一种含炔基的亚胺衍生物及其制备方法和应用与流程
本发明属于有机合成技术领域,尤其涉及一种含炔基的亚胺衍生物及其制备方法和应用。
背景技术:
在合成化学中,为了追求更加绿色高效、步骤经济性的合成方法,对烷基csp3-h键的直接官能团化反应受到越来越到的关注。然而,为了实现惰性csp3-h键的位置选择性和反应活性的兼得,该类转化往往使用双齿配体来实现反应的高效性。但是这些向分子中预先引入的双齿配体往往难以脱除、且价格昂贵,这也限制了该类反应的实用性。
亚胺作为用胺来保护羰基化合物的一类含氮化合物,其特点在于易合成、易转化;更为重要的是,相比于原先羰基化合物底物,亚胺具有更好的与金属催化剂配位效果,这对于实现惰性烷基csp3-h键的选择性活化起着至关重要的作用。而相比于以往经典的双齿配体,亚胺可以作为“无痕导向基”,在促进底物进行目标转化后,可以很方便地通过水解或还原得到羰基或胺类化合物,从而极大地增强了反应在具有生物活性分子改造中的应用。
若用亚胺作为酮羰基的衍生物来实现烷基csp3-h键的炔基化反应。该类转化具有以下特点:1)亚胺作为易引入的官能团,相比于活性较低的烷基羰基化合物,它可以很好地促进csp3-h键的选择性官能团化反应;2)亚胺可以很好的转化,如通过水解或还原反应得到酮或胺,因此,如果可以实现亚胺直接的选择性氮原子的γ位csp3-h键炔基化反应,结合亚胺和炔烃的易转化的特点,该类含亚胺的炔烃化合物可以很方便地通过催化的方式转化为杂环化合物。
但是,亚胺氮原子γ位含炔基片段的化合物是很难合成的,即便通过含亚胺片段的烷基卤与炔基化试剂的偶联反应,也非常具有挑战性。因此,合成亚胺氮原子γ位含炔基片段的化合物有待于进一步开发,目前亚胺氮原子γ位含炔基片段的化合物种类有限,还需进行拓宽。
技术实现要素:
有鉴于此,本发明提供了一种含炔基的亚胺衍生物及其制备方法和应用,该含炔基的亚胺衍生物在亚胺氮原子的γ位含炔基片段,提供一种新的含炔基的亚胺衍生物,拓宽含炔基的亚胺衍生物的种类。
本发明的具体技术方案如下:
一种含炔基的亚胺衍生物,所述含炔基的亚胺衍生物的结构式如式(ⅰ)所示:
其中,r1选自氢、c1~c20的烃基、c5~c30的芳基或c5~c30的取代芳基,r2、r3和r4独立的选自氢、c1~c20的烃基、c5~c30的芳基、c5~c30的取代芳基或官能团中的一种或r2与r4形成环烷基或取代环烷基,r5为取代硅基。
本发明中,含炔基的亚胺衍生物在亚胺氮原子的γ位含炔基片段,该含炔基的亚胺衍生物可通过亚胺的转化如水解或还原,进一步得到含炔基的烷基酮或含炔基的烷基胺化合物,并且通过金属催化反应或自由基反应,氮原子可以作为分子内的亲核试剂对炔基片段进攻,从而获得含氮杂环化合物,产品的实用性高。
优选的,r1选自氢、烷基、苯基或取代苯基,r2、r3和r4独立的选自氢、烷基、环烷基、卤素、三氟甲基或酯基中的一种或r2与r4形成环烷基或取代环烷基,r5为三异丙基硅基、二甲基叔丁基硅基或含环己基的氧硅醚。
进一步的,r1选自的烷基优选为甲基、乙基、异丙基或叔丁基,r1选自的取代苯基优选为苄基;r2、r3和r4独立的选自的烷基优选为甲基、乙基、异丙基或叔丁基,r2与r4形成的环烷基优选为环丙烷、环丁烷、环戊烷或环己烷。
优选的,式(ⅰ)所示含炔基的亚胺衍生物选自
本发明还提供了一种含炔基的亚胺衍生物的制备方法,包括以下步骤:
将式(ⅱ)所示化合物和式(ⅲ)所示化合物在催化剂下进行反应,得到式(ⅰ)所示的含炔基的亚胺衍生物;
其中,
r1选自氢、c1~c20的烃基、c5~c30的芳基或c5~c30的取代芳基,r2、r3和r4独立的选自氢、c1~c20的烃基、c5~c30的芳基、c5~c30的取代芳基或官能团中的一种或r2与r4形成环烷基或取代环烷基,r5为取代硅基,x为氢、溴、氯、碘或含碘杂环基团,含碘杂环基团优选为
所述催化剂为金属催化剂,所述金属催化剂的金属选自钯、钌、铑和铱中的一种或多种。
通常,亚胺极其容易发生水解或其它转化,使其通常只作为反应中原位生成的中间体。而在本发明含炔基的亚胺衍生物的制备方法中,式(ⅱ)所示化合物具有兼顾活性和稳定性的亚胺基团,可作为羰基的保护基来实现原本烷基酮难以实现的γ位csp3-h键直接的炔基化反应,
本发明制备方法中,直接使用惰性烷基csp3-h键的官能团化反应策略来实现制备氮原子γ位含有炔基的亚胺衍生物,具有良好的原子经济性和步骤经济性。更为重要的是,制备得到的产物有望通过催化反应得到含炔基的酮或胺以及含氮杂环,该制备方法的实用性高,具有高效高选择性。
本发明中,所述金属催化剂更优选为金属为二价钌或三价铑的金属催化剂。
优选的,所述金属催化剂选自醋酸钯、氯化钯、三氯化钌、二氯(对甲基异丙基苯基)钌(ii)二聚体、五甲基环戊二烯氯化铑二聚体、三乙腈-五甲基环戊二烯基氯化铑二聚体和五甲基环戊二烯基氯化铱(iii)二聚体中的一种或多种,更优选为三乙腈-五甲基环戊二烯基-二(六氟锑酸)铱、五甲基环戊二烯基氯化铑二聚体或五甲基环戊二烯基氯化铱二聚体。
优选的,所述将式(ⅱ)所示化合物和式(ⅲ)所示化合物在催化剂下进行反应具体为:
将式(ⅱ)所示化合物和式(ⅲ)所示化合物溶于惰性溶剂中,在氧化剂和金属催化剂的作用下,在碱性条件下进行反应。
优选的,所述氧化剂选自醋酸银、碳酸银、三氟磺酸银、硝酸银、醋酸铜、卤化亚铜、卤化铜、三卤化铁和硝酸铁中的一种或多种,更优选为醋酸铜;
调节所述碱性条件的碱选自醋酸钠、醋酸铯、醋酸钾、碳酸钠和磷酸钾中的一种或多种,更优选为醋酸钠。
优选的,所述反应的温度为60℃~150℃,优选为80℃~120℃,更优选为100℃;
所述反应的时间为8h~48h,优选为8h~36h。
本发明中,惰性溶剂选自甲苯、四氢呋喃、1,4-二氧六环、n,n’-二甲基甲酰胺、n,n’-二甲基乙酰胺、n-甲基吡咯烷酮、二甲亚砜、乙腈、1,2-二氯乙烷、乙醇或丙酮,更优选为1,2-二氯乙烷。
优选的,式(ⅱ)所示化合物和式(ⅲ)所示化合物的摩尔比为1∶1~1∶4;
所述金属催化剂的用量为所述式(ⅱ)所示化合物用量的1mol%~5mol%,更优选为2mol%。
本发明中,碱的用量为式(ⅱ)所示化合物用量的5~50mol%,更优选为15mol%;
氧化剂的用量为式(ⅱ)所示化合物用量的10~300mol%,更优选为30mol%。
式(ⅱ)所示化合物在惰性溶剂的浓度为0.1mol/l~3.0mol/l,优选为0.2mol/l;式(ⅲ)所示化合物在惰性溶剂的浓度为0.5mol/l~3.0mol/l,优选为1.0mol/l。
本发明中,制备方法优选包括以下步骤:在空气氛围下,在反应器中依次加入式(ⅱ)所示化合物(0.1mmol)、五甲基环戊二烯基氯化铱(iii)二聚体(2.0mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、醋酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含式(iii)所示化合物(0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于100℃下反应12h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为50:1至5:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物。
本发明还提供了上述技术方案所述含炔基的亚胺衍生物和/或上述技术方案所述制备方法制得的含炔基的亚胺衍生物在药物制备中的应用。
综上所述,本发明提供了一种含炔基的亚胺衍生物,其特征在于,所述含炔基的亚胺衍生物的结构式如式(ⅰ)所示,其中,r1选自氢、c1~c20的烃基、c5~c30的芳基或c5~c30的取代芳基,r2、r3和r4独立的选自c1~c20的烃基、c5~c30的芳基、c5~c30的取代芳基或官能团中的一种或r2与r4形成环烷基或取代环烷基,r5为取代硅基。本发明中,含炔基的亚胺衍生物在亚胺氮原子的γ位含炔基片段,该含炔基的亚胺衍生物可通过亚胺的转化如水解或还原,进一步得到含炔基的烷基酮或含炔基的烷基胺化合物,并且通过金属催化反应或自由基反应,氮原子可以作为分子内的亲核试剂对炔基片段进攻,从而获得含氮杂环化合物,产品的实用性高。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
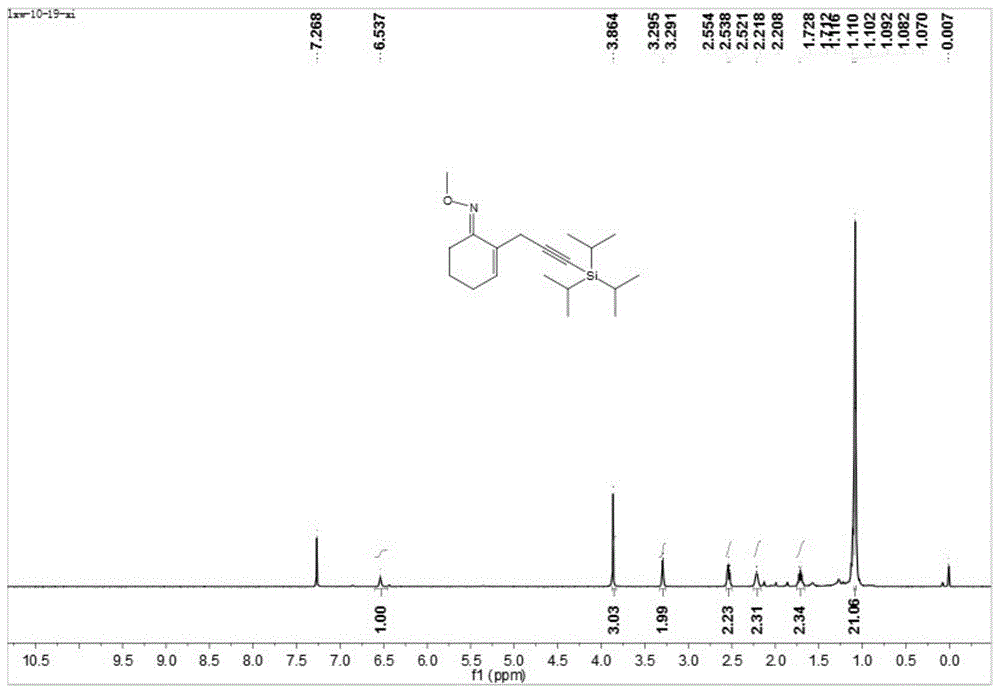
图1为本发明实施例1提供的(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1a)的核磁共振1h谱图;
图2为本发明实施例1提供的(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1a)的核磁共振13c谱图;
图3为本发明实施例2提供的(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-乙基肟醚(1b)的核磁共振1h谱图;
图4为本发明实施例2提供的(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-乙基肟醚(1b)的核磁共振13c谱图;
图5为本发明实施例3提供的(z)-5-甲基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1c)的核磁共振1h谱图;
图6为本发明实施例3提供的(z)-5-甲基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1c)的核磁共振13c谱图;
图7为本发明实施例4提供的(r,e)-5-异丙基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1d)的核磁共振1h谱图;
图8为本发明实施例4提供的(r,e)-5-异丙基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1d)的核磁共振13c谱图;
图9为本发明实施例5提供的(e)-1-(三异丙基硅基)-1-壬炔-5-酮-o-苄基肟醚(1e)的核磁共振1h谱图;
图10为本发明实施例5提供的(e)-1-(三异丙基硅基)-1-壬炔-5-酮-o-苄基肟醚(1e)的核磁共振13c谱图;
图11为本发明实施例6提供的(r,e)-5-(丙基-1-烯-2-基)-2-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-2-烯-1-酮o-甲基肟醚(1f)的核磁共振1h谱图;
图12为本发明实施例6提供的(r,e)-5-(丙基-1-烯-2-基)-2-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-2-烯-1-酮o-甲基肟醚(1f)的核磁共振13c谱图;
图13为本发明实施例7提供的(e)-2-(甲氧亚胺基)-1-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-1-甲酸乙酯(1g)的核磁共振1h谱图;
图14为本发明实施例7提供的(e)-2-(甲氧亚胺基)-1-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-1-甲酸乙酯(1g)的核磁共振13c谱图。
具体实施方式
本发明提供了一种含炔基的亚胺衍生物及其制备方法和应用,用于提供一种新的含炔基的亚胺衍生物,拓宽含炔基的亚胺衍生物的种类。
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本实施例进行(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1a)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2a(13.9mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(2.0mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含炔烃化合物3a(54.0mg,0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应24h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1a),24.9mg,纯度为95%,产率为78%。
对(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1a)进行核磁共振检测,请参阅图1至图2,结果为:1hnmr(400mhz,cdcl3):δ4.27-4.23(m,1h),2.64(dd,j=4.0hz,16.8hz,1h),2.46-2.40(m,3h),2.18(t,j=6.0hz,2h),1.66-1.65(m,2h),1.59-1.58(m,4h),1.34(d,j=6.4hz,3h),1.07-1.05(m,21h);13cnmr(100mhz,cdcl3):δ160.3,105.4,81.8,76.4,32.3,27.1,26.8,25.9,25.8,25.4,18.6,18.5,11.3。
本实施例表明可以通过亚胺化合物经氮-氧键的协助实现酮的β位csp3-h键的炔基化反应,反应具有极好的位置选择性,且csp2-h键在反应体系中兼容。
实施例2
本实施例进行(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-乙基肟醚(1b)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2b(15.3mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(2.0mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含炔烃化合物3a(54.0mg,0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应24h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-乙基肟醚(1b),21.0mg,纯度为95%,产率为63%。
对(e)-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-乙基肟醚(1b)进行核磁共振检测,请参阅图3至图4,结果为:1hnmr(400mhz,cdcl3):δ4.27-4.23(m,1h),2.64(dd,j=4.0hz,16.8hz,1h),2.46-2.40(m,3h),2.18(t,j=6.0hz,2h),1.66-1.65(m,2h),1.59-1.58(m,4h),1.34(d,j=6.4hz,3h),1.07-1.05(m,21h);13cnmr(100mhz,cdcl3):δ160.3,105.4,81.8,76.4,32.3,27.1,26.8,25.9,25.8,25.4,18.6,18.5,11.3。
本实施例表明可以通过亚胺化合物经氮-氧键的协助实现酮的β位csp3-h键的炔基化反应,且肟醚的取代基可以是各种烷基醚。
实施例3
本实施例进行(z)-5-甲基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1c)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2c(15.3mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(2.0mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含炔烃化合物3b(78mg,0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应24h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(z)-5-甲基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1c),25.3mg,纯度为95%,产率为76%。
对(z)-5-甲基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1c)进行核磁共振检测,请参阅图5至图6,结果为:1hnmr(400mhz,cdcl3):δ=2.57(s,2h),2.49-2.40(m,2h),2.18(t,j=6.0hz,2h),1.70-1.60(m,2h),1.58-1.56(m,4h),1.37(s,6h),1.08-1.07(m,21h);13cnmr(100mhz,cdcl3):δ=158.2,105.4,80.4,77.5,31.4,30.7,26.1,25.0,24.8,24.2,17.6,10.3。
本实施例表明可以通过亚胺化合物经氮-氧键的协助实现酮的β位csp3-h键的炔基化反应,且反应对活性烯烃csp2-h键不变。
实施例4
本实施例进行(r,e)-5-异丙基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1d)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2d(18.1mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(2.0mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含炔烃化合物3b(78mg,0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应24h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(r,e)-5-异丙基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1d),23.5mg,纯度为95%,产率为65%。
对(r,e)-5-异丙基-2-(3-(三异丙基硅基)-2-丙炔-1-基)环己基-2-烯-1-酮o-甲基肟醚(1d)进行核磁共振检测,请参阅图7至图8,结果为:1hnmr(400mhz,cdcl3):δ=4.08-4.02(m,1h),2.61(dd,j=4.4hz,16.8hz,1h),2.51-2.43(m,3h),2.18(m,2h),1.73-1.64(m,2h),1.59-1.58(m,6h),1.11-1.04(m,21h),0.95(t,j=7.2hz,3h);13cnmr(100mhz,cdcl3):δ=160.4,105.5,81.7,81.3,32.3,27.1,25.9,25.8,25.43,25.41,24.6,18.6,18.5,17.7,11.3,9.7。
本实施例反应可以通过亚胺化合物经氮-氧键的协助进行天然产物香芹酮衍生物的后期衍生化,且烯烃csp2-h键保持不变。
实施例5
本实施例进行(e)-1-(三异丙基硅基)-1-壬炔-5-酮-o-苄基肟醚(1e)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2e(21.9mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(3.2mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含炔烃化合物3b(78mg,0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应18h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(e)-1-(三异丙基硅基)-1-壬炔-5-酮o-苄基肟醚(1e),32.7mg,纯度为95%,产率为82%。
对(e)-1-(三异丙基硅基)-1-壬炔-5-酮o-苄基肟醚(1e)进行核磁共振检测,请参阅图9至图10,结果为:1hnmr(400mhz,cdcl3):δ7.27-7.26(m,4h),7.23-7.18(m,1h),4.37-4.31(m,1h),3.11(dd,j=5.6hz,8.8hz,1h),3.02(dd,j=6.8hz,14.0hz,1h),2.58-2.49(m,2h),2.47-2.43(m,2h),2.19-2.16(m,2h),1.65-1.56(m,6h),1.25(s,2h),1.10-1.09(m,19h);13cnmr(100mhz,cdcl3):δ=160.6,138.4,129.7,128.1,126.1,105.3,82.5,80.8,38.4,32.2,29.7,27.1,25.8,24.2,18.7,18.6,18.5,18.4,17.7,12.3,11.4,11.2。
本实施例表明可以通过亚胺化合物经氮-氧键的协助实现酮的区域选择性地β位csp3-h键的炔基化反应,反应可以实现柔性烷基链上的一级csp3-h键发生转化,且二级csp3-h键保持不变,表现出良好的化学选择性。
实施例6
本实施例进行(r,e)-5-(丙基-1-烯-2-基)-2-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-2-烯-1-酮o-甲基肟醚(1f)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2f(17.9mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(2.0mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射炔烃化合物3c(95.6mg,0.2mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应20h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(1f),21.9mg,纯度为95%,产率为61%。
对(r,e)-5-(丙基-1-烯-2-基)-2-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-2-烯-1-酮o-甲基肟醚(1f)进行核磁共振检测,请参阅图11至图12,结果为:1hnmr(400mhz,cdcl3):δ7.33-7.31(m,2h),7.16-7.13(m,2h),4.45-4.41(m,2h),3.26(dd,j=4.8hz,14.0hz,1h),3.11(dd,j=8.0hz,14.0hz,1h),2.62(d,j=5.2hz,2h),2.49-2.35(m,2h),2.13(d,j=6.0hz,2h),1.63-1.54(m,6h),1.09-1.08(m,21h);13cnmr(100mhz,cdcl3):δ=159.6,135.5,133.5,130.9,128.3,126.6,125.3,104.0,81.5,78.1,35.5,31.1,26.0,24.8,24.7,24.5,24.0,17.6,17.54,17.48,16.7,11.3,10.3。
本实施例表明可以通过亚胺化合物经氮-氧键的协助实现酮的位置选择性地β位csp3-h键的炔基化反应,反应可以实现天然产物香芹酮衍生物的后期衍生化。
实施例7
本实施例进行(e)-2-(甲氧亚胺基)-1-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-1-甲酸乙酯(1g)的制备,其反应式如下所示:
在空气氛围下,在反应器中依次加入亚胺化合物2g(21.3mg,0.1mmol)、二氯(五甲基环戊二烯基)合铱(iii)二聚体(3.2mg)、双三氟甲烷磺酰亚胺银盐(5.8mg)、碳酸锂(14.8mg)和醋酸银(33.4mg),用注射器注射含炔烃化合物3b(78mg,0.3mmol)的1,2-二氯乙烷溶液(1.0ml)到反应器中置于120℃下反应16h,经薄层色谱分析确定反应结束,将反应液经硅藻土抽滤后用400目硅胶经旋蒸浓缩制成干粉,再采用柱层析分离反应产物,400目硅胶5克,展开剂为体积比为100:1至20:1的石油醚与乙酸乙酯,得到含炔基的亚胺衍生物(e)-2-(甲氧亚胺基)-1-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-1-甲酸乙酯(1g),31.0mg,纯度为95%,产率为79%。
对(e)-2-(甲氧亚胺基)-1-(3-(三异丙基硅基)-2-丙炔基-1-基)环己基-1-甲酸乙酯(1g)进行核磁共振检测,请参阅图13至图14,结果为:1hnmr(400mhz,cdcl3):δ7.21(dd,j=5.6hz,8.4hz,1h),6.95(t,j=8.8hz,2h),4.33-4.27(m,1h),3.09(dd,j=5.6hz,9.6hz,1h),2.98(dd,j=6.8hz,14.0hz,1h),2.56(dd,j=4.0hz,16.8hz,1h),2.45(dd,j=7.2hz,16.8hz,3h),2.19-2.16(m,2h),1.60-1.57(m,6h),1.10-1.07(m,21h);13cnmr(100mhz,cdcl3):δ=160.8,134.0,133.9,131.1,115.0,114.8,105.1,82.6,80.7,37.5,32.2,29.7,27.1,25.83,25.78,25.6,24.2,18.7,18.5,17.8,17.7,12.3,11.3。
本实施例表明可以通过亚胺化合物经氮-氧键的协助实现酮的区域选择性地β位csp3-h键的炔基化反应,且可以在季碳中心的一级碳位置发生反应。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 聚酯系树脂组合物及使用了该聚酯系树脂组合物的成型品的制作方法
- 聚酯系树脂组合物、该聚酯系组合物的制备方法及使用了该聚酯系树脂组合物的成型品的制作方法
- 经交联、热重排的聚(苯并*唑-共-酰亚胺),包含其的气体分离膜以及其制备方法
- 一种醇胺类金属螯合型粉末涂料交联剂的制作方法
- 酶交联型壳聚糖/聚乙烯亚胺接枝磁性明胶材料及其制备方法与应用
- 包含经交联、热重排的聚(苯并*唑-共-酰亚胺)的用于烟道气分离的膜以及其制备方法
- 一种实验室用生物质热解碳化反应装置的制造方法
- 猪真皮基质-氧化羧甲基壳聚糖-羟基磷灰石口腔修复材料及其制备方法
- 猪真皮基质-氧化羧甲基壳聚糖-羟基磷灰石口腔修复材料及其制备方法
- 一种膨胀型无卤电缆料的制作方法