替诺福韦艾拉酚胺半富马酸盐的制备方法与流程
本发明属于医药技术领域,具体涉及一种替诺福韦艾拉酚胺半富马酸盐的制备方法。
背景技术:
目前临床用于乙肝治疗的药物主要以核苷类药物为主,包括拉米夫定(lamivudine,lam)、阿德福韦酯(adeofvir,adv)、替比夫定(telbivudine,ldt)、替诺福韦酯(tenofovirdisoproxil,tdf)和恩替卡韦(enteeavir,etv)。
其中tdf与大多数核苷类似物不同,无需转化成三磷酸形式,只要二磷酸形式就能抑制hbv-dna聚合酶,这引起了人们的广泛关注。但是tdf存在一定的肾毒性和骨骼毒性,因此,吉利德(gilead)公司将tdf改造为替诺福韦艾拉酚胺半富马酸(tenofoviralafenamide,taf)。
在临床试验中,taf已被证明在相当于tdf十分之一剂量时,就具有非常高的抗病毒效果,同时可降低对肾功能和骨骼方面的副作用。目前,替诺福韦艾拉酚胺单方制剂和替诺福韦艾拉酚胺的复方制剂已分别于在美国,中国及欧洲上市销售,获得了治疗艾滋病和乙型肝炎的上市许可。其结构式如下:
专利文献cn1443189a和cn1706855a实施例报道了替诺福韦艾拉酚胺及其富马酸盐的制备方法,专利cn105085571a报道了替诺福韦艾拉酚胺一系列的包括硫酸,盐酸以及酒石酸和一系列氨基酸形成的不同盐的形式,专利cn105237571报道了替诺福韦艾拉酚胺半琥珀酸盐和半酒石酸盐形式,专利cn103732594a报道了其半富马酸盐的形式。
上述文献报道的taf的制备路线主要为如下反应式:
现有技术中一条路线是使用的dcc/tea,其中dcc为n,n′-二环己基碳二亚胺,dcc会生成大量副产品,难以纯化,即使纯化也会产生大量固废,工业上不易使用。另一条路线ch3cn/tea反应时间长(不少于48h),两者都存在产品纯度低,物料成本高等缺陷。针对这些问题,我们对taf的制备方法进行了改进。
技术实现要素:
本发明的目的是提供一种替诺福韦艾拉酚胺半富马酸盐的制备方法,优化后的工艺具有反应时间短、产品纯度高、收率高的优点,适合于大规模工业生产。
本发明所提供的替诺福韦艾拉酚胺半富马酸盐(taf)的制备方法包括以下步骤:
在上述taf中间体iii的制备方法包括以下步骤:在芳烃类、氮杂环类和腈类中的一种或氮杂环类与芳烃类或腈类的混合溶剂中加入替诺福韦(ii)和亚磷酸三苯酯,加入/不加入三乙胺和dmap,加热至一定温度至反应完成。
在所述的taf中间体iii的制备方法中,所述的芳烃类溶剂可在苯、甲苯、二甲苯中选择,优选甲苯;
在所述的taf中间体iii的制备方法中,所述的氮杂环类溶剂可在吡啶、四氢吡咯、哌啶中选择,优选吡啶;
在所述的taf中间体iii的制备方法中,所述的腈类溶剂可在乙腈、丙腈、正丁腈中选择,优选乙腈;
在所述的taf中间体iii的制备方法中,所述的氮杂环类和芳烃类的混合溶剂的体积比值优选1:1~1:10;进一步优选1:1~1:3;
在所述的taf中间体iii的制备方法中,所述的氮杂环类和腈类的混合溶剂的体积比值优选1:1~1:10;进一步优选1:3~1:5;
在所述的taf中间体iii的制备方法中,所述的ii、亚磷酸三苯酯、dmap和三乙胺的摩尔比为1:1:0~2:0~5,碱性溶剂优选1:1:0:0,非碱性溶剂优选1:1:0.8~1.5:3~4;
在所述的taf中间体iii的制备方法中,溶剂为芳烃类、氮杂环类或氮杂环类和芳烃类时,所述的反应温度为70~130℃,优选100~110℃;
在所述的taf中间体iii的制备方法中,溶剂为腈类或氮杂环类和腈类时,所述的反应温度为60~90℃,优选70~80℃。
在所述的taf中间体iii的制备方法中,反应完成后将反应液冷至室温,加入一定量的纯化水,用盐酸调ph值至2~3,过滤后滤饼用水或50%乙醇或乙醇打浆。
在所述的taf中间体iii的制备方法中,干燥方式采用90℃鼓风干燥。
在上述taf中间体iv的制备方法包括以下步骤:iii与酰化剂在芳烃类或腈类溶剂中反应,生成的酰卤和在dcm中解盐后得到的l-丙氨酸异丙酯在缚酸剂作用下酰胺化制得taf中间体iv。
在所述的taf中间体iv的制备方法中,所述的芳烃类溶剂可在苯、甲苯、二甲苯中选择,优选甲苯;
在所述的taf中间体iv的制备方法中,所述的腈类溶剂可在乙腈、丙腈、正丁腈中选择,优选乙腈;
在所述的taf中间体iv的制备方法中,所述的酰化剂可在氯化亚砜、草酰氯、乙酰氯、三氯化磷中选择,优选氯化亚砜和草酰氯;
在所述的taf中间体iv的制备方法中,所述的缚酸剂可在三乙胺、dipea(n,n-二异丙基乙基胺)、碳酸氢钠、碳酸氢钾中选择,优选三乙胺、dipea;
在所述的taf中间体iv的制备方法中,所述的l-丙氨酸异丙酯盐酸盐、缚酸剂和iii的摩尔比为1~5:2~6:1,优选3~5:3~6:1。
在所述的taf中间体iv的制备方法中,所述的酰化反应温度优选60~80℃;
在所述的taf中间体iv的制备方法中,所述的酰胺化反应温度优选-30~0℃,进一步优选-10~0℃;
在所述的taf中间体iv的制备方法中,反应液后处理需用10%磷酸二氢钠溶液、15%khco3溶液和饱和食盐水洗涤。
在上述替诺福韦艾拉酚胺游离碱v的制备方法包括以下步骤:中间体iv在一定体积的腈类溶剂中,加入晶种,之后加入拆分试剂苯酚和少量有机碱催化剂,缓慢诱导析出替诺福韦艾拉酚胺;所述的taf中间体iv的拆分方法为诱导结晶法,是利用非对映异构体溶解性的差异,诱导与晶种相同的异构体不断析出的过程;
在所述的替诺福韦艾拉酚胺游离碱v的制备方法中,所述的腈类溶剂可在乙腈、丙腈、正丁腈中选择,优选乙腈;
在所述的替诺福韦艾拉酚胺游离碱v的制备方法中,所述的有机碱可在dbu、三乙胺、dipea、三乙烯二胺、dmap、吡啶等中选择,优选dbu;
在所述的替诺福韦艾拉酚胺游离碱v的制备方法中,拆分溶液浓度为30~35%;
在所述的替诺福韦艾拉酚胺游离碱v的制备方法需要在密闭体系中完成。
在上述成品i的制备方法包括以下步骤:在酮类或腈类溶剂中,加入替诺福韦艾拉酚胺游离碱,升至一定温度使溶解,滤去不溶物后加入富马酸,溶清后加入晶种,降温析晶得到taf粗品。
在所述成品i的制备方法中,所述的酮类溶剂可在丙酮、甲乙酮、甲异丙酮、环己酮和甲己酮中选择,优选丙酮;
在所述成品i的制备方法中,所述的腈类溶剂可在乙腈、丙腈、正丁腈中选择,优选乙腈;
在所述成品i的制备方法中,所述的溶解温度为50~100℃,优选50~75℃;
在所述成品i的制备方法中,所述的析晶温度为0~30℃,优选15~25℃;
在所述成品i的制备方法中,所述的游离碱和富马酸的摩尔比为1:0.5。
与现有方法相比,本发明具有以下技术优点:
1)中间体iii的制备,以往专利中以乙腈为溶剂,反应时间不低于48小时,我们通过采用芳烃类、氮杂环类和腈类中的一种或氮杂环类与芳烃类或腈类的混合溶剂,通过调整物料配比缩短了反应时间,仅需15~24h,生成不易去除的双苯酚杂质(v)含量低,产品质量高,纯度在98%以上;
2)中间体iv的制备过程中,文献报道采用乙腈、甲苯或丙酮做溶剂,我们采用高沸点的芳烃类或腈类,反应完成后可以一次性的减压除尽对后续反应有影响的氯化亚砜,不需反复通过溶剂复溶蒸去残余的氯化亚砜。通过调整酰胺化反应的物料比例,显著降低了难以去除且影响拆分效率的二聚杂质(vi)的含量,中间体iv异构体纯度高,易于拆分,拆分收率高且游离碱中异构体含量低于0.5%;
3)文献报道游离碱v成盐制备taf时采用乙腈为溶剂,我们发现,丙酮等酮类溶剂和乙腈都可以实现成盐收率高,降解杂质少,同时对异构体杂质具有优良的去除效果,成品纯度高达99.9%。
附图说明
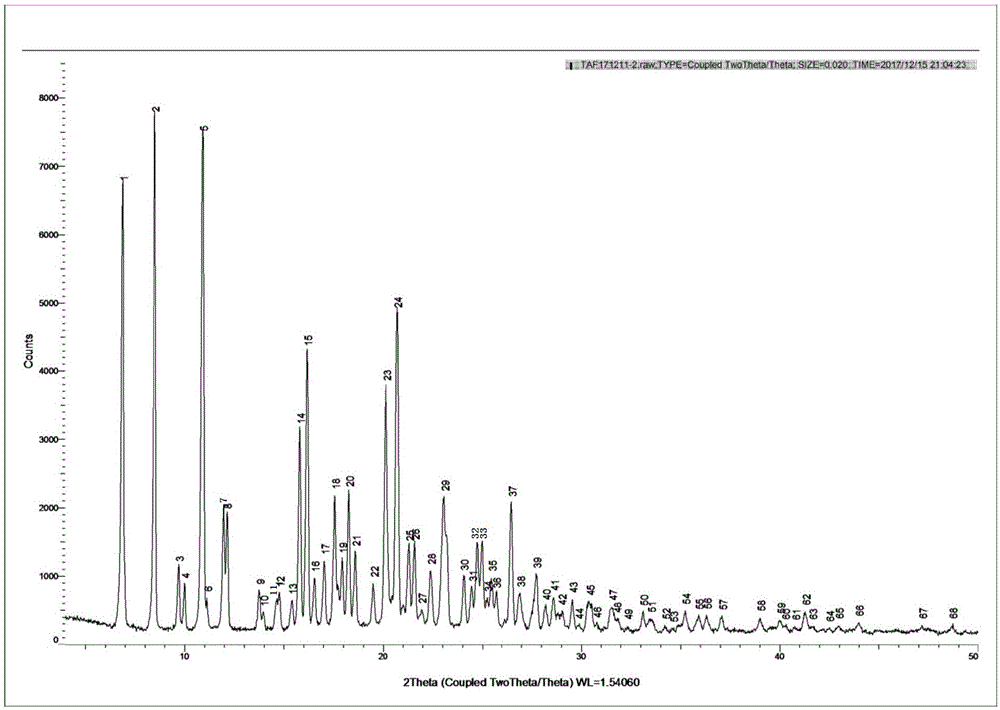
附图1为实施例1的替诺福韦艾拉酚胺半富马酸的粉末x衍射谱。
具体实施方式
下面通过具体实施例进一步阐述本发明,这些实施例仅用于说明本发明,而不用于限制本发明的范围。
实施例1:
一种替诺福韦艾拉酚胺半富马酸盐合成的工艺方法,包括以下步骤:
a.taf中间体iii的制备
将1.0eq的ii置入50l洁净干燥的反应釜中,加入4~5倍体积甲苯,开启缓慢搅拌。依次加入0.7~0.8eqdmap,1.0eq亚磷酸三苯酯,2.0eq三乙胺,升温至100-110℃搅拌反应20~24h,hplc监控ii和酸酐之和小于0.5%反应结束;
冷却到室温20~25℃;加入3倍体积纯化水,充分搅拌30分钟后分层,收集下层水相(上层甲苯回收处理);
向所得水相中,加入0.5倍体积无水乙醇,慢慢滴加浓盐酸至ph=3.8~4.0;加入少许晶种,搅拌0.5h,逐渐有固体析出;继续缓慢滴加浓盐酸ph=2.0~2.1,搅拌3~4h养晶,离心;
滤饼用1倍体积50%乙醇溶液室温打浆1h,离心;离心后固体于90℃鼓风干燥12h,得白色粉末中间体iii,收率91%,纯度≥98%,双苯酚杂质<0.1%。
b.taf中间体iv的制备
n2保护下,将1.0eq的iii置于50l洁净干燥的反应釜中,加入5倍体积乙腈,2.2eq的socl2,搅拌升温至70~75℃,保温搅拌反应2~3h,反应完全,减压浓缩至干,冷却,加入10倍体积干燥的二氯甲烷,搅拌均匀,冷却备用;
将3eq的l-丙氨酸异丙酯盐酸盐和1.5倍体积的二氯甲烷搅拌溶解,降温备用;
控制温度≤-10℃将l-丙氨酸异丙酯盐酸盐的二氯甲烷溶液加到上述浆液酰氯溶液中;滴加3~5eq的tea,控制反应液ph=4~7,维持温度-10℃以下反应2h,升至室温;
反应液用10%磷酸二氢钠溶液15倍量洗涤两次,用15%khco315倍量洗涤两次,再用饱和食盐水洗涤,分液,无水硫酸钠干燥,过滤,50℃减压浓缩至干得黄色油状物。
c.taf游离碱v的制备
中间体iv中加入4倍量乙腈,搅拌溶解完全;
浓缩至约30~35%溶液浓度,在室温搅拌下加入晶种0.01eq,搅拌结晶1小时;再加入苯酚0.05eq,dbu0.05eq,维持室温20~25℃密闭搅拌结晶约24h;
冰水浴中冷却到0℃,搅拌10h,离心,固体用冰乙腈1eq打浆,50℃鼓风干燥得替诺福韦艾拉酚胺游离碱v,收率62%,纯度98%以上,二聚杂质<0.2%,非对应异构体0.5%以下。
d.成品i的制备
于50l反应釜中,将1.0eq的游离碱、0.01倍药用炭加入到10倍体积乙腈中,加热至微回流状态1h,压滤至结晶釜中,加入0.5eq富马酸,继续加热至微回流状态至溶解完全;
降温至65℃,加入晶种0.01eq,搅拌0.5h;然后降至室温并搅拌16h,离心得到白色粉末状固体;
55℃真空干燥6h,得到白色结晶性粉末i;收率96%;hplc有关物质纯度99.96%,异构体纯度100.0%;
i的x-射线粉末衍射数据,特征在于2θ角度位置具有特征峰:6.86、8.47、9.69、9.99、10.91、11.11、11.94、12.11、13.75、13.94、14.67、14.78、15.40、15.80、16.17、16.54、17.03、17.56、17.92、18.25、18.58、19.49、20.13、20.70、21.29、21.57、21.92、22.39、23.07、24.03、24.47、24.74、24.97、25.22、25.44、25.66、26.47、26.90、27.73、28.20、28.60、28.99、29.55、30.40、30.74、31.52、31.76、33.11、33.50。
实施例2:
一种替诺福韦艾拉酚胺半富马酸盐合成的工艺方法,包括以下步骤:
a.中间体iii的制备
向三口烧瓶中称取1.0eq的ii,加入10倍量吡啶,室温搅拌,滴加亚磷酸三苯酯1.0eq;
将反应液在100℃加热搅拌15h,降至室温搅拌30min,再在冰浴下搅拌1h,过滤,滤饼中加入10倍量水,用浓盐酸调节ph≈2,冰浴搅拌30min,过滤;
滤饼用5倍量乙醇打浆,抽滤,干燥后得到白色固体iii,收率85%,纯度>97%。
b.中间体iii的制备
向三口烧瓶中称取1.0eq的ii,加入5倍量吡啶和5倍量甲苯,室温搅拌,滴加亚磷酸三苯酯1.0eq;
将反应液在100℃加热搅拌20h,降至室温搅拌30min,再在冰浴下搅拌1h,过滤,滤饼中加入10倍量水,用浓盐酸调节ph=2~3,冰浴搅拌30min,过滤;
滤饼用5倍量乙醇打浆,抽滤,干燥后得到白色固体iii,收率87%,纯度>97%。
c.中间体iv和v的制备
向反应瓶中加入1.0eqiii和5倍量甲苯,2.2eqsocl2,搅拌均匀;升温至70~80℃,保温搅拌3h,减压浓缩至干,加8倍量二氯甲烷,搅拌10min后降温至-10~0℃;
向另一反应瓶中加入5.0eql-丙氨酸异丙酯盐酸盐及5倍量二氯甲烷,搅拌至溶清,加入三乙胺后降温至-10~0℃,加入至酰氯溶液中,加料完毕后保温搅拌0.5~1h,反应完毕,升温至20~30℃;
反应液用10%磷酸二氢钠溶液10倍量洗涤两次,用15%khco310倍量洗涤两次,用饱和食盐水洗涤,分液,有机相用无水硫酸钠干燥,过滤,50℃减压浓缩至干得黄色油状物;
在中间体iv中加入4倍量乙腈,搅拌溶解完全;
浓缩至约30~35%溶液浓度,在室温搅拌下加入晶种0.01eq,搅拌结晶1小时;再加入苯酚0.05eq,dbu0.05eq,维持室温20~25℃密闭搅拌结晶约24h;
冰水浴中冷却到0℃,搅拌10h,离心,固体用冰乙腈1eq打浆,50℃鼓风干燥得替诺福韦艾拉酚胺游离碱v,收率65%,纯度98.5%,非对应异构体0.5%以下。
d.成品i的制备
向反应瓶中加入1.0eq游离碱,加入15倍体积丙酮,升温至50~60℃,搅拌至溶清;
热滤,滤液升温至50~60℃;加入0.5eq富马酸,搅拌至溶清;
降温至40~50℃,加入晶种,保温搅拌0.5h;降温至20~25℃,搅拌8h;抽滤,用丙酮淋洗滤饼;
滤饼于50~55℃真空干燥得白色粉末i,收率92%,hplc有关物质纯度99.95%,异构体纯度100.0%。
- 还没有人留言评论。精彩留言会获得点赞!