合成方法和中间体与流程
本申请要求2016年2月25日提交的印度申请号201641006639的优先权。前述的全部内容以引用方式并入本文。
发明背景
国际专利申请公布号wo2014/074932报道了一系列可用作抗生素剂的可溶性化合物。这些化合物之一,式(i)的化合物:
目前正在被评估其作为人体抗菌剂的潜在用途。
目前,需要改进的合成方法和新的合成中间体,其可用于制备商业量的式(i)的化合物。
技术实现要素:
本发明提供了可用于制备式(i)的化合物或其盐的合成方法和合成中间体。这些方法和中间体允许以成本有效且环境可接受的方式制备商业量的化合物。因此,这些方法和中间体将促进式(i)的化合物的商业开发。
在一个实施方案中,本发明提供了一种用于制备式(i)的化合物的方法:
其包括使式e的酚:
与式h的氯化物:
反应,以提供式(i)的化合物。
在一个实施方案中,本发明提供了一种用于制备式h的氯化物的方法,
其通过使式g的氨基吡啶:
与氯乙酸:
反应,以提供式h的氯化物。
在一个实施方案中,本发明提供了一种用于制备式g的氨基吡啶的方法,其通过将相应的式f的硝基吡啶:
转化为式g的氨基吡啶。
在一个实施方案中,本发明提供了一种用于制备式f的硝基吡啶的方法,其通过使2-氯-3-硝基-5-(三氟甲基)吡啶与硫脲反应,以提供式f的硝基吡啶。
在一个实施方案中,本发明提供了一种用于制备式e的酚的方法:
其包括使相应的式d的苄基醚:
脱保护,以提供式e的酚。
在一个实施方案中,本发明提供了一种用于制备式d的苄基醚的方法:
其通过使式i的酰氯:
或其盐与式c的酰胺:
偶联,以提供式d的苄基醚。
在一个实施方案中,本发明提供了一种用于制备式c的酰胺的方法,其通过将式b的酸:
转化为式c的酰胺。
在一个实施方案中,本发明提供了一种用于制备式b的酸的方法,其通过将式a的化合物:
转化为式b的酸。
在一个实施方案中,本发明提供了一种用于制备式a的化合物的方法,其通过将2,4-二氟苯酚转化为式a的化合物。
在一个实施方案中,本发明提供了一种用于制备式(i)的化合物的方法:
其包括使式j的酚:
与下式的酰氯:
反应,以提供式(i)的化合物。
在一个实施方案中,本发明提供了一种用于制备式j的化合物的方法:
其包括使式h的氯化物:
与下式的化合物:
反应,以提供式j的化合物。
在一个实施方案中,本发明提供了一种选自:
的化合物及其盐。
附图说明
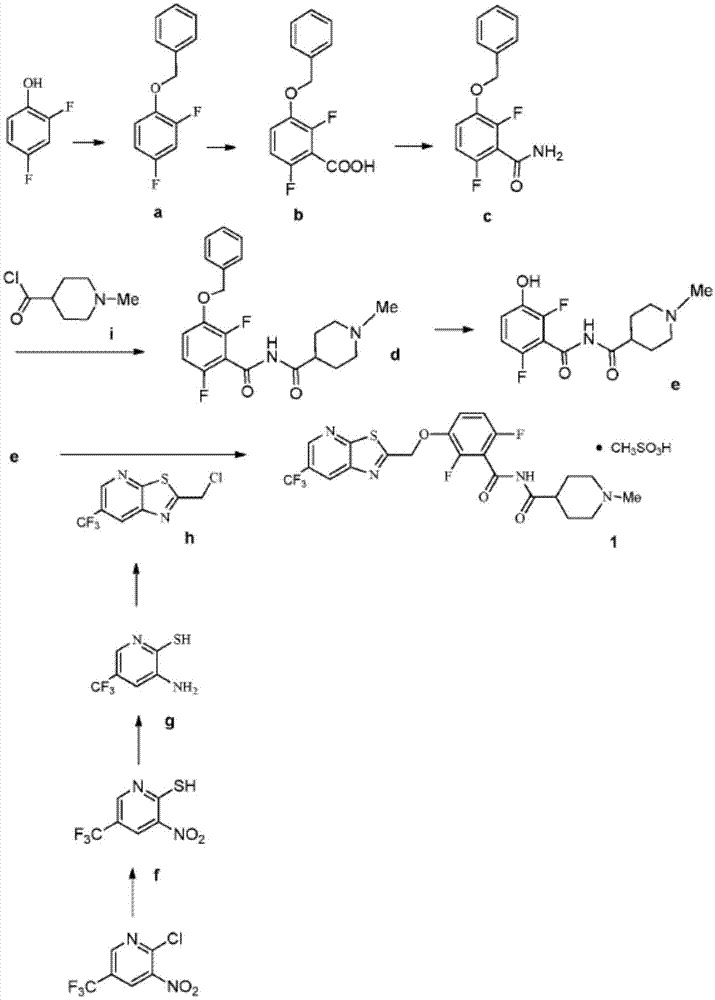
图1示出了本发明的合成路线和本发明的合成中间体。
图2示出了本发明的合成路线和本发明的合成中间体。
具体实施方式
在一个实施方案中,本发明提供了一种用于制备式(i)的化合物的方法:
其包括使式e的酚:
与式h的氯化物:
反应,以提供式(i)的化合物。所述反应通常可在极性溶剂中在约25℃至约30℃范围内或约20℃至约35℃范围内的温度下进行。合适的溶剂包括极性溶剂(例如dmf或dmso)及其混合物。合适的碱包括无机碱,诸如受阻碱(例如碳酸钾、碳酸钠)。对于盐的制备,使用酸包括甲磺酸、草酸和酒石酸。在一个实施方案中,所述反应可在约25℃至约30℃范围内或约20℃至约35℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式h的氯化物的方法,其通过使式g的氨基吡啶:
与氯乙酸:
反应,以提供式h的氯化物。所述反应通常可在极性溶剂中在约50℃至约55℃范围内或约45℃至约60℃范围内的温度下进行。合适的溶剂包括乙酸乙酯、氯代烃(例如二氯甲烷)和芳香烃(例如甲苯),及其混合物。在一个实施方案中,所述反应可在约50℃至约55℃范围内或约45℃至约60℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式g的氨基吡啶的方法,其通过将相应的式f的硝基吡啶:
转化为式g的氨基吡啶。所述反应通常可在极性溶剂中在约65℃至约70℃范围内或约60℃至约80℃范围内的温度下进行。合适的溶剂包括乙酸乙酯及其混合物。合适的还原剂包括铁/乙酸、锌/氯化铵。在一个实施方案中,所述反应可在约65℃至约70℃范围内或约60℃至约80℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式f的硝基吡啶的方法,其通过使2-氯-3-硝基-5-(三氟甲基)吡啶与硫脲反应,以提供式f的硝基吡啶。所述反应通常可在质子溶剂中在约50℃至约55℃范围内或约40℃至约60℃范围内的温度下进行。合适的溶剂包括质子溶剂(例如甲醇、异丙醇或乙醇),及其混合物。在一个实施方案中,所述反应可在约50℃至约55℃范围内或约40℃至约60℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式e的酚的方法:
其包括使相应的式d的苄基醚:
脱保护,以提供式e的酚。所述反应通常可在极性溶剂中在约25℃至约30℃范围内或约20℃至约35℃范围内的温度下进行。合适的溶剂包括极性溶剂(例如dmf或dmso)、质子溶剂(例如甲醇、乙醇)及其混合物。合适的还原剂包括pd/c、pd(oh)2和硝酸铈铵。在一个实施方案中,所述反应可在约25℃至约30℃范围内或约20℃至约35℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式d的苄基醚的方法:
其通过使式i的酰氯:
或其盐与式c的酰胺:
偶联,以提供式d的苄基醚。所述反应通常可在极性溶剂中在约25℃至约30℃范围内或约0℃至约35℃范围内的温度下进行。合适的溶剂包括烃类(例如thf)、极性溶剂(例如dmf、dmso)及其混合物。合适的碱包括无机碱(例如nah)。在一个实施方案中,所述反应可在约25℃至约30℃范围内或约0℃至约35℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式c的酰胺的方法,其通过将式b的酸:
转化为式c的酰胺。所述反应通常可在极性溶剂中在约-75℃至约0℃范围内或约-80℃至约0℃范围内的温度下进行。合适的溶剂包括醚(例如thf、乙醚和mtbe)及其混合物。合适的碱包括n-buli。在一个实施方案中,所述反应可在约-75℃至约0℃范围内或约-80℃至约0℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式b的酸的方法,其通过将式a的化合物:
转化为式b的酸。所述反应通常可在极性溶剂中在约25℃至约30℃范围内或约20℃至约40℃范围内的温度下进行。合适的溶剂包括极性溶剂(例如dmf、thf)及其混合物。合适的碱包括氨水。在一个实施方案中,所述反应可在约25℃至约30℃范围内或约20℃至约40℃范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式a的化合物的方法,其通过将2,4-二氟苯酚转化为式a的化合物。所述反应通常可在极性溶剂中在约55℃至约60℃范围内或约40℃至约65℃范围内的温度下进行。合适的溶剂包括极性溶剂(例如丙酮、乙腈)、质子溶剂(例如甲醇、乙醇)及其混合物。合适的碱包括无机碱,诸如碳酸钾、碳酸钠。在一个实施方案中,所述反应可在约55℃至约60℃范围内或约40℃至约65℃范围内的温度下进行。在一个实施方案中,本发明提供了一种用于制备式(i)的化合物的方法:
其包括使式j的酰胺:
与下式的酰氯:
反应,以提供式(i)的化合物。所述反应通常可在极性溶剂中在约0℃-50℃范围内的温度下进行。合适的溶剂包括thf、dmf、acn、dmso及其混合物。合适的碱包括胺碱,诸如受阻胺碱(例如n,n-二异丙基-n-乙胺)、无机碱诸如nah、kh、naoh、koh、naotbu在一个实施方案中,所述反应可在dmf中以nah作为碱在约0℃-室温范围内的温度下进行。
在一个实施方案中,本发明提供了一种用于制备式j的化合物的方法:
其包括使式h的氯化物:
与下式的化合物:
反应,以提供式j的化合物。所述反应通常可在极性溶剂中在约45℃至约50℃范围内或约40℃至约60℃范围内的温度下进行。合适的溶剂包括极性溶剂(例如dmf、thf)及其混合物。合适的碱包括无机碱,诸如碳酸氢钠。在一个实施方案中,所述反应可在约45℃至约50℃范围内或约40℃至约60℃范围内的温度下进行。
本发明现在将通过以下非限制性实施例来说明。
实施例
实施例1化合物的制备
txa709.甲磺酸酯的制备:将txa709游离碱溶于thf(1.5l,7.5体积)中,并且在35℃-40℃下加入甲磺酸(66g,686.78mmol)。将内容物在25℃-30℃下搅拌16h、冷却至0℃-5℃并且搅拌1h并过滤,以得到为棕色固体的粗产物。
txa709甲磺酸酯的纯化:在55℃-65℃下,向溶于丙酮:甲醇(5.5:7.0)混合物(2.5l,12.5体积)中的粗(txa709游离碱)溶液中加入活性炭(0.5g),搅拌15min并且在热条件下通过hyflo过滤。将滤液冷却至25℃-30℃,随后进一步冷却至0℃-5℃。将内容物搅拌2h、过滤并在50℃-55℃下干燥8h,以得到为浅棕色固体的纯txa709甲磺酸酯产物。(114.0g,27.89%收率)。1hnmr(300mhz,dmso-d6)δ:11.74(bs,1h),9.07(s,1h),8.95(s,1h),7.50-7.58(m,1h),7.17-7.24(m,1h),5.81(s,2h),3.45–3.50(d,2h),2.91-3.02(m,2h),2.77-2.84(d,4h),2..31(s,3h),2.04–2.08(d,2h),1.65-1.77(m,2h)。ms:515.08(m+1)。
txa709.游离碱的制备:在氮气氛下,向配备有机械搅拌器的10l四颈圆底烧瓶中装入n-[(2,6-二氟-3-羟基苯基)羰基]-1-甲基哌啶-4-甲酰胺(200g,670.49mmol)、dmf(5.0l,25体积)、碳酸钾(102g,738.06mmol)和2-(氯甲基)-6-(三氟甲基)[1,3]噻唑并[5,4-b]吡啶(254g,1005.38mmol)。将反应混合物在25℃-30℃下搅拌6h。通过hplc监测反应转化率。在反应完成后,将反应混合物物质用1nhcl猝灭,并且将ph调节至7.0至7.5。过滤沉淀的固体并且用水洗涤,以得到棕色固体。将获得的粗固体用水(2.0l,10体积)纯化,并且在60℃-65℃下干燥18h。
实施例1中使用的中间体化合物n-[(2,6-二氟-3-羟基苯基)羰基]-1-甲基哌啶-4-甲酰胺如下制备。
a.1-(苄氧基)-2,4-二氟苯的制备
在25℃-30℃下,向配备有机械搅拌器的5.0l四颈圆底烧瓶中装入2,4-二氟苯酚(500g,3843.49mmol)、苄基溴(665g,3880mmol)、碳酸钾(635g,4590mmol)和丙酮(3000ml,6.0体积)。将内容物在55℃-60℃下搅拌1h。通过hplc监测反应完成在反应完成后,在低于50℃下将丙酮完全蒸馏并且冷却至25℃-30℃。在25℃-30℃下缓慢加入水(5.0l,10.0体积)并且进一步冷却至0℃-10℃。将内容物搅拌1h,并且过滤固体并干燥,以得到粗固体。将粗产物在25℃-30℃下用水(2.5l,5.0体积)和10%异丙醇:水混合物(2.5l,5.0体积)洗涤,并在30℃-35℃下减压干燥8h,以得到为白色固体的纯产物(805g,95.26%收率)。1hnmr(300mhz,dmso-d6)δ:7.23-7.47(m,7h),6.97-7.05(m,1h),5.16(s,2h)。
b.3-(苄氧基)-2,6-二氟苯甲酸的制备
在氮气氛下,向配备有机械搅拌器的20l四颈圆底烧瓶中装入thf(7.9l,10.0体积)和二异丙基胺(474g,4680mmol)。将反应混合物冷却至0℃至-10℃,缓慢加入n-buli(1.6m己烷溶液)(2.7l,4310mmol)并且在0℃至-10℃下搅拌混合物1h。将内容物冷却至-60℃至-75℃,在-60℃至-75℃下缓慢滴加1-(苄氧基)-2,4-二氟苯(790g)的thf(3.95l,5.0体积)溶液并且搅拌1h。在-55℃至-75℃下将干燥的co2气体吹扫到反应混合物中持续1.5h。通过tlc监测反应完成。在反应完成后,将物质温度升至0℃-20℃并且用hcl水溶液(3.16l,4.0体积)调节ph0-2,加入水(2.4l,3.0体积)。分离各层并且用mdc萃取水层。合并有机层,并且在低于50℃下减压浓缩,以得到粗固体。通过使用用10%naoh溶液进行的碱-酸处理进一步纯化获得的粗产物,并且随后用10%乙酸乙酯:环己烷混合物(2.7l,3.7体积)洗涤,过滤产物并在60℃-65℃下干燥8h,以得到为白色固体的纯产物。(814g,85.86%收率)。1hnmr(300mhz,dmso-d6)δ:13.98(bs,1h),7.33-7.47(m,6h),7.10-716(m,1h),5.20(s,2h)。ms:265.12(m+1)。
c.3-(苄氧基)-2,6-二氟苯甲酰胺的制备
在氮气氛下,向配备有机械搅拌器的10l四颈圆底烧瓶中装入3-(苄氧基)-2,6-二氟苯甲酸(800g,3027.66mmol)、dmf(80.0ml,0.10体积)和干燥thf(2.4l,3.0体积)。在25℃-30℃下缓慢加入亚硫酰氯(540g,4538.95mmol)。将反应混合物在25℃-30℃下搅拌4h。通过tlc监测反应完成。在反应完成后,将反应混合物在低于20℃下的氨水溶液(8.0l,10体积)中淬灭并搅拌2h。在低于50℃下将thf溶剂完全减压蒸馏。过滤沉淀的固体并用水洗涤,以得到粗固体。通过使用10%乙酸乙酯:环己烷(2.4l,3.0体积)混合物纯化粗产物,并在60℃-65℃下干燥8h,以得到为白色固体的纯产物。(735g,92.8%收率)。1hnmr(300mhz,dmso-d6)δ:8.14(bs,1h),7.85(bs,1h),7.25-7.47(m,6h),7.03-7.09(m,1h),5.15(s,2h)。
d.n-{[3-(苄氧基)-2,6-二氟苯基]羰基}-1-甲基哌啶-4-甲酰胺的制备
在氮气氛下,向配备有机械搅拌器的20l四颈圆底烧瓶中装入3-(苄氧基)-2,6-二氟苯甲酰胺(733g,2784.4mmol)和thf(7.33l,10.0体积)。将内容物搅拌10min并冷却至0℃-5℃,在0℃-10℃下在15min内分批加入氢化钠(141.7g,5872.55mmol,60%在矿物油中的分散体)。随后在0℃-20℃下分批缓慢加入n-甲基哌啶酰氯hcl(2.0当量)并搅拌30min。将所得混合物在25℃-30℃下搅拌4h。通过hplc监测反应完成。在反应完成后,将内容物冷却至0℃-5℃,在0℃-10℃下缓慢加入水(7.3l,10.0体积),并且在0℃-10℃下用1:1的hcl水溶液(2.2l,3.0体积)调节ph1.0-2.0。分离各层并且用乙酸乙酯(6.6l,9.0体积)洗涤水层,并在低于50℃下减压浓缩有机挥发物。最后,通过在25℃-30℃下用20%na2co3水溶液调节ph8.0-8.5来分离固体。过滤沉淀的固体并且用水洗涤,以得到为白色固体的最终产物。(740g,68.42%收率)。1hnmr(300mhz,dmso-d6)δ:11.48(bs,1h),7.33-7.47(m,6h),7.11-7.14(m,1h),5.19(s,2h),2.74-2.78(d,2h),2.50-2.51(m,1h),2.13(s,3h),1.72-1.87(m,4h),1.48-1.53(m,2h)。
e.n-[(2,6-二氟-3-羟基苯基)羰基]-1-甲基哌啶-4-甲酰胺的制备
向5l高压釜中装入n-{[3-(苄氧基)-2,6-二氟苯基]羰基}-1-甲基哌啶-4-甲酰胺(350g,901.08mmol)、dmf(1.75l,10.0体积)和5%湿pd/c(52.5g,15%w/w),其中氢气压力为约0.5kg/cm2。将反应混合物在25℃-30℃下搅拌1h。通过hplc监测反应完成。在完成后,将反应混合物通过hy-flo过滤并且冷却至0℃-5℃。在低于15℃下将水(8.4l,24.0体积)缓慢加入到反应混合物中,并且过滤沉淀的固体并在60℃-65℃下干燥8h,以得到为棕色固体的纯产物。(210.0g,78.36%)。1hnmr(300mhz,dmso-d6)δ:11.41(bs,1h),6.92-6.95(m,2h),2.73-2.79(m,2h),2.39-2.44(m,1h),2.14(s,3h),1.73-1.87(m,4h),1.53-1.57(m,2h)。ms:299.25(m+1)。
实施例1中使用的中间体化合物2-(氯甲基)-6-(三氟甲基)[1,3]噻唑并[5,4-b]吡啶如下制备。
f.3-硝基-2-硫代-5-(三氟甲基)吡啶的制备
在25℃-30℃下,向配备有机械搅拌器的5.0l四颈圆底烧瓶中装入2-氯-3-硝基-5-(三氟甲基)吡啶(400g,1765.69mmol)、甲醇(2.4l,6.0体积)和硫脲(150.0g,1970mmol)。将反应混合物在50℃-55℃下搅拌4h。通过hplc监测反应完成。在完成后,在低于45℃下将甲醇从反应混合物中完全减压蒸馏出来。在25℃-30℃下将水(2.0l,5.0体积)加入到反应混合物中,并且随后在25℃-30℃下缓慢加入naoh水溶液。用甲苯洗涤水层,并且通过在0℃-5℃下用1:1hcl溶液调节ph1-2使产物沉淀。收集沉淀产物,过滤并干燥,以便以良好收率得到为棕色固体的产物。(356.0g,90%收率)。1hnmr(300mhz,dmso-d6)δ:14.99(bs,1h,),8.56-8.57(s,1h),8.37(s,1h)。ms:223.13(m-1)。
g.3-氨基-2-硫代-5-(三氟甲基)吡啶的制备
向配备有机械搅拌器的5.0l四颈圆底烧瓶中装入3-硝基-2-硫代-5-(三氟甲基)吡啶(350g,1562.5mmol)、铁粉(261.8g,4687.55mmol)、乙酸乙酯(910ml,2.6体积)和水(910ml,2.6体积)。在25℃-30℃下缓慢加入乙酸(910ml,2.6体积)。将反应混合物在65℃-70℃下搅拌1h。通过hplc监测反应完成。在反应完成后,将反应混合物冷却至25℃-30℃并且加入乙酸乙酯(1050ml,3.0体积)和水(1050ml,3.0体积)。通过hyflo床过滤反应物质,并且分离各层。将合并的有机层用碳酸氢钠、水和饱和nacl溶液洗涤,并且分离各层。浓缩有机层,并且通过在5℃-10℃下使用mdc(2l,5体积)使产物沉淀。将分离的固体过滤并干燥,以得到为棕色固体的最终产物。(255.0g,84.15%收率)。1hnmr(300mhz,dmso-d6)δ:13.85(bs,1h),7.41(s,1h),6.82-6.83(s,1h),6.16(s,2h)。ms:195.08(m+1)。
h.2-(氯甲基)-6-(三氟甲基)[1,3]噻唑并[5,4-b]吡啶的制备
在25℃-30℃下,向配备有机械搅拌器的5.0l四颈圆底烧瓶中装入3-氨基-2-硫代-5-(三氟甲基)吡啶(245g,102.9mmol)和乙酸乙酯(3.68l,15体积)。将内容物冷却至10℃-15℃,并且在10℃-15℃下将氯乙酰氯(287g,2542mmol)缓慢加入反应混合物中。将反应混合物在50℃-55℃下搅拌15h。通过hplc监测反应完成。在反应完成后,在25℃-30℃下缓慢加入水(1.225l,5.0体积)。分离有机层并且用碳酸氢钠、水和饱和nacl溶液洗涤。浓缩有机层并与异丙醇共蒸馏。将产物用异丙醇(735ml,3.0体积)在5℃-10℃下洗涤、过滤并干燥,以得到为棕色固体的最终产物。(265g,83.15%收率)。1hnmr(300mhz,cdcl3)δ:8.88(s,1h),8.47-8.48(s,1h),4.96(s,2h)。ms:252.99(m+1)。
以上步骤d中使用的中间体化合物n-甲基哌啶-4-碳酰氯如下制备。
i.n-甲基哌啶-4-碳酰氯的制备
在氮气氛下,向配备有机械搅拌器的1.0l四颈圆底烧瓶中装入亚硫酰氯(300ml,3.0体积)、n-甲基哌啶酸hcl(100g,556.6mmol)。将反应混合物在65℃-75℃下加热1h。在反应完成后,将亚硫酰氯完全蒸馏出来并与环己烷共蒸馏。最后,将产物用环己烷洗涤、过滤并且在氮气氛下干燥为hcl盐(101g,92%收率)。
实施例2化合物1的制备
向配备有磁力搅拌器的100ml圆底烧瓶中装入化合物j(1.0g,2.57mmol,化合物6(1.0g,5.05mmol)和thf(20ml)。在搅拌下,在5min内分批加入nah(600mg,15mmol,60%在矿物油中的分散体)。将所得反应混合物搅拌10分钟,然后通过移液管在5min内加入水(40μl)的thf(2ml)溶液。反应混合物从悬浮液变为棕色溶液。在反应完成后,通过加入几滴水使其猝灭,并且用二氯甲烷稀释。分离有机相,用盐水洗涤并且经na2so4干燥。真空除去溶剂,并且使用在dcm+1%nh4oh中的10%meoh通过isco纯化所得残余物以提供浅棕色固体,将其用etoac研磨,以得到米色固体(590mg,44%收率)。1hnmr(300mhz,cdcl3)δ:8.58(s,1h),8.31(broads,1h),8.24(s,1h),7.24-7.14(m,1h),6.94-6.87(m,1h),5.50(s,2h),2.94-2.80(m,3h),2.28(s,3h),2.10-1.74(m,6h)。13cnmr(100mhz,dmso-d6)δ174.9,171.1,161.2,160.3,153.5,151.1,149.1,146.6,144.5,143.8,141.8,141.7,127.8,125.0,124.1,123.8,123.5,123.2,122.3,129.6,117.6,117.5,115.9,117.7,115.6,111.4,111.1,69.1,54.8,54.4,45.9,41.9,27.6。c22h19f5n4o3s的hrms计算(m+h)+,515.1171;实测值,515.1181。
中间体化合物j,2,6-二氟-3-((6-(三氟甲基)噻唑并[5,4-b]吡啶-2-基)甲氧基)苯甲酰胺如下制备。
a.化合物j的制备
向配备有磁力搅拌器的25ml圆底烧瓶中装入2-(氯甲基)-6-(三氟甲基)噻唑并[5,4-b]吡啶(350mg,1.39mmol)、dmf(2.0ml)、nahco3(277mg,3.30mmol)和2,6-二氟-3-羟基苯甲酰胺(230mg,1.32mmol)。在50℃下将反应混合物搅拌过夜。通过tlc监测反应完成。在反应完成后,将反应混合物冷却至环境温度(25℃-30℃),加入水并且通过过滤收集沉淀的物质并干燥,以得到棕色固体。在干燥后,将粗产物用mdc研磨,以便以良好收率提供为浅棕色固体的所需产物(380mg,71%收率)。1hnmr(300mhz,dmso-d6)δ:9.03-9.04(s,1h),8.88-8.89(s,1h),7.35-7.43(m,1h),7.06-7.13(m,1h),5.74(s,2h)。ms:390.10(m+1)。
实施例3代表性盐的制备
txa709.甲磺酸酯的制备:将txa709游离碱溶于thf(1.5l,7.5体积)中,并且在35℃-40℃下加入甲磺酸(66g,686.78mmol)。将内容物在25℃-30℃下搅拌16h、冷却至0℃-5℃并且搅拌1h并过滤,以得到为棕色固体的粗产物。
txa709甲磺酸酯的纯化:在55℃-65℃下,向溶于丙酮:甲醇(5.5:7.0)混合物(2.5l,12.5体积)中的粗(txa709游离碱)溶液中加入活性炭(0.5g),搅拌15min并且在热条件下通过hy-flo过滤。将滤液冷却至25℃-30℃,随后进一步冷却至0℃-5℃。将内容物搅拌2h、过滤并在50℃-55℃下干燥8h,以得到为浅棕色固体的纯txa709甲磺酸酯产物。(114.0g,27.89%收率)。1hnmr(300mhz,dmso-d6)δ:11.74(bs,1h),9.07(s,1h),8.95(s,1h),7.50-7.58(m,1h),7.17-7.24(m,1h),5.81(s,2h),3.45–3.50(d,2h),2.91-3.02(m,2h),2.77-2.84(d,4h),2.31(s,3h),2.04–2.08(d,2h),1.65-1.77(m,2h)。ms:515.08(m+1)。
所有公布、专利和专利文件均以引用方式并入本文,恰如以引用的方式单独地并入一般。本发明已参考各种具体和优选实施方案和技术加以描述。然而,应理解可在保持处于本发明的精神和范围内的同时作出许多变化和修改。
- 还没有人留言评论。精彩留言会获得点赞!