15.a为乙腈,b为水。收集保留时间为11.08~14.68min组分。
- 还没有人留言评论。精彩留言会获得点赞!