沃雷生的中间体的手性拆分及其共晶的制作方法
本发明涉及通过作为苏沃雷生(suvorexant)的中间体的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯的拆分制备苏沃雷生或其盐的方法。本发明还涉及可用于这种制备方法的新共晶。
背景技术:
苏沃雷生(mk-4305)是[(7r)-4-(5-氯-1,3-苯并噁唑-2-基)-7-甲基-1,4-二氮杂环庚-1-基][5-甲基-2-(2h-1,2,3-三唑-2-基)苯基]甲酮的国际非专有名称(inn),其cas号为1030377-33-3。其目前作为belsomra销售,并且是用于治疗失眠的选择性双重阿立新素受体拮抗剂。
苏沃雷生的结构对应于下式(i)。
苏沃雷生具有一个构型(r)的手性中心。苏沃雷生的合成描述于wo2008069997a1的专利族和cox等人,j.med.chem.,2010年,第53卷,第5320-5332页中。具体而言,描述了基于外消旋1,4-二氮杂环庚烷衍生物((rac)-(viiia))的手性固相hplc拆分的合成(参见方案1)。
方案1:
然而,通过手性hplc的1,4-二氮杂环庚烷衍生物((rac)-(viiia))的对映体的手性拆分不适用于工业过程,并且具有几个缺点:中等产量,大量溶剂,高成本和大量的废物。
在wo2016020404a1中,基于与酒石酸衍生物如2,3-二甲苯酰基酒石酸、2,3-二苯甲酰基酒石酸、2,3-茴香酰基酒石酸和2,3-二苯甲酰基酒石酸单(二甲基酰胺)形成非对映体盐,公开了苏沃雷生不同的可能的中间体的拆分。在这些可能的中间体中,通过与2,3-二苯甲酰基d-酒石酸(dbta)形成非对映体盐来进行(rac)-(iii)的拆分(参见方案2)。
方案2:
描述了利用dbta拆分(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯的两个实例(参见wo2016020404a1的第69-70页)。一个实例以小规模(1.25g)进行,其中(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯和dbta(1eq.)在丙酮中的结晶提供5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·dbta盐,具有18%的产率和93.4%ee(对映体过量)。另一个实例以较大规模(7.26g)进行,(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯和dbta(0.5eq.)在丙酮中的结晶提供5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·dbta盐,具有31%的较高产率和76.6%的较低ee。这后一种化合物在乙醇中的再结晶提供具有64%的产率和95.4%ee的化合物。获得具有95.4%ee的化合物的总产率为19.8%。因此,在两个实例中,(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·dbta都以低产率(18-19%)和<96%ee获得。由于这些实例的对映体过量保持较低,应进行一次或多次另外的再结晶以达到98-99%ee,甚至使该拆分的低产率进一步降低。
因此,需要提供一种具有改善的产率和提高的%ee的拆分(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯(其为苏沃雷生的中间体)的有效方法。
技术实现要素:
发明人开发了通过形成(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇((r)-ted)的共晶而拆分(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯((rac)-(iii))或其盐(例如盐酸盐)的方法。
根据发明人的知识,在现有技术中尚未公开使用共晶来拆分苏沃雷生中间体。将共晶用作拆分剂的认定不是常规操作,并且理论上不能预测共晶的形成。发明人也测试了其他手性1,2-二醇,但未成功。因此,认为式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶的形成是对现有技术的贡献。
因此,本发明的一个方面涉及式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶,其中cbz是苄氧基羰基,其中(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的摩尔比为1:1,
本发明的另一方面涉及一种拆分方法,所述方法包括:通过包括下述步骤的方法制备式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐((r)-(iii)·hci)与(r)-ted的共晶,其中cbz是苄氧基羰基:a1)将a1a)式(rac)-(iii)的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯的盐酸盐与(r)-ted的混合物或者a1b)式(rac)-(iii)的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯、(r)-ted和盐酸并入到溶剂中,所述溶剂选自由乙腈、异丙醇、乙酸乙酯、丙酮、四氢呋喃、叔丁基甲基醚和甲苯组成的组;a2)加热混合物直至完全溶解,或者使其在室温和回流之间浆化;a3)必要时冷却该混合物;和a4)将由此获得的步骤a2)或a3)的共晶分离。
步骤a2)或a3)中获得的共晶是式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶。
在前述方法的具体实施方式中,所述盐是盐酸盐。
前述方法允许进行式(rac)-(iii)的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯或其盐的拆分。所述方法可包括将步骤a4)中获得的共晶转化为(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯或其盐。
(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯或其盐(特别是其盐酸盐)的拆分方法可包括:a)通过上述方法制备式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐和(r)-ted的共晶,可选地通过再结晶或在有机溶剂中浆化而将由此获得的共晶纯化;b)使由此获得的共晶解离以产生(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐,必要时将其转化成其式(r)-(iii)的游离碱;和必要时c)通过使所得游离碱与适当的酸反应而将其转化成其盐。
前述方法在包括一次再结晶时提供了非常高的%ee(>99%ee,仅一次再结晶)和令人满意的约25-30%的产率。此外,可对拆分方法的两个步骤(例如通过结晶和随后再结晶而形成共晶)使用相同的溶剂。额外的优点是解离步骤期间容易回收ted。拆分方法可由(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯或其盐酸盐、或者由任何这些化合物的(r)和(s)对映体的其他比例的混合物进行。
本发明的另一方面涉及上面定义的式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶作为制备(r)-苏沃雷生或其药学上可接受的盐的中间体的应用。
本发明的另一部分是制备式(i)的苏沃雷生或其药学上可接受的盐的方法,其包括a)进行如上所述的拆分方法,并通过下面详细公开的技术中公开的方法将由此获得的化合物转化成苏沃雷生或其药学上可接受的盐。与制备苏沃雷生的已知方法相比,这些方法具有更好的工业实用性和效用,并克服了现有方法的缺点。
附图说明
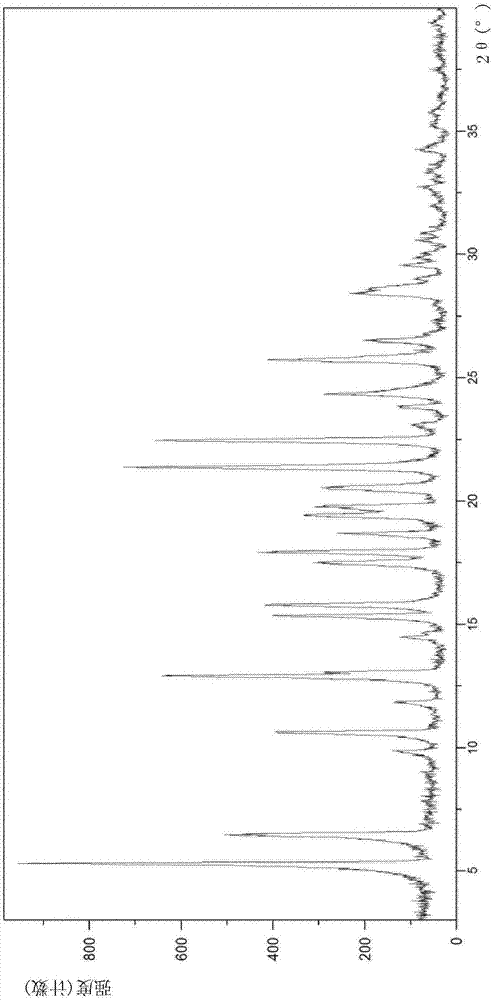
图1示出了名为a型的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·hcl的共晶的xrpd。
图2示出了a型的1hnmr。
图3示出了a型的dsc。
图4示出了a型的tga。
具体实施方式
为了便于理解,包括以下定义并且预期适用于整个说明书、权利要求和附图。
除非另有说明,否则在本发明的上下文中,术语“苏沃雷生”涉及(r)-苏沃雷生。这两个术语可互换使用。
术语“共晶”在本文中是指在室温(20-25℃)下具有构成晶胞并通过弱相互作用而相互作用的至少两种不同组分的结晶实体。因此,在共晶中,靶分子与一种或多种中性组分结晶。共晶可包括晶格中的一个或多个溶剂分子。
术语“弱相互作用”在本文中是指既不是离子也不是共价的相互作用,并且包括例如:氢键、范德华相互作用和π-π堆积。
当指定本发明的共晶组分的比例时,则其是指形成共晶的两种组分之间的摩尔比。术语“摩尔比”已用于表示共晶的每种组分的摩尔的化学计量的量。
当给出x射线衍射图的特征峰值时,则称其为“近似”值。应该理解的是,这些值是以cu-kα辐射,
如本文所公开的术语“室温”是指环境温度(不加热或冷却),并且通常为20至25℃。
出于本发明的目的,给出的任何范围包括该范围的下端点和上端点。除非另有说明,否则给出的范围(例如温度和时间等)应视为近似值。
对映体过量(ee)是用于手性物质的纯度的量度。它反映了样品含有一种对映体的含量大于另一种对映体的程度。术语“对映体纯度”(或光学纯度)定义为一种对映体相对于另一种对映体的分数过量。
这里使用表述“通过......可获得的共晶”来通过其获得方法限定本发明的每种特定共晶,并且是指可通过本文公开的任何相应方法获得的产物。出于本发明的目的,表述“可获得的”,“获得的”和等同的表达可互换使用,并且在任何情况下,表述“可获得的”包括表述“获得的”。
术语“湿研磨”和“液体辅助研磨”是等同的,并且是指这样的技术:包括将产物或混合物与加入的几滴溶剂碾磨或研磨。纯研磨和液体辅助研磨是可以用于生产共晶的技术。在纯(干)研磨中,使用研杵和研钵、使用球磨机或使用振荡研磨机手动将共晶形成物研磨在一起。在液体辅助研磨或捏合中,将少量液体(溶剂)如几滴液体加入研磨混合物中。
如上所述,本发明的一部分是提供式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-l,2-乙二醇((r)-ted)的共晶,其中(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇的摩尔比为1:1。
在式(ii)中,cbz表示苄氧基羰基。
在优选实施方式中,式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶是名为a型的晶型。该a型共晶易于操作并在室温下显示出结晶稳定性。
本发明共晶的特定晶型可通过x射线粉末衍射(xrpd)、质子核磁共振分析(1hnmr)、差示扫描量热法(dsc)和热重分析(tga)表征。
在环境条件下在具有反射θ-θ几何形状的panalyticalx'pertpro衍射仪上进行衍射测量,其配备有cu-kα辐射和pixcel检测器,在45kv和40ma下操作。将每种粉末安装在零背景硅支架上,并在数据收集期间以0.25转/秒的速度旋转。测量角度范围为3.0-40.0°(2θ),步长为0.013°。扫描速度为0.32826°/秒。
质子和碳核磁共振分析记录在varianmercury400光谱仪中的氘化氯仿中,该光谱仪配备有5mm的宽带探针atb1h/19f/x。将5-10mg样品溶解在0.6ml氘化溶剂中,获得光谱。
用mettlerdsc2记录dsc分析。将2.4900mg的样品称入具有针孔盖的40μl铝坩埚中,并在氮气(50ml/min)下以10℃/min从25℃加热至300℃。
在mettlertga/sdta851e热重分析仪中记录热重分析(tga)。将4.3400mg样品称入100μl铝坩埚中并用盖子密封。在氮气(50ml/min)下将样品以10℃/min从25℃加热至550℃。
在优选实施方式中,式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶a型的特征在于具有这样的x射线衍射图,所述x射线衍射图包括在cu-kα辐射,
更具体地说,这种新的共晶a型的特征在于在x射线粉末衍射图中显示如表1所示的峰图案,以2θ表示(单位是度),2θ(°)。
表1:选定峰的列表(仅指示相对强度大于或等于1%的峰):
该共晶a型还可由图1所示的x射线衍射图表征。
该共晶a型还可由下述1hnmr谱1hnmr(cdci3,400mhz)表征:δ=9.78(sbr,2h),7.73-7.67(m,2harom.),7.43-7.27(m,8harom.),7.21-7.03(m,10harom.),5.66-5.62(m,1h),5.19-5.09(m,2h,ph-ch2-co),4.06-3.89(m,1h),3.79-3.49(m,3h),3.46-3.25(m,2h),3.16(s,1h,-oh)3.12-2.90(m,1h),2.52(d,1h,-oh),2.33-2.18(m,1h),2.15-1.97(m,1h),1.57-1.51(m,3h,-ch3)。该共晶a型还可由图2所示的1hnmr光谱表征。
共晶a型还可由通过dsc分析测得的对应于熔点的吸热尖峰表征,该峰初起于约152℃(熔化焓-107.89j/g)。该共晶a型还可由图3所示的dsc分析表征。
共晶中(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·hci与(r)-ted的比可通过1hnmr、滴定或元素分析确定。
用于制备如上定义的式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯的盐酸盐与(r)-ted的共晶的初步方法包括:在溶剂中对a)式(rac)-(iii)的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐和(r)-ted的混合物;或者b)(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯、(r)-ted和盐酸的混合物进行湿研磨,所述溶剂选自由乙腈、异丙醇、乙酸乙酯、丙酮、四氢呋喃、叔丁基甲基醚、二氯甲烷和甲苯组成的组。在具体实施方式中,所用溶剂是乙腈。
如上所述,本发明的一部分是制备式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶的方法,其包括:a1)将a1a)式(rac)-(iii)的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯的盐酸盐与(r)-ted的混合物或者a1b)式(rac)-(iii)的(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯、(r)-ted和盐酸组合在溶剂中,所述溶剂选自由乙腈、异丙醇、乙酸乙酯、丙酮、四氢呋喃、叔丁基甲基醚和甲苯组成的组;a2)加热混合物直至完全溶解,或者将其在室温和回流之间浆化;a3)必要时冷却该混合物;a4)将由此获得的步骤a2)或a3)的共晶分离。
由于盐酸可以是盐水水溶液,溶剂体系可以是任何上述溶剂(乙腈、异丙醇、乙酸乙酯、丙酮、四氢呋喃、叔丁基甲基醚和甲苯)与水的混合物,优选acn和水的混合物。具体而言,溶剂/水的混合物中水的含量可至多10%v/v。在具体实施方式中,水的含量为5-10%v/v。
在具体实施方式中,所用溶剂是乙腈。在另一具体实施方式中,通过在乙腈中浆化获得共晶。在另一具体实施方式中,通过由乙腈中结晶获得共晶。因此,优选在回流温度下将热溶液缓慢冷却以使共晶结晶。可接种溶液以促进结晶。
可使用cox等,j.med.chem.2010年,第53卷,5320-5332页或wo2008069997a1(参见该专利申请中的方案e)中描述的方法制备(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯((rac)-(iii))。由此获得的(rac)-(iii)可通过已知方法转化成其盐酸盐,例如使其与盐酸在适当溶剂中反应。(r)-ted是市售的(cas号95061-46-4),也可以通过本领域已知的方法由作为廉价原料的扁桃酸容易且经济地获得。
本发明的共晶可通过再结晶纯化。因此,在其制备之后,可使共晶进行进一步的再结晶。在优选实施方式中,拆分方法还包括一个再结晶步骤。在具体实施方式中,本发明的共晶是a型。
还可对其进行浆化过程。在两种情况中(再结晶或浆化),溶剂选自由乙腈、异丙醇、乙酸乙酯、丙酮和甲苯组成的组。在具体实施方式中,所用溶剂是乙腈。可对制备步骤和再结晶步骤或浆化步骤使用相同的溶剂。
在优选实施方式中,使用乙腈作为溶剂制备式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶的a型。
本发明的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶也可通过其制备方法定义。因此,本发明的该方面可涵盖为可通过上述方法获得的如上定义的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶,所述方法可选地包括所述方法的任意优选或具体的实施方式,以及上面公开的一些工艺特征的可能的组合。
本发明的另一部分是式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶作为制备(r)-苏沃雷生或其药学上可接受的盐的中间体的应用。可以获得≥99%ee的苏沃雷生。本发明的共晶允许拆分苏沃雷生制备中的关键中间体。
因此,(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯或其盐酸盐的拆分方法也是本发明的一部分,并且包括:通过上述方法制备式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶,可选地将由此获得的共晶再结晶或浆化;和b)使由此获得的共晶解离以产生(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐,必要时将所述盐转化成其式(r)-(iii)的游离碱,并且必要时通过使所得游离碱与适当的酸反应而将其转化成其盐。
在优选实施方式中,拆分方法由作为原材料的外消旋5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯游离碱进行。在另一优选实施方式中,拆分方法由作为原材料的外消旋5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯的盐酸盐进行。
在拆分方法的具体实施方式中,解离步骤包括:(1)将式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶在室温的水中浆化;(2)将(r)-ted与介质分离;和(3)将水相碱化,用适当的有机溶剂萃取(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯。由此获得的溶液可直接用于下一步骤。作为选择,可从溶液中分离(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯游离碱。作为实例,可将合并的有机相在室温于真空下浓缩至干燥,获得为无水油状物的(r)-(iii)游离碱。必要时可将该游离碱与适当的酸反应从而制备(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯游离碱的盐。
含有(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐的水相可碱化,例如用1m氢氧化钠(ph12)碱化,并且可例如用二氯甲烷萃取,优选萃取两次。
在另一具体实施方式中,解离步骤包括:(1)将式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-ted的共晶在室温的水中浆化;(2)将(r)-ted与介质分离;和(3)浓缩水相以分离(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐。
当(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·hci与(r)-ted的共晶a型通过在室温的水中浆化而解离时,(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐保持溶于水,而不溶于水的(r)-ted结晶。结晶(r)-ted可从反应介质中回收,例如通过过滤或在有机溶剂中萃取而从反应介质中回收。
(r)-5-甲基-1,4-二氮杂环庚烷-l-羧酸苄酯游离碱的对映体过量可如实施例部分的描述测定。结果示出本发明的方法(参见例如实施例4、5和8)可比wo2016020404a1中描述的拆分方法提供更高的总产率(多达30%而非20%)以及优异的对映体过量(>99%ee而非96%ee)。
另一个对映体(s)-(iii)或其盐酸盐可以使用ted的另一个对映体(即(s)-ted)以相同的方式拆分。其与其对映体具有相同的x射线、dsc和tga。
每种所述化合物的手性可如实施例9所示进行测定。
如上定义且通过本发明的方法获得的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯(r)-(iii)或其盐可容易通过本领域已知的方法转化成苏沃雷生或其药学上可接受的盐。这种转换可以遵循两种不同的方法。
一种方法包括a)用式(iv)的苯甲酸衍生物将(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯(r)-(iii)或其盐n酰化,其中x是oh或cl;b)进行n脱保护以提供化合物(vi);和c)与苯并噁唑衍生物化合物(vii)偶联。该方法应用于例如wo20080699997或j.med.chem.2010,53,5320-5332中。
因此,本发明的另一部分是制备式(i)的苏沃雷生或其药学上可接受的盐的方法,其包括:a)进行如上定义的拆分方法;b)用式(iv)的苯甲酸衍生物将由此获得的式(r)-(iii)的化合物n酰化以产生式(v)的化合物,其中x是oh或cl;c)将由此获得的式(v)的化合物的氨基进行脱保护以提供式(vi)的化合物;d)将步骤c)中获得的所述化合物与式(vii)的苯并噁唑衍生物偶联以产生苏沃雷生(i)或其药学上可接受的盐,其中y是h、cl或br;和必要时通过将所得苏沃雷生游离碱与可接受的酸反应而将其转化成其药学上可接受的盐。
前述方法的步骤b)可在碱的存在下进行,特别当原材料为(r)-(iii)盐酸盐时。适当的碱的实例为叔胺(例如三乙胺或二异丙基乙胺)或无机碱(例如碱金属碳酸盐,例如碳酸钾)。
当式(iv)的化合物中的x是oh时,偶联可通过本领域技术人员公知的偶联剂实现。
用于进行偶联的实例程序(其中y是h、cl或br)可分别见于wo2015008218、wo2008069997和wo2013169610。
该方法在下述方案中示出:
在式(r)-(iii)和(v)中,cbz表示苄氧基羰基。
另一种方法包括用正交保护基pg对上面定义的化合物(r)-(iii)进行n保护,然后进行n-cbz切除。然后,可通过涉及:a)与式(vii)的苯并噁唑衍生物偶联;b)进行n脱保护以获得混合物(xi),和c)与苯甲酸衍生物(iv)偶联的方法获得苏沃雷生。该方法的各方面例如应用于wo2016020404a1、wo2015006218a2和wo2012148553a1,其中x和y如上所定义。
因此,本发明的另一部分涉及制备式(i)的苏沃雷生或其药学上可接受的盐的方法,其包括:a)进行如上定义的拆分方法;b)用正交保护基pg(即可在cbz的存在下选择性去除的保护基)将由此获得的式(r)-(iii)的化合物n保护以产生式(viii)的化合物;c)将cbz保护基切割以产生式(ix)的化合物;将由此获得的化合物与式(vii)的苯并噁唑衍生物偶联以产生式(x)的化合物,其中y是h、cl或br;e)将式(x)的化合物n脱保护以产生式(xi)的化合物;f)将式(xi)的化合物与式(iv)的苯甲酸衍生物偶联以产生苏沃雷生或其药学上可接受的盐,其中x是oh或cl(其中x=oh时,可用本领域技术人员公知的偶联剂进行偶联);必要时g)通过将所得苏沃雷生游离碱与药学上可接受的酸反应而将其转化成药学上可接受的盐。
前述方法的步骤b)可在碱的存在下进行,特别当原材料为(r)-(iii)盐酸盐时。适当的碱的实例为叔胺(例如三乙胺或二异丙基乙胺)或无机碱(例如碱金属碳酸盐,例如碳酸钾)。
该方法在下述方案中示出:
在上面公开的制备苏沃雷生的方法的具体实施方式中,化合物(iv)中的x是氯。在制备苏沃雷生的方法的另一具体实施方式中,化合物(vii)中的y是溴或氯。
cbz基团的脱保护可例如通过用h2、pd(oh)2、etoac氢解进行。该氨基保护基可通过本领域已知的其它程序(参见t.w.greene和g.m.wuts,protectivegroupsinorganicsynthesis,第三版,wiley,n.y.,1999,531-535页)引入和去除。
基团pg是合适的正交氨基保护基。本文所用的术语“合适的正交氨基保护基”表示涵盖除保护基cbz之外的任何氨基保护基以保护式(r)-(iii)的化合物的其它氨基,其对选择的cbz切除条件(例如氢解)稳定。用于氨基的代表性保护基是本领域技术人员公知的,描述于例如t.w.greene和g.m.wuts,protectivegroupsinorganicsynthesis,第三版,wiley,n.y.,1999(见第7章)中。用于pg的优选的保护基包括但不限于形成氨基甲酸酯的基团如boc(叔丁氧基羰基)、fmoc(9-芴基甲基氧基羰基)、氨基甲酸甲酯和氨基甲酸乙酯;形成磺酰胺的基团如甲苯磺酰基;和形成酰胺的基团如甲酰基、取代或未取代的乙酰基和苯甲酰基。在优选实施方式中,pg是boc。
化合物(ix)与化合物(vii,y=cl或br)的偶联和式(xi)的化合物与式(iv,x=cl)的苯甲酸衍生物的偶联可例如在碱(例如三乙胺)和适当的溶剂(例如二氯甲烷)的存在下进行。
化合物(ix)与化合物(vii,y=h)的偶联可例如根据wo2015008218a在乙酸和乙腈中的cu(oac)2存在下进行。
化合物(xi)与式(iv,x=oh)的苯甲酸衍生物的偶联可在本领域技术人员已知的标准酰胺形成偶联剂存在下进行。
术语“药学上可接受的盐”是指通过本领域已知的方法由包括无机或有机酸的药学上可接受的无毒酸制备的盐。这种酸包括乙酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、乙磺酸、富马酸、葡糖酸、谷氨酸、氢溴酸、盐酸、羟乙磺酸、乳酸、马来酸、苹果酸、扁桃酸、甲磺酸、粘液酸、硝酸、双羟萘酸、泛酸、磷酸、琥珀酸、硫酸、酒石酸或对甲苯磺酸。
在整个说明书和权利要求书中,词语“包括”和该词语的变体并不旨在排除其他技术特征、添加剂、组件或步骤。此外,词语“包括”涵盖“由......组成”的情况。通过阅读说明书,本发明的其他目的、优点和特征对于本领域技术人员而言将变得显而易见,或者可以通过本发明的实践来学习。通过举例说明的方式提供以下实施例和附图,并且它们不旨在限制本发明。此外,本发明涵盖本文所述的特定和优选实施方式的所有可能组合。
实施例
实施例1:检测(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型和(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶
所用的原材料是(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·ηcl和(r)-ted(1:1)或(s)-ted(1:1)。
(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶通过在乙腈(acn)中浆化和湿研磨获得。
(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶也通过用作为溶剂的异丙醇、乙酸乙酯、丙酮和甲苯浆化以及利用异丙醇、乙酸乙酯、丙酮、四氢呋喃、叔丁基甲基醚、二氯甲烷和甲苯湿研磨获得。
(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型可通过在acn中浆化和研磨(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯·hci与(r)-ted(1:1)获得。
(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型也可通过用作为溶剂的异丙醇、乙酸乙酯、丙酮和甲苯浆化以及利用异丙醇、乙酸乙酯、丙酮、四氢呋喃、叔丁基甲基醚、二氯甲烷和甲苯湿研磨获得。
实施例2:通过在乙腈(acn)中浆化制备(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶和(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型
在eppendorf管中,将外消旋5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐(19.8mg,0.07mmol)和(s)-1,1,2-三苯基-1,2-乙二醇(20.1mg,0.07mmol)悬浮在乙腈(0.2ml)中。将所得悬浮液置于室温下搅拌15小时(过夜)。然后,通过离心分离回收固体并在室温下高真空干燥。
可由代替(s)-1,1,2-三苯基-1,2-乙二醇的(r)-1,1,2-三苯基-1,2-乙二醇开始通过相同方法获得(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇a型。
实施例3:通过在acn中湿研磨制备(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶和(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型
在eppendorf管中,将(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐(20.3mg,0.07mmol)、(s)-1,1,2-三苯基-1,2-乙二醇(20.2mg,0.07mmol)、两滴乙腈和三枚钢球合并。将所得混合物在研钵中以30mhz研磨三次,每次10分钟,并在室温下在高真空下干燥固体。
可由代替(s)-1,1,2-三苯基-1,2-乙二醇的(r)-1,1,2-三苯基-1,2-乙二醇开始通过相同方法获得(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇a型。
实施例4:通过由acn中结晶制备式(ii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型
在圆底10ml烧瓶中,将(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐(1g,3.52mmol)和(r)-1,1,2-三苯基-1,2-乙二醇(1.027g,3.53mmol,1.0当量)悬浮在乙腈(6.0ml)中。加热所得混合物至回流。将得到的澄清溶液在回流下搅拌10分钟,然后缓慢冷却至室温。在冷却期间,每10℃用a型接种混合物,白色固体在60℃开始结晶。一旦在室温下,将所得悬浮液在室温下搅拌3小时并在0-5℃下用冰浴搅拌1小时,然后分离固体。白色固体在烧结漏斗(3号)中过滤,用冷乙腈洗涤两次(2x1.0ml),并在室温下于高真空下干燥,从而提供为白色固体的a型(835mg,41%产率,88%ee)。
实施例5:由acn中对(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型进行再结晶
在配备有磁性搅拌的圆底10ml烧瓶中,将(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型(801.4mg,1.39mmol,88%ee)悬浮在乙腈(4.8ml)中。加热所得混合物至回流。将得到的澄清溶液在回流下搅拌10分钟,然后缓慢冷却至室温。在冷却期间,每10℃用a型接种混合物,白色固体在60℃开始结晶。一旦在室温下,将所得悬浮液在室温下搅拌3小时并在0-5℃下用冰浴搅拌1小时,然后分离固体。白色固体在烧结漏斗(3号)中过滤,用冷乙腈洗涤两次(2x0.8ml),并在室温下于高真空下干燥,从而提供为白色固体的a型(639mg,80%产率,>99%ee)。
晶种晶体可通过上述任何方法获得。它们可另外从上面公开的湿研磨方法获得,但使用式(r)-(iii)的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯作为原材料。
实施例6:由acn中结晶(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型和(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶
进行了几次测定,以7volacn结晶共晶的条件如表1所示。
表1:
实施例7:由acn中对(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型和(s)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(s)-(-)-1,1,2-三苯基-1,2-乙二醇的共晶进行再结晶
进行了几次测定,以6volacn再结晶共晶的条件如表2所示。
表2
实施例8:通过解离(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯盐酸盐与(r)-(+)-1,1,2-三苯基-1,2-乙二醇共晶a型制备(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯游离碱
在配备有磁性搅拌的圆底10ml烧瓶中,将a型(94mg,0.164mmol,>99%ee)悬浮在冷水(2.0ml)中并在冰浴中于0-4℃下搅拌1小时。将(r)-ted用烧结漏斗(3号)过滤,用冷水洗涤两次(2x0.5ml),并在室温下于高真空下干燥,得到为白色结晶固体的(r)-1,1,2-三苯基-1,2-乙二醇(44mg,92%回收率)。
将合并的水性母液用tbme洗涤(2x1ml)然后添加二氯甲烷(3ml)和1m碳酸钠溶液直至ph10。在剧烈搅拌后,分离相并将水相用二氯甲烷萃取两次(2x3ml)。将合并的有机相用水(3ml)和盐水(3ml)洗涤,在无水硫酸钠上干燥。在室温下真空蒸馏溶剂以获得为淡黄色油状物的(r)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯游离碱(40mg,84%产率,99%ee)。
实施例9:(r)-5-甲基-1,4-二氮杂环庚烷-l-羧酸苄酯游离碱的手性构型的测定
(r)-5-甲基-1,4-二氮杂环庚烷-l-羧酸苄酯游离碱的手性构型通过以下方式测定:
a)再现sandoz专利(wo2016020404a1,参见例如第17页起和第70页起)中描述的用dbta拆分(rac)-5-甲基-1,4-二氮杂环庚烷-1-羧酸苄酯,允许通过手性hplc分析比较(r)-iii的保留时间和a型的保留时间,并确认(r)-ted与(r)-iii·hci形成共晶。
在chiralpakia柱(5μm,4.6mmx250mm)上用作为洗脱液的庚烷/etoh-0.2%dea的90:10混合物并以1ml/min的流速进行分析式hplc分析(((r)-iii是第一洗脱对映体,tr(r)-iii12.0分钟,(s)-iii)14.0分钟)。
b)用boc基团保护(r)-iii游离碱的另一胺,测量旋光率([α]d-24.0(c1.10,氯仿)),并将其与参考文献值([α]d-24.3(c1.0,氯仿),参见cox等,j.med.chem.2010年,第53卷,5320-5332页)比较。
引用列表
专利文献
-wo2008069997a1
-wo2015008218a2
-wo2012148553a1
非专利文献
-j.med.chem.2010年,第53卷,5320-5332页
-
 0访客 来自[中国] 2021年06月16日 20:14苏沃雷生能买到吗?老母亲患发作性睡病60年了!
0访客 来自[中国] 2021年06月16日 20:14苏沃雷生能买到吗?老母亲患发作性睡病60年了!