化合物或其盐、抗炎症剂、针对肺癌的抗癌剂、化合物或其盐的制造方法、炎症性疾病的治疗方法及肺癌的治疗方法与流程
本发明涉及化合物或其盐、抗炎症剂、针对肺癌的抗癌剂、化合物或其盐的制造方法、炎症性疾病的治疗方法及肺癌的治疗方法。
背景技术:
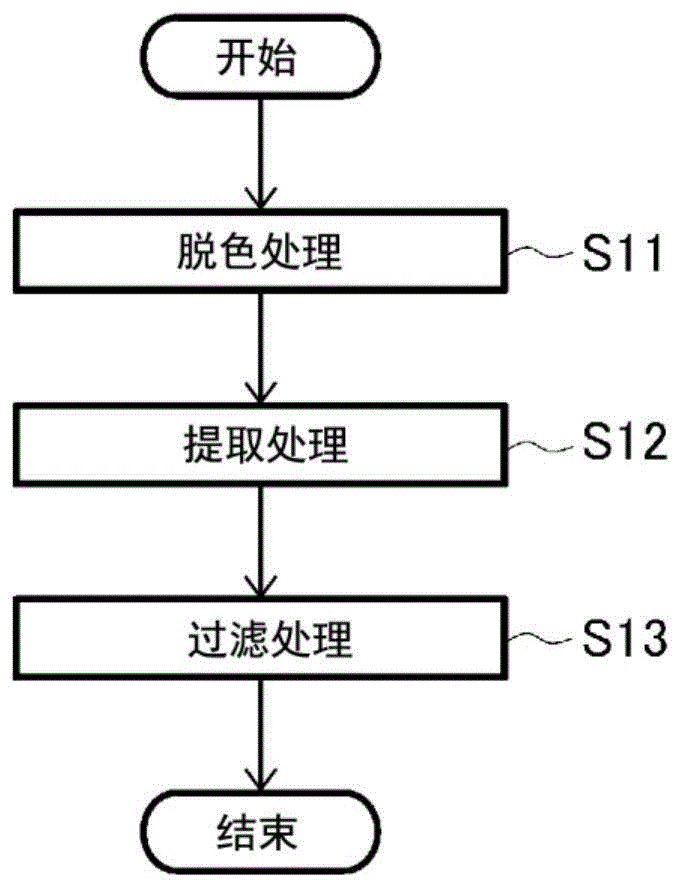
:作为抗炎症剂及针对肺癌的抗癌剂,已提出了各种物质(例如,参照非专利文献1及2)。现有技术文献非专利文献非专利文献1:和田孝一郎、上崎善规著、“pparγアゴニストの抗炎症作用―基礎·臨床研究の最新動向-(pparγ激动剂的抗炎症作用―基础和临床研究的最新动向―)”、日本临床、2010年、第68卷第2号、p.278-283非专利文献2:vincentt.devita,jr.andedwardchu,“ahistoryofcancerchemotherapy”,cancerres,november1,2008,vol.68,issue.21,p.8643-8653技术实现要素:发明要解决的问题但是,需要能够作为抗炎症剂及针对肺癌的抗癌剂等使用的、安全性优异的新化合物及其制造方法。因此,本发明的目的在于,提供能够作为抗炎症剂及针对肺癌的抗癌剂等使用的、安全性优异的新化合物及其制造方法。用于解决问题的方案为了达到上述目的,本发明的化合物或其盐的特征在于,其为式(a)或式(b)所示的化合物或其盐。式(a)及式(b)中,x1为己糖或羟基,x2为磷酸基或羟基。本发明的化合物或其盐的制造方法的特征在于,包括下述工序:从固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)及其培养物中的至少一者中提取包含式(a)或式(b)所示的化合物或其盐的粗提取液的粗提取工序;和从上述粗提取液中分离式(a)或式(b)所示的化合物或其盐的纯化工序。发明的效果为了达到上述目的,本发明人们反复进行了一系列研究,发现从固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)或其培养物中得到的式(a)或式(b)所示的化合物或其盐具有抗炎症效果及针对肺癌的抗癌效果,从而完成了本发明。式(a)或式(b)所示的化合物或其盐的安全性高,能够长期给药。附图说明图1是示出本发明的制造方法中的粗提取工序的一例的流程图。图2是示出本发明的制造方法中的纯化工序的一例的流程图。图3是示出实施例3中的被检液的il-8浓度的图表。图4是示出实施例4中的被检液的吸光度的图表。图5是示出实施例5中的被检液的吸光度的图表。图6是示出实施例6中的被检液的tnf-α浓度的图表。图7是示出实施例7中的被检液的tnf-α浓度的图表。图8是示出实施例8-1中的被检液的il-6浓度的图表。图9是示出实施例9中的小鼠的肿瘤体积的图表。图10是在实施例2中将甲酯化处理物供于gc-ms分析时的质谱图。图11是在实施例2中将水解甲酯化处理物供于gc-ms分析时的质谱图。图12是在实施例2中将吡咯烷基化处理物供于gc-ms分析时的质谱图。图13是在实施例2中将水解吡咯烷基化处理物供于gc-ms分析时的质谱图。图14是在实施例2中将图12中的峰4的化合物供于gc-ms分析时的质谱图。图15是在实施例2中将图13中的峰d的化合物供于gc-ms分析时的质谱图。图16a是在实施例2中将试样溶液供于1hnmr测定时的光谱的一部分。图16b是在实施例2中将试样溶液供于1hnmr测定时的光谱的一部分。图16c是在实施例2中将试样溶液供于1hnmr测定时的光谱的一部分。图16d是在实施例2中将试样溶液供于1hnmr测定时的光谱的一部分。图17a是在实施例2中将试样溶液供于13cnmr测定时的光谱的一部分。图17b是在实施例2中将试样溶液供于13cnmr测定时的光谱的一部分。图17c是在实施例2中将试样溶液供于13cnmr测定时的光谱的一部分。图17d是在实施例2中将试样溶液供于13cnmr测定时的光谱的一部分。图18是在实施例2中将试样溶液供于31pnmr测定时的光谱图。图19是在实施例2中将试样溶液供于gc-ms分析时的质谱图。图20是在实施例2中将图19中的m/z=1417.93的峰的化合物供于gc-ms/ms分析时的质谱图。图21是将试样溶液供于gc-ms分析时的质谱图。图22是在实施例2中将图21中的m/z=538.15的峰的化合物供于gc-ms/ms分析时的质谱图。图23是在实施例2中将图22中的m/z=474.27的峰的化合物供于gc-ms/ms/ms分析时的质谱图。图24是在实施例2中将图22中的m/z=601.17的峰的化合物供于gc-ms/ms/ms分析时的质谱图。图25是在实施例2中将图24中的m/z=557.19的峰的化合物供于gc-ms/ms/ms/ms分析时的质谱图。图26是在实施例2中将图24中的m/z=645.12的峰的化合物供于gc-ms/ms/ms/ms分析时的质谱图。图27是示出在实施例8-2中pparγ基因的表达量的图表。图28是示出在实施例2中肼分解物的经4mol/l的koh处理而游离出的糖链部分的凝胶过滤谱的图表。图29是在实施例2中将试样a供于1hnmr测定时的光谱。图30是在实施例2中将试样a供于2ddqf-cosy测定时的光谱。图31是在实施例2中将试样a供于2dnoesy测定时的光谱。图32是在实施例2中将试样a供于2d1h-31phmbc测定时的光谱。图33是实施例2中的试样a的示意图。具体实施方式在本发明的制造方法中,上述粗提取工序中的上述提取处理可以是利用使蛋白质不溶化的有机溶剂的提取处理。在这种情况下,上述提取溶剂可以是苯酚。在本发明的制造方法中,在上述粗提取工序中,在上述提取处理之前对上述固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)及其培养物中的至少一者可以进行色素的脱色处理。在这种情况下,上述色素的脱色处理可以是利用选自由丙酮、甲醇及氯仿组成的组中的至少一种的脱色处理。在本发明的制造方法中,在上述粗提取工序中,对于上述粗提取液可以进行过滤处理。在本发明的制造方法中,在上述纯化工序中,对于上述粗提取液可以进行酶处理和利用使蛋白质不溶化的有机溶剂的提取处理。在这种情况下,上述酶处理可以是利用核酸分解酶及蛋白质水解酶中的至少一者的酶处理,上述有机溶剂可以是苯酚。在本发明的制造方法中,在上述纯化工序中,对于上述提取处理后的提取液可以进行过滤处理。以下对本发明进行详细说明。<新型化合物>如上所述,本发明的化合物或其盐的特征在于,其为式(a)或式(b)所示的化合物或其盐。在式(a)及式(b)中,x1与x2的组合为己糖与磷酸基、羟基与羟基、己糖与羟基、羟基与磷酸基的4种组合,优选己糖与磷酸基、羟基与羟基的组合。x1为己糖时,例如,位于其2位的碳的羟基的氧与作为x1的键合对象的己糖的4位的碳键合。就本发明的化合物或其盐而言,如果式(a)或式(b)中x1及x2分别为己糖及磷酸基则成为式(a1)或式(b1)所示的化合物或其盐,如果式(a)或式(b)中x1及x2均为羟基则成为式(a2)或式(b2)所示的化合物或其盐。本发明的化合物或其盐以下也称为“本发明的新型化合物”。本发明的新型化合物例如可以通过后述的制造方法得到。但是,后述的制造方法不过是例示,不对本发明进行限定。在本发明的新型化合物中,己糖可以为例如葡萄糖。本发明的新型化合物可以用于任何用途,例如,能够作为后述的抗炎症剂、针对肺癌的抗癌剂的材料等来利用。如后述的实施例中已证实那样,本发明的新型化合物具有抑制nf-κb(nuclearfactor-kappab,核因子κb)、tnf-α(tumornecrosisfactor-α,肿瘤坏死因子α)及il-6(interleukin-6,白介素-6)等炎症性细胞因子的生产的功能。另外,如后述的实施例中已证实那样,本发明的新型化合物还具有活化作为toll样受体之一的tlr4(toll-likereceptor4,toll样受体4)的功能。已知tlr4的活化会促进具有抗炎症作用的i型干扰素的产生(ninamaeshimaandrachelc.fernandez,“recognitionoflipidavariantsbythetlr4-md-2receptorcomplex”,frontiersincellularandinfectionmicrobiology,february2013,volume3,article3,p.2,figure2)。因此,本发明的新型化合物具有抗炎症效果。进而,如后述的实施例中已证实那样,本发明的新型化合物还具有抑制肺癌增殖的功能。<抗炎症剂及炎症性疾病的治疗方法>本发明的抗炎症剂是通过本发明的化合物或其盐(上述新型化合物)所具有的抗炎症效果来抑制炎症的抗炎症剂,除了包含本发明的新型化合物以外没有任何限制。作为通过本发明的抗炎症剂抑制炎症的疾病,可列举例如:溃疡性结肠炎及克罗恩病等炎症性肠疾病;牛皮癣及皮炎等炎症性皮肤疾病;脑炎、肝炎、肾炎、肺炎、支气管炎、脉管炎、脑膜炎、甲状腺炎、糖尿病、炎症性胆汁疾病、伴有炎症的癌症等,没有特别限定。根据本发明的抗炎症剂,通过本发明的新型化合物所具有的抗炎症效果还发挥炎症所伴随的疼痛的镇痛效果。上述抗炎症剂的剂形可列举例如散剂、细颗粒剂、颗粒剂、片剂、包衣片剂、胶囊剂、含片剂、液剂等,没有特别限制。另外,上述抗炎症剂的组成没有特别限制,除了本发明的新型化合物以外,还可以含有例如赋形剂、结合剂、润滑剂、崩解剂、吸收促进剂、乳化剂、稳定化剂、防腐剂等各种添加剂等。另外,上述抗炎症剂可以利用通常使用的制剂化技术等来制造。本发明的炎症性疾病的治疗方法包括:给药包含上述新型化合物的本发明的抗炎症剂的工序。在本发明中,“治疗”包含例如:改善症状(好转)、消除症状(治愈)、防止症状恶化、预防等,在后述的肺癌的治疗方法中也同样。作为给药上述抗炎症剂的动物种属,没有特别限制,可列举例如:人或猴、牛、猪、狗、猫等非人哺乳类;鸡等禽类;鱼贝类等。作为上述给药方法,没有特别限制,可列举例如经口给药或非经口给药,上述非经口给药可列举例如经皮吸收、注射、栓剂给药等。上述抗炎症剂的给药量例如可以根据动物种属、年龄等适宜设定,没有特别限制。<针对肺癌的抗癌剂及肺癌的治疗方法>本发明的针对肺癌的抗癌剂是通过本发明的化合物或其盐(上述新型化合物)所具有的针对肺癌的抗癌效果来抑制肺癌增殖的抗癌剂,除了包含本发明的新型化合物以外没有任何限制。另外,本发明的肺癌的治疗方法包括:给药包含上述新型化合物的本发明的针对肺癌的抗癌剂的工序。上述抗癌剂的剂形、给药上述抗癌剂的动物种属及上述抗癌剂的给药方法与上述抗炎症剂的剂形、给药上述抗炎症剂的动物种属及上述抗炎症剂的给药方法同样。<新型化合物的制造方法>如上所述,本发明的化合物或其盐(上述新型化合物)的制造方法的特征在于,包括下述工序:从固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)及其培养物中的至少一者中提取包含式(a)或式(b)所示的化合物或其盐的粗提取液的粗提取工序;和从上述粗提取液分离式(a)或式(b)所示的化合物或其盐的纯化工序。〔粗提取工序〕如上所述,上述粗提取工序是从固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)及其培养物中的至少一者中提取包含式(a)或式(b)所示的化合物或其盐的粗提取液的工序。首先对上述固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)及其培养物中的至少一者进行说明。上述固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)及其培养物中的至少一者优选具有下述(1)~(30)的菌学特征。需要说明的是,上述bp0899株在国家高级工业科学技术学院,国际专利生物保藏中心(日本千叶县木更津市上总镰足2-5-8)以保藏编号nitep-644进行了保藏(保藏日:2008年9月12日),进而,以保藏编号nitebp-644进行了国际保藏(移管日:2010年10月27日)。(1)细胞的形状:杆状形或卵形(2)多形性:无(3)细胞的大小:0.8μm×1.0μm(4)运动性的有无:有(5)孢子的有无:无(6)普通琼脂培养时的光泽:有(7)普通琼脂培养时的色素产生:有(8)普通肉汤培养时的表面生长的有无:无(9)普通肉汤培养时的培养基混浊的有无:有(10)明胶穿刺培养时的明胶液化:阴性(11)石蕊·牛乳培养时的凝固:无(12)石蕊·牛乳培养时的液化:无(13)革兰氏染色性:阴性(14)硝酸盐的还原:无(15)脱氮反应:无或有(16)mr试验:阴性(17)吲哚产生:无(18)硫化氢的生成:无(19)淀粉的水解:无(20)柠檬酸的利用(christensen):无(21)无机氮源的利用(铵盐):有(22)过氧化氢酶的生成:阳性(23)氧化酶的生成:阳性(24)厌氧生长性:有(25)o-f试验(氧化/发酵):阴性/阴性(26)β-半乳糖苷酶活性:阴性(27)精氨酸双水解酶活性:阴性(28)赖氨酸脱羧酶活性:阴性(29)色氨酸脱氨酶活性:阴性(30)明胶酶活性:阴性上述bp0899株的16srrna的碱基序列优选为序列号1所示的碱基序列。上述bp0899株及其培养物中的至少一者在暗处的好氧培养条件下进而可以显示出例如下述(31)的表所示的性质。需要说明的是,该表中,“-”表示无产生,“+”表示有产生。(31)由糖类产酸及产气上述菌学特征例如还可以在预培养后进一步基于主培养的结果进行评价。上述预培养还可以例如在普通琼脂培养基中接种上述bp0899株,在30℃下进行24小时培养。上述主培养的条件可以根据各菌学特征的评价方法进行适宜设定。具体而言,上述(1)~(5)的培养条件是例如使用普通琼脂培养基、在30℃、暗处的好氧培养,上述(6)~(7)的培养条件是例如使用普通肉汤培养基、在30℃、明处的厌氧培养,上述(8)~(12)的培养条件是例如使用各培养基、在30℃、暗处的好氧培养,上述(13)、(14)、(16)、(17)、(19)~(23)、(25)的氧化试验、(26)、(29)、(30)及(31)是例如在暗处的好氧培养,(15)、(18)、(24)、(25)的发酵试验、(27)及(28)是例如在暗处的厌氧培养。作为这些菌学特征的试验方法,没有特别限制,可以采用现有公知的方法。具体而言,例如可列举:barrowg.i.及felthamr.k.a.著“cowanandsteel‘smanualfortheidentificationofmedicalbacteria.”(英国)3rdeditioncambridgeuniversitypress1993年、坂崎等著《新細菌培地学講座·下》(新细菌培养基学讲座·下)(东京)第二版近大出版1988年、长谷川编著《微生物の分類と同定(下)》(微生物的分类和鉴定(下))(东京)学会出版中心1985年、土壤微生物研究会编《新編土壌微生物学実験》(新编土壤微生物学实验)(东京)养贤堂1992年等中记载的方法。上述(15)的试验方法例如可以采用:上述《微生物の分類と同定(下)》(微生物的分类和鉴定(下))中记载的驹形等的方法、使用了giltay培养基的上述《新編土壌微生物学実験》(新编土壤微生物学实验)中记载的方法、使用了pyn培养基的下水法等。上述驹形等的方法是:在使用了1%硝酸钠肉汤的厌氧培养条件下,将观察到生长和气体形成的样品判定为脱氮反应阳性。上述使用了giltay培养基的方法是:在使用了装有德拉姆管(durhamtube)的上述giltay培养基(ph7.0~7.2)的厌氧培养条件下,将产气和呈现深蓝色的样品判定为脱氮反应阳性。需要说明的是,上述giltay培养基是混合了a液(kno31g、天冬酰胺1g、1%溴百里酚蓝·醇溶液5ml及蒸馏水500ml)及b液(柠檬酸钠8.5g、mgso4·7h2o1g、fecl3·6h2o0.05g、kh2po41g、cacl2·6h2o0.2g及蒸馏水500ml)的培养基。另外,上述试验方法例如还可以使用市售的细菌鉴定试剂盒。作为上述试剂盒,没有特别限制,例如可以使用细菌鉴定试剂盒api20e(biomeleucorporation制)等。上述bp0899株及其培养物中的至少一者例如可以进一步具有下述(32)~(40)的菌学特征。(32)菌落的颜色:红色(33)明胶穿刺培养:不生长(34)vp试验:阴性(35)柠檬酸的利用(koser):有(36)无机氮源的利用(硝酸盐):有(37)脲酶活性:阴性(38)生长的ph范围:5~9(39)由d-甘露糖醇产酸:产生(40)由d-甘露糖醇产气:不产生作为上述(32)~(40)的菌学特征的试验方法,没有特别限制,可以采用现有公知的方法。具体而言,例如可列举上述文献等中记载的方法。另外,上述试验方法还可以使用例如市售的细菌鉴定试剂盒。作为上述试剂盒,没有特别限制,可以使用例如上述细菌鉴定试剂盒等。作为上述bp0899株的采集源,没有特别限定,可列举例如:土壤、海水、河水、湖水、沼泽水等。另外,作为上述土壤,可列举例如:陆地、海底、河底、湖底及沼泽底的土、砂和泥土等,没有特别限定作为上述bp0899株的分离方法,例如可以使用现有公知的采集法、培养法等,没有特别限制。作为上述分离方法,例如,采集源为湖水的情况,可以通过用过滤器等对采集的湖水进行过滤,将其滤液用琼脂培养基等进行培养,从得到的菌落中分离上述bp0899株。另外,例如,采集源为泥土的情况,可以利用缓冲液等将所采集的泥土悬浮后,将该悬浮液离心分离,将得到的上清液用琼脂培养基等进行培养,从得到的菌落中分离上述bp0899株。上述分离出的bp0899株例如可以进一步在液体培养基中进行培养。在上述bp0899株的培养中,对于培养基没有特别限定,可列举例如:低级脂肪酸添加培养基、苹果酸添加培养基、l-干燥标准品复原用培养基802“daigo”(日本制药株式会社制)、mys培养基(平石及北川、bulletinofthejapanesesocietyofscientificfisheries、1984年、50卷、11号、p.1929-1937)、改良mys培养基、生长用培养基等,优选为低级脂肪酸添加培养基、苹果酸添加培养基、l-干燥标准品复原用培养基802“daigo”(日本制药株式会社制)。作为上述低级脂肪酸添加培养基及上述苹果酸添加培养基,可列举例如:在下述表1的基础培养基中添加了生物素、维生素b1、烟酸、低级脂肪酸或苹果酸的钠盐的培养基。作为上述低级脂肪酸,没有特别限制,优选例如乙酸、丙酸、乳酸等。[表1](基础培养基)作为上述改良mys培养基、生长用培养基,可列举例如下述表2及表3的组成的培养基。[表2](改良mys培养基的组成)[表3](生长用培养基的组成)上述培养中,对于温度范围没有特别限定,例如为23~39℃、30℃。另外,上述培养中,对于ph范围没有特别限定,例如为ph5.5~8.5、6.0~8.5、7.0。上述培养例如可以在好氧条件下进行,还可以在厌氧条件下进行,没有特别限制,优选为厌氧条件下。另外,上述培养时的光照条件也没有特别限制,例如,既可以是黑暗条件、也可以是照明条件,优选为2000勒克斯~10000勒克斯的照度下。上述培养例如还可以在密闭照明式培养槽内进行。另外,还可以边使用在上述密闭照明式培养槽内所具备的搅拌装置搅拌培养液边进行培养。上述培养时间没有特别限制,例如可以是上述bp0899株的增殖达到稳定期为止的时间。上述bp0899株的增殖在约72小时以内达到稳定期的培养条件下的情况,上述培养时间例如可以是72小时。如上所述,上述bp0899株的16srrna的碱基序列优选为序列号1所示的碱基序列。上述16srrna的碱基序列例如可以利用上述方法等从所分离和培养的上述bp0899株中提取dna,使用引物等来确定。上述dna的提取和上述碱基序列的确定方法例如可以使用常规方法,没有特别限制。另外,上述引物没有特别限制,例如可列举以下的引物等。(引物)9f(序列号2)5’-gagtttgatcctggctcag-3’339f(序列号3)5’-ctcctacgggaggcagcag-3’785f(序列号4)5’-ggattagataccctggtagtc-3’1099f(序列号5)5’-gcaacgagcgcaaccc-3’536r(序列号6)5’-gtattaccgcggctgctg-3’802r(序列号7)5’-taccagggtatctaatcc-3’1242r(序列号8)5’-ccattgtagcacgtgt-3’1541r(序列号9)5’-aaggaggtgatccagcc-3’作为上述bp0899株的培养物,可列举例如:上述bp0899株的菌体、上述bp0899株的培养上清液、上述bp0899株的菌体提取物等,没有特别限定。上述培养物可以是例如上述菌体的处理物、上述培养上清液的处理物、上述菌体提取物的处理物等,没有特别限定。作为上述处理物,没有特别限定,可列举例如:上述培养物的浓缩物、干燥物、冷冻干燥物、溶剂处理物、表面活性剂处理物、酶处理物、蛋白质分级物、超声波处理物、磨碎处理物等。另外,上述培养物可以是例如上述菌体、上述培养上清液、上述菌体提取物、上述菌体的处理物、上述培养上清液的处理物、上述菌体提取物的处理物等的混合物。作为上述混合物,可以以任意的组合和比率混合,没有特别限制。作为上述组合,没有特别限制,可列举例如:上述菌体与上述培养上清液的混合物等。图1的流程图中示出上述粗提取工序的一例。如图所示,本例的上述粗提取工序包括脱色处理(步骤s11)、提取处理(步骤s12)和过滤处理(步骤s13)。(1)脱色处理(步骤s11)首先,对上述固氮红细菌(rhodobacterazotoformans)bp0899株(保藏编号nitebp-644)或其培养物进行色素的脱色处理。对于上述色素的脱色处理没有特别限制,可列举例如利用有机溶剂的脱色处理。上述有机溶剂可列举例如丙酮、甲醇、氯仿及它们的混合溶剂等。上述脱色处理例如可通过将上述菌体或上述培养物与上述有机溶剂混合来进行。具体而言,例如,相对于烧杯内的上述bp0899株的冷冻干燥菌体10g~60g加入丙酮25ml~150ml,使用搅拌器充分进行搅拌。接下来将上述搅拌后的溶液的上清液转移到50ml锥形管中,以2000rpm~5000rpm、5分钟~10分钟的条件进行离心分离,将得到的上清液除去,向沉淀物中加入丙酮20ml~40ml,返回到上述烧杯中。重复进行该操作直至通过目视观察不到上述bp0899株的色素的颜色(褐色)为止,然后将上述沉淀物使用抽吸器减压干燥至质量恒定为止,由此得到脱色后的干燥菌体。(2)提取处理(步骤s12)接下来,将上述脱色处理后的菌体或培养物用使蛋白质不溶化的有机溶剂进行处理,进行蛋白质的除去。上述有机溶剂可列举例如苯酚等。就上述提取处理而言,例如,将上述菌体或培养物与上述有机溶剂和水性溶剂混合,将由于上述有机溶剂而不溶化的蛋白质分配到上述有机溶剂相中,使作为目标的上述化合物分配到上述水性溶剂相中。具体而言,例如,向上述烧杯内的脱色干燥菌体10g~60g中加入注射用水而使上述脱色干燥菌体的浓度为60mg/ml~90mg/ml。接下来,加入与上述注射用水等量的90%苯酚,在加热搅拌器上在65℃~70℃下搅拌20分钟~40分钟,将该操作作为初次提取。然后将上述搅拌后的溶液冷却到10℃以下,然后使用离心分离管按照8000rpm~20000rpm、20分钟~60分钟、2℃~10℃的条件进行离心分离,由此分离为苯酚相和水相,将得到的上述水相回收到50ml锥形管中,向残留在上述离心分离管中的苯酚相中加入与所回收的水相等量的注射用水,重复进行与上述的初次提取同样的操作(第2次提取)。进而,将与上述初次提取同样的操作再重复进行一次(第3次提取)。通过如此提取3次而回收500ml~1000ml的所得到的水相。(3)过滤处理(步骤s13)接下来,对通过提取处理得到的水相实施过滤处理,将混入上述水相的有机溶剂(上述提取处理中使用的苯酚等有机溶剂)除去。作为上述过滤,可列举例如超滤等。上述过滤中的分级分子量为例如7000,优选将小于上述分级分子量的分子除去。具体而言,例如,将上述回收的水相装入分级分子量为7000的透析管,将外液设为蒸馏水1l~10l而进行透析。反复进行上述透析,直至外液确认不到作为苯酚的吸收波长的270nm处的光吸收为止,将内液回收,作为包含上述本发明的新型化合物的粗提取液。〔纯化工序〕在图2的流程图中示出上述纯化工序的一例。如图所示,本例的上述纯化工序包括酶处理(步骤s21)、提取处理(步骤s22)和过滤处理(步骤s23)。(1)酶处理(步骤s21)对于上述粗提取工序中得到的包含上述本发明的新型化合物的粗提取液,进行酶处理。对上述酶处理没有特别限制,可列举例如利用核酸分解酶的处理、利用蛋白质水解酶的处理,可以进行任一种处理,也可以进行两种处理。在后者的情况下,对其顺序没有特别限制,例如,可以在进行利用核酸分解酶的处理后,进行利用蛋白质水解酶的处理。首先,对上述粗提取液实施利用核酸分解酶的处理。对上述核酸分解酶没有特别限制,可列举例如rna分解酶、dna分解酶。作为上述rna分解酶,没有特别限定,可使用例如sigma公司制的核糖核酸酶a、和光纯药工业(株)制的核糖核酸酶a、罗氏公司制的核糖核酸酶a等。作为上述dna分解酶,没有特别限定,可使用例如sigma公司制的脱氧核糖核酸酶i、和光纯药工业(株)制的脱氧核糖核酸酶i、罗氏公司制的脱氧核糖核酸酶i等。具体而言,例如,向上述粗提取液中加入0.2mg/ml~1mg/ml的rna分解酶和1μg/ml~10μg/ml的dna分解酶并在30℃~40℃下孵育4小时~24小时。接下来,对上述粗提取液实施利用蛋白质水解酶的处理。作为上述蛋白质水解酶,没有特别限定,可使用例如sigma公司制的蛋白水解酶k、和光纯药工业(株)制的蛋白水解酶k、罗氏公司制的蛋白水解酶k等。具体而言,例如,向上述粗提取液中添加100μg/ml~300μg/ml的蛋白质水解酶并在40℃~50℃下孵育2小时~24小时。(2)提取处理(步骤s22)接下来,将上述粗提取液用使蛋白质不溶化的有机溶剂进行处理,进行蛋白质的除去。上述有机溶剂可列举例如苯酚等。具体而言,例如,将上述酶处理后的提取液按照2000rpm~5000rpm、20分钟~60分钟的条件进行离心分离。然后,将得到的沉淀级分约1ml~10ml和上清液级分约50ml~100ml中的上述沉淀级分装入分级分子量50000~100000的超滤管中,将外液设为蒸馏水5ml~15ml而进行超滤。向得到的内液中加入注射用水10ml~60ml和90%苯酚10ml~60ml,在加热搅拌器上在65℃~70℃下搅拌20分钟~40分钟,将该操作作为初次提取。然后将上述搅拌后的溶液冷却到10℃以下,然后使用离心分离管按照8000rpm~20000rpm、20分钟~60分钟、2℃~10℃的条件进行离心分离,分离为苯酚相和水相,得到的上述水相回收到50ml锥形管中,向残留在上述离心分离管中的苯酚相中加入与所回收的水相等量的注射用水,重复进行与上述初次提取同样的操作(第2次提取)。进而,将与上述初次提取同样的操作再重复进行一次(第3次提取)。如此操作总计回收60ml~120ml的3次的提取操作的水相。(3)过滤处理(步骤s23)接下来,对通过提取处理得到的水相实施过滤处理,将混入上述水相的有机溶剂(上述提取处理中使用的苯酚等有机溶剂)除去。作为上述过滤,可列举例如超滤等。上述过滤中的分级分子量为例如50000~100000,优选将小于上述分级分子量的分子除去。具体而言,例如,将上述回收的水相装入分级分子量为7000的透析管,将外液设为蒸馏水0.5l~1l而进行24小时~96小时的透析。将得到的内液装入分级分子量为50000~100000的超滤管,将外液设为蒸馏水5ml~15ml而进行超滤。将得到的内液冷冻干燥,由此得到作为本发明的新型化合物的式(a)或式(b)所示的化合物或其盐。实施例接下来,对本发明的实施例进行说明。但是,本发明不受下述的实施例限定。只要没有特别声明,则市售的试剂基于它们的说明来使用。〔实施例1〕利用下述方法制造式(a)所示的化合物及式(b)所示的化合物。〔1.粗提取工序〕(1-1)脱色处理(步骤s11)相对于烧杯内的上述bp0899株的冷冻干燥菌体20.03g加入丙酮50ml,使用搅拌器搅拌10分钟。接下来,将上述搅拌后的溶液的上清液转移到50ml锥形管中,按照2000rpm、5分钟的条件进行离心分离,除去所得到的上清液,对沉淀物加入丙酮20ml,返回到上述烧杯中。重复进行该操作,直至通过目视确认不到上述bp0899株的色素的颜色(褐色)为止。然后,将脱色后的沉淀物用抽吸器进行减压干燥,直至质量恒定为止,由此得到脱色干燥菌体。(1-2)提取处理(步骤s12)向装入烧杯的上述脱色干燥菌体16g加入注射用水,使上述脱色干燥菌体的浓度为75mg/ml。接下来,加入与上述注射用水等量的90%苯酚,在加热搅拌器上在65℃~70℃下搅拌30分钟,将其作为初次提取。然后,将上述搅拌后的溶液冷却到10℃以下,然后使用离心分离管按照15000rpm、40分钟、4℃的条件进行离心分离,由此分离为苯酚相和水相,所得到的上述水相回收到50ml锥形管中,向残留在上述离心分离管中的苯酚相中加入与所回收的水相等量的注射用水,重复进行与上述初次提取同样的操作(第2次提取)。进而,将与上述初次提取同样的操作再重复进行一次(第3次提取)。由此回收450ml的3次提取操作的水相。(1-3)过滤处理(步骤s13)将上述回收的水相450ml装入分级分子量7000的透析管,将外液设为蒸馏水2.5l而进行透析。对外液进行上述透析22次,直至确认不到作为苯酚的吸收波长的270nm处的光吸收为止,回收含有式(a)所示的化合物及式(b)所示的化合物的作为粗提取液的内液75ml。[2.纯化工序](2-1)酶处理(步骤s21)首先,向上述粗提取工序中得到的含有式(a)所示的化合物及式(b)所示的化合物的粗提取液中添加0.5mg/ml的rna分解酶(商品名:sigma公司制的核糖核酸酶a)和5μg/ml的dna分解酶(sigma公司制的脱氧核糖核酸酶i),在37℃下孵育6小时。接下来,向上述粗提取液中添加200μg/ml的蛋白质水解酶(sigma公司制的蛋白水解酶k),在50℃下孵育4小时后,按照3000rpm、30分钟的条件进行离心分离。(2-2)提取处理(步骤s22)将上述酶处理中通过离心分离得到的、沉淀级分约3ml以下和上清液级分约72ml中的上述沉淀级分装入分级分子量100000的超滤管,将外液设为蒸馏水15ml而进行超滤。向得到的内液中加入注射用水30ml和90%苯酚30ml,在加热搅拌器上在65℃~70℃下搅拌30分钟,将其作为初次提取。然后将上述搅拌后的溶液冷却到10℃以下,然后使用离心分离管按照15000rpm、40分钟、4℃的条件进行离心分离,由此分离为苯酚相和水相,得到的上述水相回收到50ml锥形管中,向残留在上述离心分离管中的苯酚相中加入与所回收的水相等量的注射用水,重复进行与上述初次提取同样的操作(第2次提取)。进而,将与上述初次提取同样的操作再重复进行一次(第3次提取)。由此回收80ml的3次提取操作的水相。(2-3)过滤处理(步骤s23)将上述回收的水相装入分级分子量7000的透析管,将外液设为蒸馏水1l而进行72小时的透析。将得到的内液装入分级分子量100000的超滤管,将外液设为蒸馏水15ml而进行超滤。将得到的内液进行冷冻干燥,由此得到纯化物164.53mg。〔实施例2〕将上述纯化物供于质谱(massspectrometry)及核磁共振(nuclearmagneticresonance、nmr)以确定其结构。(1)纯化物的分解物的质谱将上述纯化物按照以下方式分解,由此制备上述纯化物的分解物。首先,将上述纯化物溶解于0.1mol/l的盐酸而达到10mg/ml的浓度,将上述溶解液在水浴中加热90分钟,得到沉淀物和无色透明的上清液。接下来,回收上述沉淀物,供于氯仿提取,回收氯仿级分。向上述氯仿级分中追加氯仿而达到38mg/ml的浓度,将该溶解液6μl供于薄层层析(tlc)。需要说明的是,作为tlc板,使用了10cm×10cm的硅胶60f254tlc板(merck公司制),作为展开溶剂,使用了氯仿:甲醇:蒸馏水:三乙胺=30:13:2:0.1(体积比)的溶液。然后,对展开后的上述tlc板喷雾50%的硫酸,刮取与染色最强的斑点对应的胶,再次溶解于上述展开溶剂。然后,在除去溶剂后,回收溶剂以外的级分并冷冻干燥,将其作为上述纯化物的分解物(以下也称为“分解物”。)。将上述分解物分别供于以下的处理,由此制备甲酯化处理物、吡咯烷基化处理物、水解甲酯化处理物及水解吡咯烷基化处理物这4种试样溶液。<甲酯化处理物的制备>(i)向按照上述分解物浓度为30mg/ml的方式制备的氯仿溶液0.1ml中添加0.2mg/mlbht(二丁基羟基甲苯)氯仿溶液1ml,干固。(ii)对上述干固物添加5%盐酸·甲醇1ml,在85℃下反应24小时。(iii)将上述反应物自然冷却后,添加己烷1ml、水0.5ml,回收己烷相。(iv)将上述回收的己烷相在氮气气流下馏去溶剂,再溶解于氯仿1ml,得到甲酯化处理物的试样溶液。<吡咯烷基化处理物的制备>(i)取上述甲酯化处理物的试样溶液200μl至玻璃试管中。(ii)向上述玻璃试管中添加吡咯烷400μl及乙酸40μl,在100℃下反应30分钟。(iii)反应结束后,向上述反应物加入二氯甲烷2ml和5%乙酸水溶液2ml并振荡混合后,回收二氯甲烷相。(iv)在氮气气流下从上述回收的二氯甲烷相中除去溶剂后,再溶解于氯仿200μl,得到吡咯烷基化处理物的试样溶液。<水解甲酯化处理物的制备>(i)对于上述分解物,进行与上述甲酯化处理物的制备中的(i)同样的处理,得到干固物。(ii)向上述干固物中添加0.5mol/lnaoh·甲醇1ml,在50℃下反应1小时。(iii)将上述反应物自然冷却后,添加己烷1ml、1mol/l盐酸试液0.5ml,回收己烷相。(iv)对上述回收的己烷相,进行与上述甲酯化处理物的制备中的(iv)同样的处理,得到水解甲酯化处理物的试样溶液。<水解吡咯烷基化处理物的制备>对上述水解甲酯化处理物,进行与上述吡咯烷基化处理物的制备中的(i)~(iv)同样的处理,得到水解吡咯烷基化处理物的试样溶液。(1-1)甲酯化处理物及水解甲酯化处理物的gc-ms将上述甲酯化处理物及上述水解甲酯化处理物按照下述条件供于gc-ms(气相色谱质谱联用)分析。<gc-ms条件>设备:jms-700v(日本电子(株)制)色谱柱:spb-130m×0.25mm膜厚0.25μm柱温度:50℃(保持1分钟)→300℃(以+8℃/分钟升温、保持30分钟)注入口温度:250℃检测器:氢火焰离子化检测器(fid)300℃进样量:1μl(无分流进样)载气:氦气(线速度30cm/sec、恒定流量模式)检测器:ms离子化法:ei(electronionization)离子化电流:300μa离子化能量:70ev离子化室温度:300℃电子加速电压:10kv扫描范围:m/z=35~500(sec/scan)将其结果示于图10及图11。图10是将上述甲酯化处理物供于gc-ms分析时的质谱图,图11是将上述水解甲酯化处理物供于gc-ms分析时的质谱图。在图10及图11中,纵轴表示检测强度,横轴表示检测时间(min)。如图10所示,由上述甲酯化处理物得到峰1~11。将这些中的检测强度高的峰1、2、4、6及7的化合物分别进一步以与上述相同的条件供于gc-ms分析。对于得到的质谱图,利用数据库(thenistmassspectralseachprogramforthenist/epa/nihmassspectrallibrary)的自动模式对具有相似形状的质谱图的结构物进行谱库检索。其结果是,峰1、2、4及7的化合物分别与式(2)~(5)的化合物显示高的相似度。另外,本发明人们发现,峰6的化合物的质谱图与strittmatter等(strittmatterw.etal(1983),journalofbacteriology,vol.155,no.1,p.153-p.158,fig2)记载的3-氧代-十四烷酸甲酯的质谱图非常相似。因此推定,峰6的化合物为式(6)的3-氧代-十四烷酸甲酯。另外,如图11所示,由上述水解甲酯化处理物得到峰a~i。在此,比较了图10的质谱图和图11的质谱图,确认图11中的峰a、b及d分别为对应于图10的峰1、2及4的峰。将这些峰a、b及d分别进一步按照与上述相同的条件供于gc-ms分析。对于得到的质谱图,将具有相似形状的质谱图的结构物使用上述数据库进行检索。其结果是,峰a、b及d分别与式(7)、式(3)及式(4)的化合物显示高的相似度。需要说明的是,在图11中没有确认到对应于图10的峰6及7的峰,我们认为其原因如下。即,图10是未将上述分解物水解而直接供于gc-ms分析的结果,而图11是将上述分解物水解、仅将通过水解而分离出的物质供于gc-ms分析的结果。因此认为,图11中确认到的峰是上述分解物中具有可水解的酯键的化合物的峰。因此推定,在图11中没有确认到对应的峰的、图10的峰6及7的化合物是不具有可水解的酯键的化合物。综合图10及图11的结果来推定图10中检测强度高的峰1、2、4及7的化合物,则如以下的说明及表4所示。首先,如上所述,根据谱库检索的结果推定图10的峰1的化合物为式(2)的化合物。但是,如果峰1的化合物为式(2)的化合物,则与图10中的峰1相比应该在更短的保留时间(即,左侧)观察到峰,存在矛盾。另一方面,如上所述,根据谱库检索的结果推定对应于图10的峰1的图11的峰a的化合物为式(7)的化合物。峰a的化合物为式(7)的化合物时,与图11中的峰a的结果也一致。因此推定图10中的峰1的化合物为式(7)的化合物。接下来,如上所述,根据谱库检索的结果推定图10中的峰2的化合物为式(3)的bht(二丁基羟基甲苯)。另外,如上所述,根据谱库检索的结果推定对应于图10的峰2的图11的峰b的化合物也为式(3)的bht(二丁基羟基甲苯),图10与图11的结果一致。因此推定图10中的峰2的化合物为式(3)的bht(二丁基羟基甲苯),其不是上述分解物中包含的化合物,推定是在制备试样溶液时添加的bht(二丁基羟基甲苯)。并且,如上所述,根据谱库检索的结果推定图10中的峰4的化合物为式(4)的化合物。另外,如上所述,根据谱库检索的结果推定对应于图10的峰4的图11的峰d的化合物也为式(4)的化合物。因此推定图10中的峰4的化合物为式(4)的化合物。需要说明的是,关于确定式(4)的化合物的碳间双键的考察将在以下的(1-2)中详述。进而,如上所述,根据谱库检索的结果推定图10的峰7的化合物为式(5)的化合物。[表4](1-2)吡咯烷基化处理物及水解吡咯烷基化处理物的gc-ms接下来,为了确定式(4)的化合物中的碳间双键位置,将上述吡咯烷基化处理物及上述水解吡咯烷基化处理物按照下述条件供于gc-ms分析。<gc-ms条件>设备:jms-700v(日本电子(株)制)色谱柱:spb-130m×0.25mm膜厚0.25μm柱温度:100℃(保持1分钟)→300℃(以+10℃/分钟升温、保持30分钟)注入口温度:280℃检测器:氢火焰离子化检测器(fid)300℃进样量:1μl(无分流进样)载气:氦气(线速度30cm/sec、恒定流量模式)检测器:ms离子化法:ei离子化电流:300μa离子化能量:70ev离子化室温度:300℃电子加速电压:10kv扫描范围:m/z=35~500(sec/scan)将其结果示于图12及图13。图12是将上述吡咯烷基化处理物供于gc-ms分析时的质谱图,图13是将上述水解吡咯烷基化处理物供于gc-ms分析时的质谱图。图12及图13中,纵轴表示检测强度,横轴表示时间(min)。将图12中的峰4及图13中的峰d的化合物分别进一步按照与上述相同的条件供于gc-ms分析。其结果是,由峰4的化合物得到图14所示的质谱图,由峰d的化合物得到图15所示的质谱图。图14及图15中,纵轴表示检测强度,横轴表示m/z值。其中,在如式(8)所示那样从肼所键合的碳数起第4位和第5位的碳间断裂时,分子量为140。因此,在从肼所键合的碳数起第7位和第8位的碳间断裂时,如果不是双键则应该检测到m/z=182,但该位置存在双键,因此检测到m/z=180。根据以上而推定式(4)的化合物中的碳间双键存在于从羰基所键合的碳数起第7位和第8位之间。综合以上的上述(1-1)及上述(1-2)的结果,推定上述分解物中包含式(4)~(7)的4种化合物。另外,如上所述,仅将使上述分解物通过水解而分离出的物质进行gc-ms分析时在图11中未确认到峰,因此推测式(5)及式(6)的化合物是不具有酯键的化合物。(2)纯化物的分解物的结构分析从按照上述(1)中得到的分解物的浓度为30mg/ml的方式制备的氯仿溶液0.1ml中除去溶剂,添加氘代dmso600μl,转移到5mm试管中,将其作为试样溶液。<1hnmr测定及13cnmr测定>将上述试样溶液按照下述测定条件供于1hnmr测定及13cnmr测定。(测定条件)装置:unityinova500型(varian公司制)观察频率:499.8mhz(1h核)125.7mhz(13c核)溶剂:氘代dmso基准(※):溶剂:1h核(2.49ppm)、13c核(39.7ppm)温度:设为70℃测定方法:13cnmr、dept、noesy、roesy、cosy、tocsy、hsqc、hmbc※70℃下的氘代dmso下的化学位移值使用alba等(albas.etal(2004),glycobiology,vol.14,no.9,p.805-p.815)记载的值。<31pnmr测定>将加入了200μl的85%磷酸的3mm试管插入到装有上述试样溶液的上述5mm试管中。此时,使观测到的来自85%磷酸的信号与0.000ppm对齐,然后,拔出上述3mm试管,将上述试样溶液按照下述测定条件供于31pnmr测定。(测定条件)装置:unityinova500型(varian公司制)观测频率:499.8mhz(31p核)溶剂:氘代dmso基准:85%磷酸(外标):0.000ppm温度:设为25℃将其结果示于图16~图18。图16a~图16d是1hnmr测定的光谱,图17a~图17d是13cnmr测定的光谱,图18是31pnmr测定的光谱。图16~图18中,纵轴表示检测强度,横轴表示化学位移值(ppm)。根据图16~图18的光谱推定上述分解物为包含上述4种化合物和2分子的葡糖胺的式(9)的化合物。在式(9)中,键合在2分子的葡糖胺上的上述4种化合物从左侧起依次为式(7)的化合物、式(5)的化合物、式(4)的化合物、式(7)的化合物及式(6)的化合物。需要说明的是,式(9)中的数字对应于图16的1hnmr测定的光谱中的峰的数字,式(9)中的字母(大写)对应于图17的13cnmr测定的光谱中的峰的字母(大写),式(9)中的字母(小写)对应于图18的31pnmr测定的光谱中的峰的字母(小写)。(9)(3)纯化物的分解物的质量分析为了进一步证明上述分解物为式(9)的化合物,进行了以下的分析。将按照上述(1)中得到的分解物的浓度为30mg/ml的方式制备的氯仿溶液用甲醇稀释40000倍,将所得的溶液作为试样溶液,按照下述条件供于gc-ms分析。<gc-ms条件>设备:在amazonetd(brukerdaltonics公司制)上安装esiinterface测定模式:esi-it-ms、negativemode将其结果示于图19。图19是将上述试样溶液供于gc-ms分析时的质谱图。图19中,纵轴表示检测强度,横轴表示m/z值。如图19所示,确认到多个峰,但最接近作为上述分解物的推定化合物的式(9)的化合物的分子量的峰为m/z=1417.93的峰。因此,判断m/z=1417.93为上述分解物的峰,将该峰进一步按照同样的条件供于gc-ms/ms分析。将其结果示于图20。图20是将图19中的m/z=1417.93的峰的化合物供于gc-ms/ms分析时的质谱图。图20中,纵轴表示检测强度,横轴表示m/z值,括弧内的数值表示m/z值。根据图20进行了以下推测。如上所述,推定峰1的化合物为式(9)的化合物。将峰1的m/z值(1417)减去峰2的m/z值(1229)而得的差值为188,因此推定峰2的化合物是从上述分解物解离掉分子量为约188的式(7)的化合物而得的化合物。将峰1的m/z值(1417)减去峰3的m/z值(1194)而得的差值为223,因此推定峰3的化合物是从上述分解物解离掉分子量为约223的式(4)的化合物的化合物。将峰1的m/z值(1417)减去峰4的m/z值(1042)而得的差值为375,因此推定峰4的化合物是从上述分解物解离掉2个分子量为约188的式(7)的化合物而得的化合物。由此可以确认,从式(9)的化合物解离掉式(7)的化合物或式(4)的化合物而得的化合物为峰2~4。由此可以确认,上述分解物中至少包含式(7)的化合物及式(4)的化合物这两者,由此得到如下结果:进一步支持上述(2)中推定的上述分解物的推定结构式(9)。(4)纯化物的质量分析将上述纯化物按照leone等(serenaleoneetal,“structuralelucidationofthecore-lipidabackbonefromthelipopolysaccharideofacinetobacterradioresistenss13,anorganicsolventtolerantgram-negativebacterium”,carbohydrateresearch,april10,2006,vol.341,issue.5,p.582-590)记载的方法进行肼分解,得到肼分解物105.4mg。按照上述肼分解物浓度为2.1mg/ml的方式溶解于蒸馏水,用甲醇稀释200倍,将所得的溶液作为试样溶液,按照下述条件供于gc-ms分析。<gc-ms条件>设备:在amazonetd(brukerdaltonics公司制)上安装electrosprayionization(esi)interface测定模式:esi-it-ms、negativemode将其结果示于图21。图21是将上述试样溶液供于gc-ms分析时的质谱图。图21中,纵轴表示检测强度,横轴表示m/z值,括弧中的数值表示所检测的离子的价数。将图21所示的多个峰中最大的峰(m/z=538.15)的化合物按照同样的条件供于gc-ms/ms分析,得到图22所示的质谱图。进而,将图22所示的多个峰中m/z=474.27的峰的化合物及m/z=601.17的峰的化合物按照同样的条件分别进一步供于gc-ms/ms/ms分析,得到图23及图24所示的质谱图。进而,将图24所示的多个峰中的m/z=557.19的峰的化合物及m/z=645.12的峰的化合物按照同样的条件分别进一步供于gc-ms/ms/ms/ms分析,得到图25及图26所示的质谱图。图22~图26中,纵轴表示检测强度,横轴表示m/z值,括弧中的数值表示所检测的离子的价数。图22~图26中除了示出质谱图以外,还示出推定对应于各峰的化合物的示意图。上述示意图中,p表示式(10)的结构式,hex表示式(11)的结构式,kdo表示式(12)的结构式,hexu表示式(13)的结构式,hexn表示式(14)的结构式,f表示上述(2)中推定的4种化合物中的任一者。需要说明的是,图22~图26中,hex为葡萄糖(glc)。(5)小结总结上述(1)~(4)的结果,确定上述纯化物包含在式(a)及式(b)中x1及x2分别为己糖及磷酸基的化合物(式(a1)所示的化合物及式(b1)所示的化合物)。需要说明的是,推测如式(9)所示那样,在上述(2)中在β-1,6-二葡糖胺骨架的1位的碳上键合有羟基。另一方面,推定如图21的示意图所示那样,在上述(4)中在β-1,6-二葡糖胺骨架的1位的碳上键合有磷酸基。对此,基于以下理由确定后者的磷酸基是正确的结构。即,理由在于,在上述(2)中对上述纯化物实施规定的处理而得到了上述分解物,通常已知的是,在该过程中使用弱酸时,2分子葡糖胺中的右侧的葡糖胺上所键合的磷酸基脱落而置换为-oh基的频度高。(6)糖链部分的nmr解析将上述(4)中得到的肼分解物用4mol/l的koh进行处理,将游离的糖链部分利用凝胶过滤柱色谱(bio-rad公司制的bio-gelp4mediaextrafine<45μm(wet):#150-4128)进行分级·纯化。图28的图表中示出其凝胶过滤谱。将图28中的级分号14-18合并(干燥重量4.8mg),将其溶解于500μl的d2o(99.96%d),作为nmr测定用的试样(以下也称为“试样a”。)。nmr测定按照下述条件来实施。(测定条件)装置:drx500及advance600分光器(brukerbiospin公司制)探头:cryogenictxiprobe、txiprobe及bboprobe探头温度:25℃测定方法:1d1h、1d1h-selectivetocsy、1h-selectivenoesy、1h-selectiverosey、1d13c、1d31p、2d1h-1hdqf-cosy、hohana、noesy、roesy、1h-13chsqc-tocsy、1h-13chsqc-noesy、1h-13chmbc、1h-31phmbc首先鉴定来自各糖残基的端基异构体位的信号(h1),以端基异构体位的信号为基点,由dqf-cosy、hohana、1h-13chsqc-tocsy光谱鉴定残基内的信号(葡萄糖残基及葡糖胺残基的情况下为h2-h6,葡萄糖醛酸残基的情况下为h2-h5)。将其结果示于图29及图30。图29是对上述试样a进行1hnmr测定时的光谱,图30是对上述试样a进行2ddqf-cosy测定时的光谱。需要说明的是,图29及图30中,glc对应于图22~图26中的hex,kdo对应于图22~图26中的kdo,glca对应于图22~26中的hexu,glcn对应于图22~26中的hexn,此后也同样。在kdo的情况下,以3位的质子(h3ax、h3eq)为基点而进行kdo残基内的信号(h4-h8)的归属。关于糖残基间的结合,利用noesy光谱中观测到的糖残基间的noe信号(图31)及1h-13chmbc光谱中观测到的糖残基间的相关信号进行鉴定。图31是将上述试样a供于2dnoesy测定时的光谱。关于各糖残基的结合方式(α/β),利用1j(c1,h1)如下所述地进行确定。1j(c1,h1)glcn-1174hz(α)glcn-2164hz(β)glca171hz(α)glc-1173hz(α)glc-2171hz(α)关于磷酸基的存在及结合部位,通过1d31p及1h-13chmbc而明确。将其结果示于图32。图32是将试样a供于2d1h-31phmbc测定时的光谱。2个磷酸基中,一个键合在glcn-1的1位,另一个键合在glcn-2的4位。由以上的结果,确定上述试样a的结构如图33的示意图所示。由该结果确定,上述(4)中得到的肼分解物的肼分解前的化合物包含在式(a)及式(b)中x1及x2均为羟基的化合物(式(a2)所示的化合物及式(b2)所示的化合物)。这些化合物的使用了单糖类的符号的结构式如下所述。需要说明的是,图33中示出了采取椅型构象的糖链结构式。式(a2)所示的化合物式(b2)所示的化合物〔实施例3〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有抗炎症效果这一点进行了确认。(1)被检液的制备将使实施例1中得到的纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)按照达到2mg/ml的浓度的方式溶解于注射用水并在4℃下保存的溶液以37℃加热5分钟,按照37℃、1分钟的条件进行超声波处理。将上述超声波处理后的上述溶液10μl添加到下述组成的培养液990μl中并充分混合,由此制备上述纯化物的浓度为20000ng/ml的溶液,将其用上述培养液逐级稀释,由此得到2000ng/ml、200ng/ml、20ng/ml、2ng/ml及0.2ng/ml总计6种被检液。需要说明的是,上述6种被检液在向细胞中添加时被2倍稀释,因此上述纯化物的终浓度分别成为10000ng/ml、1000ng/ml、100ng/ml、10ng/ml、1ng/ml及0.1ng/ml。[培养液的组成]dmem培养基500ml胎牛血清55.5ml青霉素-链霉素-谷氨酰胺(100×)5.6ml(2)被检液的添加对导入有人tlr4基因的源自人胚胎的肾细胞(invivogen公司制)添加上述培养液,由此制备上述细胞的浓度为4×105细胞/ml的溶液。将上述溶液各100μl(即,4×104细胞/100μl/孔)接种在96孔平底板中。接种后,在37℃、5%co2下培养24小时,然后除去培养上清液,将新鲜的上述培养液向各孔中各添加100μl。接下来,将上述被检液向各孔中各添加100μl。在添加上述被检液后,在37℃、5%co2下培养24小时。(3)il-8的浓度的测定回收各孔的培养上清液,使用humanil-8elisamax(注册商标)standard(biolegend公司制)测定白介素-8(interleukin-8:il-8)的浓度。将测定结果示于图3的图表。图3中,纵轴表示il-8的浓度,横轴表示被检液的浓度。如图3所示,il-8的浓度呈被检液浓度依赖性地上升。这里,已知il-8的生产量通常为tlr4活化的指标,另外,如上所述,已报道了tlr4的活化促进具有抗炎症作用的i型干扰素的产生。即,il-8浓度的上升意味着抗炎症作用的活化。因此,由这些结果确认,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有抗炎症作用。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。〔实施例4〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有抗炎症效果这一点进行了确认。(1)被检液的制备与实施例3中的“(1)被检液的制备”同样进行而制备上述纯化物的浓度为20000ng/ml的溶液,将其用下述组成的培养液逐级稀释,由此得到100ng/ml及10000ng/ml的共两种被检液。需要说明的是,上述两种被检液向细胞中添加时被稀释100倍,因此上述纯化物的终浓度分别成为1ng/ml及100ng/ml。[培养液的组成]rpmi1640培养基500ml灭活胎牛血清55.5ml青霉素-链霉素-谷氨酰胺(100×)5.6ml(2)被检液的添加向小鼠巨噬细胞(raw264.7、atcc)中添加上述培养液,制备上述细胞的浓度为1.067×106细胞/ml的溶液。将上述溶液向6孔板中各接种3ml(即,3.2×106细胞/3ml/孔)。接种后,在37℃、5%co2下培养2小时后,将上述被检液向各孔中各添加30μl。添加上述被检液后,在37℃、5%co2下培养24小时。接下来,将上述各孔的培养上清液回收到15ml管中。然后向上述各孔中添加新鲜的上述培养液1ml,在使其遍布孔整体后回收到上述15ml管中,将该操作重复进行2次。然后,将回收到上述15ml管中的上述培养液以1000rpm、3分钟的条件进行离心分离后,除去上清液,再添加37℃的新鲜的上述培养液3ml而悬浮,再次以同样的条件进行离心分离。离心分离后除去上清液,向沉淀中添加37℃的新鲜的上述培养液2ml而悬浮。然后将上述悬浮液各2ml添加到预先添加有新鲜的上述培养液1ml的状态的各孔中。然后,添加新鲜的上述培养液30μl(以下将该工序称为“培养液添加工序”。)。添加后,在37℃、5%co2下培养30分钟后,将上述各孔的培养上清液回收到15ml管中,以1000rpm、3分钟的条件进行离心分离,将上清液用抽吸器除去。进而,向除去培养上清液后的上述6孔板的各孔中添加冷却的pbs(phosphatebufferedsaline)/磷酸酶抑制剂液3ml,通过吹打而剥离细胞,将细胞悬浮液添加到上述15ml管中。将上述15ml管以1000rpm、3分钟的条件进行离心分离,利用抽吸器除去上清液后,向上述15ml管中添加冷却的上述pbs/磷酸酶抑制剂液0.5ml而悬浮,由此制备细胞悬浮液。使用nuclearextract试剂盒(activemotif公司制)由上述细胞悬浮液提取核蛋白。(3)核内nf-κb量的测定使用transamnf-κbp65试剂盒(activemotif公司制)测定吸光度,由此确认上述核蛋白中的nf-κb量。将确认结果示于图4的图表。图4中,纵轴表示吸光度,横轴表示被检液的浓度。如图4所示,被检液的浓度越高则吸光度越为低值,因此可知在被检液的浓度越高时上述核蛋白中的nf-κb量越少。由该结果确认,上述纯化物抑制了作为炎症性细胞因子之一的nf-κb的产生,即,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有抗炎症效果。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。〔实施例5〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有抗炎症效果这一点进行了确认。本例中,除了以下两点以外与实施例4同样地进行实验,通过测定吸光度来确认上述核蛋白中的nf-κb量。即,本例中,作为被检液,使用上述纯化物的浓度为100ng/ml、10000ng/ml及1000000ng/ml的3种被检液。需要说明的是,上述3种被检液在向细胞中添加时被稀释100倍,因此上述纯化物的终浓度分别成为1ng/ml、100ng/ml及10000ng/ml。另外,本例中,在实施例3中的上述培养液添加工序中添加含有终浓度100ng/ml的由革兰氏阴性菌成团泛菌(pantoeaagglomerans)纯化而得的lps(脂多糖(lipopolysaccharide),成团泛菌(pantoeaagglomerans)、自然免疫应用技研(株)制、以下称为“lpsp”。)的上述培养液30μl,来代替上述培养液30μl。将确认结果示于图5的图表。图5中,纵轴表示吸光度,横轴表示被检液的浓度。如图5所示,被检液的浓度越高则吸光度越为低值,因此可知被检液的浓度越高则上述核蛋白中的nf-κb量越少。由该结果确认,上述纯化物抑制作为炎症性细胞因子之一的nf-κb的产生,即,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有抗炎症效果。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。〔实施例6〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有抗炎症效果这一点进行了确认。(1)被检液的制备与实施例3中的“(1)被检液的制备”同样进行而制备上述纯化物的浓度为20000ng/ml的溶液,将其用下述组成的培养液逐级稀释,由此得到2ng/ml、0.2ng/ml及0.02ng/ml总计3种被检液。需要说明的是,上述3种被检液在向细胞中添加时被2倍稀释,因此上述纯化物的终浓度分别成为1ng/ml、0.1ng/ml及0.01ng/ml。[培养液的组成](2)被检液的添加向小鼠巨噬细胞(raw264.7、atcc)中添加上述培养液,制备上述细胞的浓度为4×105细胞/ml的溶液。将上述溶液向96孔平底板中各接种100μl(即,4×104细胞/100μl/孔)。接种后,在37℃、5%co2下培养2小时直至上述细胞铺展、粘附在孔的底部为止。接下来,将上述被检液向各孔中各添加100μl。添加上述被检液后,在37℃、5%co2下培养24小时。培养后,将各孔的培养上清液除去,重新添加上述培养液150μl(以下将该工序称为“培养液添加工序”。),在37℃、5%co2下培养24小时。(3)tnf-α浓度的测定培养后,回收各孔的培养上清液50μl,转移到新的96孔平底板的各孔中,使用小鼠(mouse)tnf-α测定试剂盒(biolegend公司制)测定吸光度并换算为tnf-α浓度。将测定结果示于图6的图表。图6中,纵轴表示tnf-α浓度,横轴表示被检液的浓度。如图6所示,被检液的浓度越高则tnf-α浓度越为低值,因此可知被检液的浓度越高则tnf-α的产生越少。由该结果确认,上述纯化物抑制作为炎症性细胞因子之一的tnf-α的产生,即,确认上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有抗炎症效果。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。〔实施例7〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有抗炎症效果这一点进行确认。本例中,除了以下两点以外与实施例6同样地进行实验,回收各孔的培养上清液50μl并测定tnf-α浓度。即,本例中,作为被检液,使用上述纯化物的浓度为20μg/ml、2μg/ml、200ng/ml、20ng/ml及2ng/ml的总计5种被检液。需要说明的是,上述5种被检液在向细胞中添加时被2倍稀释,因此上述纯化物的终浓度分别成为10μg/ml、1μg/ml、100ng/ml、10ng/ml及1ng/ml。另外,本例中,在实施例5中的上述培养液添加工序中,添加含有终浓度100ng/ml的lpsp的上述培养液150μl来代替上述培养液150μl。将测定结果示于图7的图表。图7中,纵轴表示tnf-α浓度,横轴表示被检液的浓度。如图7所示,被检液的浓度越高则tnf-α浓度越为低值,因此被检液的浓度越高则tnf-α的产生越少。由该结果确认,上述纯化物抑制作为炎症性细胞因子之一的tnf-α的产生,即,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有抗炎症效果。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。〔实施例8〕〔实施例8-1〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有抗炎症效果这一点进行确认。(1)被检液的制备与实施例3中的“(1)被检液的制备”同样进行,制备上述纯化物的浓度为20000ng/ml的溶液,将其用下述组成的培养液逐级稀释,由此得到400ng/ml、4000ng/ml及40000ng/ml的总计3种被检液。需要说明的是,上述3种被检液在向细胞中添加时被4倍稀释,因此上述纯化物的终浓度分别成为100ng/ml、1000ng/ml及10000ng/ml。[培养液的组成]rpmi1640培养基500ml灭活胎牛血清55.5ml青霉素-链霉素-谷氨酰胺(100×)5.6ml(2)被检液的添加向源自人末梢血单核细胞的细胞(thp-1、dspharmabiomedicalco.,ltd.制)中添加上述培养液,制备上述细胞的浓度为4×105细胞/ml的溶液。将上述溶液在24孔板各接种500μl(即,2.0×105细胞/500μl/孔)。接种后,向各孔中添加上述培养液250μl、或含有40μmol/l的gw9662(和光纯药工业(株)制)的上述培养液250μl。需要说明的是,上述gw9662为作为核内受体之一的pparγ(过氧化物酶体增殖体激活受体γ,peroxisomeproliferator-activatedreceptorγ)的抑制剂。添加后,在37℃、5%co2下培养1小时。然后,将上述被检液向各孔中各添加250μl,在37℃、5%co2下培养22小时。培养后,将各孔的培养上清液回收到2ml管中。另外,向各孔中添加新鲜的上述培养液0.5ml,使其遍布孔整体,将其回收到上述2ml管中。向空的各孔中添加新鲜的上述培养液0.49ml。将回收到上述2ml管的上述培养液以1000rpm、5分钟的条件进行离心分离,除去上清液,进而添加新鲜的上述培养液1ml而悬浮,再次以同样的条件进行离心分离。离心分离后除去上清液,将各孔中的0.49ml的培养液添加到上述2ml管中进行悬浮,再次倒回各孔中。然后向各孔中添加培养液250μl、或含有40μmol/l的上述gw9662的培养液250μl,在37℃、5%co2下培养1小时。培养后,按照上述lpsp的终浓度达到100ng/ml的方式对各孔添加含有lpsp的培养液250μl,在37℃、5%co2下培养22小时。(3)il-8的浓度的测定培养后,将各孔的培养上清液回收到管中,离心分离,将上清液回收到1.5ml管中,使用humanil-6elisamaxdeluxe(biolegend公司)测定吸光度,换算成白介素-6(interleukin6:il-6)的浓度。将测定结果示于图8的图表。图8中,纵轴示出il-6的浓度,横轴示出被检液的浓度。如图8所示,在gw9662-的情况下、即pparγ未受抑制时,被检液的浓度越高则il-6浓度值越低,因此可知被检液的浓度越高则il-6的产生越减少。与此相对地,在gw9662+的情况下、即pparγ受到抑制时,也是被检液的浓度越高则il-6浓度值越低,因此可知被检液的浓度越高则il-6的产生越减少,但是其减少的程度不如pparγ未受抑制时显著。由该结果确认,上述纯化物抑制作为炎症性细胞因子之一的il-6的产生,即,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有抗炎症效果。另外暗示,pparγ参与了由上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)引起的il-6的产生抑制。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。〔实施例8-2〕通过理化学研究所开发的cage(capanalysisofgeneexpression)法,对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物促进pparγ基因的表达这一点进行确认。向上述thp-1细胞添加与实施例8-1(2)同样的培养液,制备上述细胞的浓度为5×105细胞/ml的溶液。通过上述cage法测定该溶液的pparγ基因的表达量,结果为3.97tpm(tagspermillion)。在上述测定之后,将该细胞溶液分成如下4组。即,向上述细胞溶液中按照终浓度为1μg/ml的方式添加上述纯化物的溶液的组(实施例8-2a)、向上述细胞溶液中按照终浓度为10μg/ml的方式添加上述纯化物的溶液的组(实施例8-2b)、未对上述细胞溶液添加任何物质的组(比较例8-2a)、及向上述细胞溶液中按照100ng/ml的量添加lpsp溶液的组(比较例8-2b)的总计4组。将上述4组培养3小时后,通过上述cage法测定pparγ基因的表达量。将其结果示于图27。图27是示出pparγ基因的表达量的图表。图27中,横轴表示培养时间,纵轴表示pparγ基因的表达量(tpm)。如图27所示,在实施例8-2a中,在3小时之内,pparγ基因的表达量从3.97tpm大幅增加到5.86tpm,在实施例8-2b中,在3小时之内,pparγ基因的表达量也从3.97tpm大幅增加到5.90tpm。与此相对地,在比较例8-2a中,在3小时之内,pparγ基因的表达量从3.97tpm稍稍减少到3.89tpm,在比较例8-2b中,在3小时之内,pparγ基因的表达量从3.97tpm增加到4.31tpm,但是增加的程度不显著。由该结果确认,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)促进pparγ基因的表达。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物也得到同等结果。〔实施例9〕对式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物具有针对肺癌的抗癌效果这一点进行了确认。(1)荷瘤小鼠的制作将源自lewis肺癌的细胞株(lewislungcarcinoma:3ll、由jcrb细胞银行获得)的悬浮液按照每只小鼠的癌细胞给药量为2.5×105细胞/200μl的方式皮下给药到5只6周龄的雄性c57bl/6j小鼠的腹侧部。给药2周后,从1只上述小鼠中摘出肿瘤,在培养皿上用pbs(-)洗涤。接下来,将洗涤后的上述肿瘤切碎至2mm见方的程度,将其作为肿瘤碎片,将上述肿瘤碎片转移到50ml管中,添加胶原酶液5ml,在37℃下温热10分钟。然后,将包含上述肿瘤碎片的胶原酶液通过吹打而制成更细小的肿瘤碎片,将包含上述肿瘤碎片的胶原酶液转移到另一50ml管中,在冰上冷却。冷却后,向包含上述肿瘤碎片的胶原酶液中进一步添加胶原酶液5ml,将同样的操作重复进行5次至观察不到肿瘤碎片为止。然后,将包含上述肿瘤碎片的胶原酶液用细胞滤网(网眼尺寸70μm、bd公司制)过滤,将过滤液按照1200rpm、7分钟的条件进行离心分离。离心分离后除去上清液,向沉淀中添加rpmi1260培养基(无血清)20ml,通过颠倒混合而悬浮后,按照1200rpm、7分钟的条件进行离心分离。离心分离后除去上清液,向沉淀中添加pbs(-)而洗涤2次后,添加pbs(-)10ml而悬浮,由此制备癌细胞悬浮液。将上述癌细胞悬浮液按照每只小鼠的癌细胞给药量为2.5×105细胞/200μl的方式皮下给药到12只上述小鼠的腹侧部。给药14天后,从10只上述小鼠摘出肿瘤,通过与上述同样的方法制备癌细胞悬浮液。将上述癌细胞悬浮液按照每只小鼠的癌细胞给药量为2.5×105细胞/50μl的方式给药到108只上述小鼠的腹部皮内。给药后,在各小鼠的肿瘤尺寸达到直径5mm程度的时刻(给药后第8天)将小鼠分成下述所示的6组(各组n=6)。[表5]给药样品cy的给药第1组(实施例9-1)实施例1中得到的纯化物(ip)-第2组(实施例9-2)实施例1中得到的纯化物(ip)+第3组(实施例9-3)实施例1中得到的纯化物(po)-第4组(实施例9-4)实施例1中得到的纯化物(po)+第5组(比较例9-1)生理盐水(ip)-第6组(参考例9-1)-+ip=腹腔内给药po=自由摄取cy=环磷酰胺(抗癌剂)(2)药剂的给药分组之后,对各组给药上述表5所示的物质。就上述纯化物的给药而言,在实施例9-1及实施例9-2的腹腔内给药(ip)中,在刚分组后按照小鼠的上述纯化物的摄取量为0.5mg/10ml/kg的方式进行1次。在实施例9-3及实施例9-4的自由摄取(po)的情况下,使用装有上述纯化物的浓度为1μg/ml的溶液的给水瓶,在刚分组后开始。需要说明的是,给水瓶每隔3天更换为新的给水瓶。比较例9-1的生理盐水的给药通过ip方式按照小鼠的生理盐水摄取量为10ml/kg的方式在刚分组后进行1次。实施例9-2、实施例9-4及参考例9-1中的cy(环磷酰胺、针对肺癌的抗癌剂、和光纯药工业(株)制)的给药通过ip方式按照小鼠的cy摄取量为100mg/10ml/kg的方式在刚分组后进行1次。(3)肿瘤体积的计测将分组当天作为第0天,在第3天、第6天及第9天用卡尺测定肿瘤的长径及短径,以其为基础算出肿瘤体积。肿瘤体积的算出按照shime等(shimeh,etal,“toll-likereceptor3signalingconvertstumor-supportingmyeloidcellstotumoricidaleffectors”,procnatlacadsciusa,february7,2012,vol.109,no.6,p.2066-2071)记载的方法利用计算式:长径(mm)×短径(mm)2×0.4=肿瘤体积(mm3)来进行。将计测结果示于图9的图表。图9中,纵轴表示肿瘤体积(mm3),横轴表示以分组当天为第0天时的、分组后的经过天数。如图9所示,给药上述纯化物的实施例9-1及实施例9-2与给药生理盐水的比较例9-1相比,肿瘤体积小。由此确认,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有针对肺癌的抗癌效果。另外,除了上述纯化物以外还给药cy的实施例9-2及实施例9-4与仅给药cy的参考例9-1相比,肿瘤体积小。由此确认,上述纯化物(式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物及式(b2)所示的化合物的混合物)具有增强cy的针对肺癌的抗癌效果的效果。需要说明的是,使用式(a1)所示的化合物、式(b1)所示的化合物、式(a2)所示的化合物或式(b2)所示的化合物代替上述纯化物也得到同等结果。以上参照实施方式及实施例说明了本发明,但本发明不限于上述实施方式及实施例。本发明的构成和详细内容可以在本发明的范围内进行本领域技术人员可理解的各种变更。本申请以2016年9月23日提出的日本申请特愿2016-186116及2016年12月8日提出的日本专利申请2016-238863为基础主张优先权,其公开的全部内容均引入本申请中。产业上的可利用性如上所述,本发明的化合物或其盐能够作为抗炎症剂及针对肺癌的抗癌剂等使用。本发明的化合物或其盐的安全性高,因此能够长期给药。sequencelisting<110>tfk株式会社(tfkco.,ltd.)<120>化合物或其盐、抗炎症剂、针对肺癌的抗癌剂、化合物或其盐的制造方法、炎症性疾病的治疗方法及肺癌的治疗方法<130>tf16007wo<150>jp2016-186116<151>2016-09-23<150>jp2016-238863<151>2016-12-08<160>9<170>patentinversion3.1<210>1<211>1427<212>dna<213>固氮红细菌(rhodobacterazotoformans)<400>1gagtttgatcctggctcagaatgaacgctggcggcaggcctaacacatgcaagtcgagcg60aagtcttcggacttagcggcggacgggtgagtaacgcgtgggaacatgcccaaaggtacg120gaatagccccgggaaactgggagtaataccgtatgtgcccttcgggggaaagatttatcg180cctttggattggcccgcgttggattaggtagttggtggggtaatggcctaccaagccgac240gatccatagctggtttgagaggatgatcagccacactgggactgagacacggcccagact300cctacgggaggcagcagtggggaatcttagacaatgggcgcaagcctgatctagccatgc360cgcgtgatcgatgaaggccttagggttgtaaagatctttcaggtgggaagataatgacgg420taccaccagaagaagccccggctaactccgtgccagcagccgcggtaatacggagggggc480tagcgttattcggaattactgggcgtaaagcgcacgtaggcggactggaaagtcaggggt540gaaatcccggggctcaaccccggaactgcctttgaaactcccagtcttgaggtcgagaga600ggtgagtggaattccgagtgtagaggtgaaattcgtagatattcggaggaacaccagtgg660cgaaggcggctcactggctcgatactgacgctgaggtgcgaaagcgtggggagcaaacag720gattagataccctggtagtccacgccgtaaacgatgaatgccagtcgtcgggcagcatgc780tgttcggtgacacacctaacggattaagcattccgcctggggagtacggccgcaaggtta840aaactcaaaggaattgacgggggcccgcacaagcggtggagcatgtggtttaattcgaag900caacgcgcagaaccttaccaacccttgacatggcgatcgcggttccagagatggttcctt960cagttcggctggatcgcacacaggtgctgcatggctgtcgtcagctcgtgtcgtgagatg1020ttcggttaagtccggcaacgagcgcaacccacgtcctcagttgccagcattcagttgggc1080actctggggaaactgccggtgataagccggaggaaggtgtggatgacgtcaagtcctcat1140ggcccttacgggttgggctacacacgtgctacaatggcagtgacaatgggttaatcccaa1200aaagctgtctcagttcggattggggtctgcaactcgaccccatgaagtcggaatcgctag1260taatcgcgtaacagcatgacgcggtgaatacgttcccgggccttgtacacaccgcccgtc1320acaccatgggaattggttctacccgaaggcggtgcgccaacctcgcaagaggaggcagcc1380gaccacggtaggatcagtgactggggtgaagtcgtaacaaggtagcc1427<210>2<211>19<212>dna<213>人工序列(artificial)<220><223>引物<400>2gagtttgatcctggctcag19<210>3<211>19<212>dna<213>人工序列(artificial)<220><223>引物<400>3ctcctacgggaggcagcag19<210>4<211>21<212>dna<213>人工序列(artificial)<220><223>引物<400>4ggattagataccctggtagtc21<210>5<211>16<212>dna<213>人工序列(artificial)<220><223>引物<400>5gcaacgagcgcaaccc16<210>6<211>18<212>dna<213>人工序列(artificial)<220><223>引物<400>6gtattaccgcggctgctg18<210>7<211>18<212>dna<213>人工序列(artificial)<220><223>引物<400>7taccagggtatctaatcc18<210>8<211>16<212>dna<213>人工序列(artificial)<220><223>引物<400>8ccattgtagcacgtgt16<210>9<211>17<212>dna<213>人工序列(artificial)<220><223>引物<400>9aaggaggtgatccagcc17当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1