一种柔性二胺单体及其制备方法和在制备聚酰亚胺中的应用与流程
本发明属于有机化学
技术领域:
,具体涉及一种柔性二胺单体及其制备方法和在制备聚酰亚胺中的应用。
背景技术:
近年来,随着高
技术领域:
的迅速发展,如光电领域中的存储、显示、波导、太阳能电池的更新换代;航空航天等领域对材料的性能要求不断提高;传统的无机材料已很难满足上述高等
技术领域:
的发展要求。因此,由高分子材料做成的可以随意拉伸、折叠、弯曲并可恢复到原样的柔性材料正在成为如今高
技术领域:
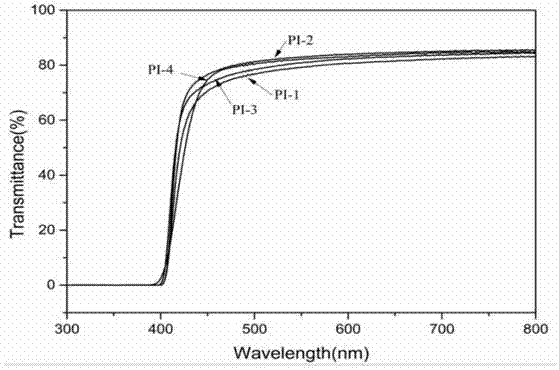
材料的发展趋势。目前,常用聚酰亚胺薄膜作为柔性基底材料。但是聚酰亚胺在制作聚酰亚胺薄膜时存在一些问题,比如聚酰亚胺的溶解性较差,增大了聚酰亚胺薄膜的制作难度;另外聚酰亚胺薄膜的光学透过率较低。为了提高聚酰亚胺的溶解性和聚酰亚胺薄膜的光学透过率,研究人员主要采用的方法为物理方法和化学方法,物理方法主要是通过掺杂无机纳米粒子降低聚酰亚胺结晶度来提高聚酰亚胺的溶解性和聚酰亚胺薄膜的光学透过率,但上述物理方法会导致聚酰亚胺的力学性能大大降低;化学方法主要通过结构改性,向聚酰亚胺分子链中引入大体积的取代基、柔性基团、非对称结构、脂环结构或非共平面结构等来改善聚酰亚胺的溶解性和聚酰亚胺薄膜的光学透过率,但上述化学方法无法同时提高聚酰亚胺薄膜的光学透过率和聚酰亚胺的力学性能。因此,聚酰亚胺在制备柔性基底材料时,存在溶解性、光学透过率及力学性能无法同时兼顾的问题。技术实现要素:本发明提供了一种三氟甲基取代联亚苯结构的柔性二胺单体及其制备方法,本发明提供的三氟甲基取代联亚苯结构的柔性二胺单体与二酐反应,能够制备得到溶解性好且力学性能好的聚酰亚胺,而且由此聚酰亚胺制备得到的聚酰亚胺薄膜的光学透过率较高。为了达到以上目的,本发明提供了以下技术方案:本发明提供了一种具有三氟甲基取代联亚苯结构的柔性二胺单体,具有式i所示结构:其中,x为n=1或2。优选的,所述柔性二胺单体包括1,4-(3-氨基-5-三氟甲基环基氧基)苯、4,4-(3-氨基-5-三氟甲基环基氧基)联苯、1,4-(3-氨基-5-三氟甲基酯基)苯或4,4’-(3-氨基-5-三氟甲基酯基)联苯。本发明提供了上述技术方案所述柔性二胺单体的制备方法:当所述x为时,所述柔性二胺单体的制备方法,包括以下步骤:(1)在氩气保护下,将3-卤-硝基三氟环己烷、二甲基铜锂、乙二胺四乙酸和二羟基化合物在极性有机溶剂中混合后,进行亲核取代反应,得到具有三氟甲基取代联亚苯结构的二硝基化合物;所述二羟基化合物包括1,4-对苯二酚或4,4’-二羟基-联苯化合物;(2)采用硫化钠对所述步骤(1)得到的二硝基化合物进行还原反应,得到x为的式i所示结构的柔性二胺单体;当所述x为时,所述柔性二胺单体的制备方法,包括以下步骤:(a)在氩气保护下,将3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸混合,进行酯化反应,得到具有三氟甲基取代联亚苯结构的酯基二硝基化合物;所述二羟基化合物包括1,4-对苯二酚或4,4’-二羟基-联苯化合物;(b)采用硫化氢和水合肼对所述步骤(a)得到的酯基二硝基化合物进行还原反应,得到x为的式i所示结构柔性二胺单体。优选的,所述步骤(1)中3-卤-硝基三氟环己烷、二甲基铜锂、乙二胺四乙酸和二羟基化合物的摩尔比为4:0.1~0.2:0.3~0.4:1.5~2。优选的,所述步骤(1)中亲核取代反应的温度为210~220℃。优选的,所述步骤(2)中还原反应的温度为105~115℃。优选的,所述步骤(a)中3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸的摩尔比为1:1.5~2:0.4~1。优选的,所述步骤(a)中酯化反应的温度为115~125℃。优选的,所述步骤(b)中还原反应的温度为85~110℃。本发明还提供了上述技术方案所述柔性二胺单体或者上述技术方案所述制备方法制备得到的柔性二胺单体在制备聚酰亚胺中的应用。本发明提供了一种具有三氟甲基取代联亚苯结构的柔性二胺单体及其制备方法和应用。本发明提供的柔性二胺单体包括三氟甲基、柔性基团和脂环结构,其中柔性基团为醚键或酯基,脂环结构为联亚苯基。当本发明提供的柔性二胺单体与二酐反应生成聚酰亚胺时,所述柔性二胺单体中的三氟甲基和柔性基团增大了聚酰亚胺分子链的自由体积和柔顺性,使溶剂容易渗入,改善了聚酰亚胺的溶解性;所述柔性二胺单体中的脂环结构可以进一步提高聚酰亚胺的溶解性和透明性;另外,本发明提供的柔性二胺单体主链中存在芳环结构,使由本发明制备得到的聚酰亚胺具有较强的分子链间作用力,进而使由本发明所述柔性二胺单体制备得到的聚酰亚胺具有较强的力学性能。实施例结果表明,由本发明提供的柔性二胺单体制备得到的聚酰亚胺具有较好的溶解性和力学性能,e(gpa)为4.5~5.1gpa,σmax(mpa)为210~230mpa,εmax(%)为14.5~18.3%,且聚酰亚胺薄膜的光透过率较高,在可见光范围内平均光透过率超过85%,而且5%的热失重温度平均超过530℃。附图说明图1为实施例1制备得到的1,4-(3-氨基-5-三氟甲基环基氧基)苯的氢核磁谱图;图2为应用例1制备得到的聚酰亚胺的氢核磁谱图;图3为应用例1~4制备得到的聚酰亚胺薄膜的紫外测试谱图;图4为应用例1~4制备得到的聚酰亚胺薄膜的热失重曲线。具体实施方式本发明提供了一种具有三氟甲基取代联亚苯结构的柔性二胺单体,具有式i所示结构:其中,x为n=1或2。在本发明中,所述柔性二胺单体优选包括1,4-(3-氨基-5-三氟甲基环基氧基)苯、4,4-(3-氨基-5-三氟甲基环基氧基)联苯、1,4-(3-氨基-5-三氟甲基酯基)苯或4,4’-(3-氨基-5-三氟甲基酯基)联苯。在本发明中,所述1,4-(3-氨基-5-三氟甲基环基氧基)苯具有式ii所示结构,所述4,4-(3-氨基-5-三氟甲基环基氧基)联苯具有式iii所示结构,所述1,4-(3-氨基-5-三氟甲基酯基)苯具有式iv所示结构,所述4,4’-(3-氨基-5-三氟甲基酯基)联苯具有式v所示结构。本发明提供的柔性二胺单体含三氟甲基、柔性基团(醚键、酯基)和脂环结构(联亚苯基),本发明提供的柔性二胺单体能够与二酐反应,生成聚酰亚胺,三氟甲基和柔性基团(醚键、酯基)的存在增大了聚酰亚胺分子链的自由体积和柔顺性,使溶剂容易渗入,改善了聚酰亚胺的溶解性;脂环结构(联亚苯基)的引入可以进一步提高聚酰亚胺的溶解性和透明性,这是因为脂环结构(联亚苯基)破坏了聚酰亚胺分子结构中的大π键,使分子链间的相互作用降低,分子链间的自由体积增大,从而使得分子间的间距增大,溶剂小分子可以进入到分子与分子之间使其溶解,在一定程度上改善了聚酰亚胺的溶解性和透明性;另外,本发明提供的柔性二胺单体主链中存在芳环结构,使由本发明制备得到的聚酰亚胺的直线型很强,具有较强的分子链间作用力,进而使由本发明所述柔性二胺单体制备得到的聚酰亚胺具有较强的力学性能。因此由本发明提供的柔性二胺单体制备的聚酰亚胺具有优异的光透过率、力学性能和溶解性能。本发明还提供了上述技术方案所述具有三氟甲基取代联亚苯结构的柔性二胺单体的制备方法。在本发明中,当所述x为时,所述柔性二胺单体的制备方法,包括以下步骤:(1)在氩气保护下,将3-卤-硝基三氟环己烷、二甲基铜锂、乙二胺四乙酸和二羟基化合物在极性有机溶剂中混合后,进行亲核取代反应,得到具有三氟甲基取代联亚苯结构的二硝基化合物;所述二羟基化合物包括1,4-对苯二酚或4,4’-二羟基-联苯化合物;(2)采用硫化钠对所述步骤(1)得到的三氟甲基取代联亚苯结构的二硝基化合物进行还原反应,得到x为的式i所示结构的柔性二胺单体。在本发明中,如无特殊说明,所述制备方法中的原料均为本领域技术人员所熟知的市售商品。本发明在氩气保护下,将3-卤-硝基三氟环己烷、二甲基铜锂、乙二胺四乙酸和二羟基化合物在极性有机溶剂中混合,得到混合物料。在本发明中,所述二甲基铜锂为催化剂,起到促进亲核取代反应发生的作用;所述乙二胺四乙酸为配体,起到调节金属催化剂中心的电性,改变其催化活性的作用。在本发明中,所述3-卤-硝基三氟环己烷、二甲基铜锂、乙二胺四乙酸和二羟基化合物的摩尔比优选为4:0.1~0.2:0.3~0.4:1.5~2,进一步优选为4:0.12~0.18:0.32~0.38:1.32~1.84,更优选为4:0.14~0.16:0.34~0.36:1.32~1.64。本发明优选将3-卤-硝基三氟环己烷、二甲基铜锂、乙二胺四乙酸和二羟基化合物的摩尔比控制在上述范围内,有利于原料之间充分反应。本发明优选根据混合物料的固含量控制极性有机溶剂的质量。在本发明中,所述混合物料的固含量优选为20%~25%,进一步优选为22%~24%。本发明优选将混合物料的固含量控制在上述范围内,有利于反应原料之间充分反应。本发明在氩气保护下将上述原料混合,目的是防止空气中的氧气、水蒸气等对反应造成影响。得到混合物料后,本发明将所述混合物料进行亲核取代反应,得到具有三氟甲基取代联亚苯结构的二硝基化合物。在本发明中,所述亲核取代反应的温度优选为210~220℃,进一步优选为212~218℃,更优选为214~216℃。在本发明中,所述亲核取代反应的时间优选通过tlc实时检测结果确定,当所述tlc实时检测的原料点消失时,停止反应。本发明优选将亲核取代反应产物冷却后,与甲醇混合;将混合溶液依次进行过滤、滤饼干燥和柱层析处理,得到具有三氟甲基取代联亚苯结构的二硝基化合物。本发明对所述亲核取代反应产物冷却的方式没有特别限制,采用本领域常用方法即可。本发明优选将冷却后的亲核取代反应产物与甲醇混合。本发明优选将亲核取代反应产物与甲醇的混合溶液依次进行过滤、滤饼干燥和柱层析处理,得到具有三氟甲基取代联亚苯结构的二硝基化合物。本发明对过滤和滤饼干燥处理的具体实施方式没有特别限制,采用本领域技术人员所熟知的方式即可。本发明优选对干燥后的反应产物进行柱层析处理,所述柱层析处理用洗脱剂优选包括石油醚和环己烷,所述石油醚和环己烷的体积比优选为2:1。本发明优选采用上述洗脱剂,有利于得到纯度较高的三氟甲基取代联亚苯结构的二硝基化合物。得到具有三氟甲基取代联亚苯结构的二硝基化合物后,本发明采用硫化钠对三氟甲基取代联亚苯结构的二硝基化合物进行还原反应,得到x为的式i所示结构的柔性二胺单体。本发明优选将所述三氟甲基取代联亚苯结构的二硝基化合物与还原反应用溶剂混合,加热回流后,再与硫化钠混合,进行还原反应。在本发明中,所述还原反应用溶剂优选包括1,4-二氧六环或无水乙醇。在本发明中,所述三氟甲基取代联亚苯结构的二硝基化合物的质量与还原反应溶剂的体积比优选为4~6g:40~75ml,进一步优选为4.5~5.5g:50~70ml。在本发明中,所述三氟甲基取代联亚苯结构的二硝基化合物与硫化钠的质量比优选为1:0.8~1,进一步优选为1:0.9~1。本发明优选将二硝基化合物与硫化钠的质量比控制在上述范围内,有利于硫化钠充分将二硝基化合物还原为二胺基化合物。在本发明中,所述还原反应的温度优选为105~115℃,进一步优选为108~112℃。在本发明中,所述还原反应的时间优选通过tlc检测结果确定,当所述tlc检测中二硝基化合物原料点消失时,停止反应。本发明优选对还原反应产物依次进行过滤、滤液浓缩、干燥和重结晶处理,得到具有三氟甲基取代联亚苯结构的柔性二胺单体。在本发明中,所述过滤的温度优选为105~115℃,本发明优选在还原反应完成后,不经冷却处理,直接进行过滤,即趁热过滤。本发明优选通过趁热过滤,有利于除去未反应完的硫化钠,而且有利于避免还原反应生成的柔性二胺单体析出。本发明对过滤、滤液浓缩和干燥的具体实施方式没有特别要求,采用本领域技术人员所常用的方式即可。本发明优选对干燥后的粗产物进行重结晶处理,得到具有三氟甲基取代联亚苯结构的柔性二胺单体;在本发明中,所述重结晶处理优选包括以下步骤:将所述干燥后的粗产物和1,4-二氧六环混合后,加热回流,得到回流反应液;向所述回流反应液中缓慢加入去离子水,直至刚有沉淀析出且不溶解,对析出的沉淀进行干燥处理,得到具有三氟甲基取代联亚苯结构的柔性二胺单体。本发明优选通过加热回流处理,使粗产物充分溶解在良性溶剂1,4-二氧六环中。在本发明中,所述加热回流的温度优选为105~115℃;进一步优选为110℃。本发明在重结晶处理中,优选先将粗产物与1,4-二氧六环混合,使粗产物充分溶解在1,4-二氧六环中,然后再逐渐加入劣溶剂去离子水,使溶解在1,4-二氧六环中的柔性二胺单体溶解度降低而逐渐析出。本发明优选通过重结晶处理,提高了三氟甲基取代联亚苯结构的柔性二胺单体的纯度。本发明优选通过过滤收集析出的沉淀,再对析出的沉淀进行干燥处理,得到具有三氟甲基取代联亚苯结构的柔性二胺单体。本发明对过滤的具体实施方式没有特别限制,采用本领域技术人员所熟知的方式即可。在本发明中,所述干燥处理的温度优选为95~105℃,进一步优选为100℃;所述干燥处理的时间优选为11~13h,进一步优选为12h。在本发明中,当所述x为时,所述三氟甲基取代联亚苯结构的柔性二胺单体的制备过程优选具体如式vi所示:在本发明中,当所述x为时,所述柔性二胺单体的制备方法,包括以下步骤:(a)在氩气保护下,将3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸混合,进行酯化反应,得到具有三氟甲基取代联亚苯结构的酯基二硝基化合物;所述二羟基化合物包括1,4-对苯二酚或4,4’-二羟基-联苯化合物;(b)采用硫化氢和水合肼对所述步骤(a)得到的三氟甲基取代联亚苯结构的酯基二硝基化合物进行还原反应,得到x为的式i所示结构的柔性二胺单体。在本发明中,如无特殊说明,所述制备方法中的原料均为本领域技术人员所熟知的市售商品。本发明在氩气保护下,将3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸混合,得到混合原料。在本发明中,所述浓硫酸作为催化剂和吸水剂,起到促进酯化反应发生的作用。本发明对所述浓硫酸的浓度没有特殊要求,采用本领域技术人员所熟知的即可。在本发明中,所述3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸的摩尔比优选为1:1.5~2:0.4~1,进一步优选为1:1.6~1.8:0.5~0.8。本发明优选将3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸的摩尔比控制在上述范围内,有利于使反应原料之间充分反应。本发明对所述3-硝基-三氟甲基环己烷甲酸、二羟基化合物和浓硫酸的混合方式没有特别限制,采用本领域技术人员所常用的混合方式即可。得到混合原料后,本发明将所述混合原料进行酯化反应,得到具有三氟甲基取代联亚苯结构的酯基二硝基化合物。在本发明中,所述酯化反应的温度优选为115~125℃,进一步优选为120℃;所述酯化反应的时间优选为0.5~1.0h,进一步优选为0.6~0.8h。所述酯化反应完成后,本发明优选将酯化反应产物依次进行冷却、蒸馏浓缩、中和、静置分层、洗涤、旋蒸和重结晶处理,得到具有三氟甲基取代联亚苯结构的酯基二硝基化合物。本发明优选将酯化反应产物进行冷却处理,得到冷却的酯化反应产物。本发明对冷却的方式没有特别限制,采用本领域技术人员所熟知的冷却方式即可。本发明优选对冷却的酯化反应产物进行蒸馏浓缩处理,得到浓缩的酯化反应产物。在本发明中,所述浓缩的酯化反应产物的体积与冷却的酯化反应产物的体积比优选为0.5~0.6:1。本发明对蒸馏浓缩的具体实施方法没有特别限制,采用本领域技术人员所熟知的蒸馏浓缩方式即可。本发明优选对浓缩的酯化反应产物进行中和处理,得到中和产物。在本发明中,所述中和试剂优选为饱和碳酸钠溶液。本发明优选通过中和处理除去未反应完的3-硝基-5-(三氟甲基)苯甲酸原料。本发明优选通过中和产物的ph值控制饱和碳酸钠溶液的加入量,当所述中和产物的ph值为中性时,停止加入饱和碳酸钠溶液。本发明优选对中和反应产物进行静置分层处理,所述静置分层后的下层优选为水溶液,上层优选为有机相。本发明优选收集上层有机相。本发明对静置分层处理的具体实施方式没有特别限制,采用本领域技术人员所常用的静置分层方式即可。本发明优选对有机相依次进行饱和食盐水洗涤、饱和氯化钙洗涤和水洗涤。本发明优选通过饱和食盐水洗涤,有利于除去残留的中和试剂碳酸钠;本发明优选通过饱和氯化钙洗涤,有利于除去反应原料中未反应完的二羟基化合物;本发明优选通过水洗涤,有利于除去残留的无机盐。所述洗涤处理完成后,本发明优选除去水相,得到洗涤后的有机相。本发明优选对洗涤后的有机相进行旋蒸处理,得到粗产品。本发明优选通过旋蒸处理,除去有机相中的有机溶剂,得到粗产品。本发明优选对所述粗产品进行重结晶处理,得到具有三氟甲基取代联亚环己烷结构的酯基二硝基化合物。在本发明中,所述重结晶处理优选包括以下步骤:将所述干燥后的粗产品和n,n-二甲基甲酰胺混合后,加热回流,得到回流反应液;向所述回流反应液中缓慢加入去离子水,直至刚有沉淀析出且不溶解,对析出的沉淀进行干燥处理,得到具有三氟甲基取代联亚苯结构的酯基二硝基化合物。本发明优选通过加热回流处理,使粗产品充分溶解在良性溶剂n,n-二甲基甲酰胺中。在本发明中,所述加热回流的温度优选为155~165℃;进一步优选为160℃。本发明在重结晶处理中,优选先将粗产品与n,n-二甲基甲酰胺混合,使粗产品充分溶解在n,n-二甲基甲酰胺中,然后再逐渐加入劣溶剂去离子水,使溶解在n,n-二甲基甲酰胺中的酯基二硝基化合物溶解度降低而逐渐析出。本发明优选通过重结晶处理,提高了三氟甲基取代联亚苯结构的酯基二硝基化合物的纯度。本发明优选通过过滤收集析出的沉淀。本发明对过滤的具体实施方式没有特别限制,采用本领域技术人员所熟知的方式即可。本发明优选对析出的沉淀进行干燥处理。在本发明中,所述干燥处理的温度优选为95~105℃,进一步优选为100℃;所述干燥处理的时间优选为11~13h,进一步优选为12h。得到具有三氟甲基取代联亚苯结构的酯基二硝基化合物后,本发明采用硫化氢和水合肼对所述酯基二硝基化合物进行还原反应,得到x为的具有三氟甲基取代联亚苯结构的柔性二胺单体。本发明优选将三氟甲基取代联亚苯结构的酯基二硝基化合物与还原反应的溶剂混合后,加热至回流,然后加入硫化氢水溶液和水合肼溶液,进行还原反应。在本发明中,所述还原反应的溶剂优选包括无水乙醇或1,4-二氧六环。所述三氟甲基取代联亚苯结构的酯基二硝基化合物与还原反应溶剂的质量比优选为10~20:80~85,进一步优选为12~18:82~84。在本发明中,所述三氟甲基取代联亚苯结构的酯基二硝基化合物与硫化氢的质量比优选为1:0.48~0.90,进一步优选为1:0.5~0.8。在本发明中,所述硫化氢水溶液的质量分数优选为80%~90%,进一步优选为85%。在本发明中,所述三氟甲基取代联亚苯结构的酯基二硝基化合物与硫化氢水溶液的质量比优选为1:0.6~1,进一步优选为1:0.7~0.9。本发明优选将酯基二硝基化合物与硫化氢水溶液的质量比控制在上述范围内,有利于硫化氢充分将酯基二硝基化合物还原为二胺基化合物。在本发明中,所述三氟甲基取代联亚苯结构的酯基二硝基化合物与水合肼的质量比优选为2:12.75~17,进一步优选为2:15~16。在本发明中,所述水合肼溶液的质量分数优选为85%。在本发明中,所述三氟甲基取代联亚苯结构的酯基二硝基化合物与水合肼溶液的质量比优选为2:15~20,进一步优选为2:16~18。本发明优选将酯基二硝基化合物与水合肼溶液的质量比控制在上述范围内,有利于水合肼充分将酯基二硝基化合物还原为二胺基化合物。本发明优选同时加入硫化氢和水合肼,有利于促进还原反应的进行,缩短反应进行的时间,提高反应效率。在本发明中,所述还原反应的温度优选为85~110℃,进一步优选为90~100℃。在本发明中,所述还原反应的时间优选通过tlc检测结果确定,当所述tlc检测至酯基二硝基化合物原料点消失,即停止反应。本发明优选对还原反应产物依次进行过滤、滤液浓缩、旋蒸和重结晶处理,得到x为的具有三氟甲基取代联亚苯结构的柔性二胺单体。本发明对过滤的具体实施方式没有特别要求,采用本领域技术人员所常用的方式即可。本发明优选将过滤后的滤液浓缩,所述浓缩后的滤液体积为原滤液体积的1/4~1/3。本发明对滤液浓缩的具体实施方式没有特别限定,采用本领域技术人员所常用的滤液浓缩方式即可。本发明优选将浓缩后的滤液进行旋蒸处理,得到粗品。本发明对旋蒸处理的具体实施方式没有特别要求,采用本领域技术人员所熟知的旋蒸方式即可。本发明优选对粗品进行重结晶处理,得到具有三氟甲基取代联亚苯结构的柔性二胺单体。在本发明中,所述重结晶处理优选包括以下步骤:将所述粗品和无水乙醇混合后,加热回流,得到回流反应液;向所述回流反应液中缓慢加入去离子水,直至刚有沉淀析出且不溶解,冷却过滤,对析出的沉淀进行干燥处理,得到具有三氟甲基取代联亚苯结构的柔性二胺单体。本发明优选通过加热回流处理,使粗品充分溶解在良性溶剂无水乙醇中。在本发明中,所述加热回流的温度优选为80~90℃;进一步优选为85℃。本发明在重结晶处理中,优选先将粗品与无水乙醇混合,使粗品充分溶解在无水乙醇中,然后再逐渐加入劣溶剂去离子水,使溶解在无水乙醇的柔性二胺单体溶解度降低而逐渐析出。本发明优选通过重结晶处理,提高了三氟甲基取代联亚苯结构的柔性二胺单体的纯度。本发明优选冷却至室温后,过滤收集析出的沉淀。本发明优选在冷却后再进行过滤,有利于使柔性二胺单体以沉淀的形式充分析出。本发明对过滤的具体实施方式没有特别限制,采用本领域技术人员所熟知的方式即可。本发明优选对析出的沉淀进行干燥处理。在本发明中,所述干燥处理的温度优选为55~65℃,进一步优选为60℃;所述干燥处理的时间优选为11~13h,进一步优选为12h。在本发明中,当所述x为时,所述三氟甲基取代联亚苯结构的柔性二胺单体的制备过程具体如式vii所示:本发明还提供了上述技术方案所述柔性二胺单体或者上述技术方案所述制备方法制备得到的柔性二胺单体在制备聚酰亚胺中的应用。在本发明中,所述聚酰亚胺的制备方法优选包括以下步骤:在氩气保护下,将所述柔性二胺单体、二酐单体、催化剂和脱水剂混合,在极性有机溶剂中进行反应,得到聚酰亚胺。在本发明中,所述二酐单体优选具有式viii所示结构:在本发明中,所述二酐单体中ar优选具有以下结构:在本发明中,所述二酐单体进一步优选包括均苯四甲酸二酐、3,3’,4,4’-联苯四酸二酐、3,3’,4,4’-二苯醚四酸二酐、3,3’,4,4’-二苯甲酮四酸二酐、4,4’-(六氟异丙烯)二酞酸酐和4,4’-(4,4’-二酚氧基丙基)-二苯甲酸酐中的一种或多种。在本发明中,所述柔性二胺单体和二酐单体的摩尔比优选为1:1。在本发明中,所述催化剂优选包括吡啶和/或氯化钙。在本发明中,所述脱水剂优选包括乙酸酐和/或亚磷酸苯酯。在本发明中,所述催化剂、脱水剂和柔性二胺单体的摩尔比优选为0.5~1.5:1.5~2.5:0.5~1.5,进一步优选为0.8~1.2:1.8~2.2:0.8~1.2,更优选为1:2:1。在本发明中,所述极性有机溶剂优选包括n,n-二甲基乙酰胺、n,n-二甲基甲酰胺和n-甲基吡咯烷酮中的一种或多种。在本发明中,所述柔性二胺单体和二酐单体的总质量与极性有机溶剂的质量比优选为10~30:70~90,进一步优选为15~25:75~85。在本发明中,所述柔性二胺单体和二酐单体在氩气保护下反应,目的是防止柔性二胺单体被空气中的氧气氧化。本发明优选在氩气下进行反应,有利于避免柔性二胺单体被氧化,进而有利于保证二酐单体和柔性二胺单体顺利反应制备得到聚酰亚胺。在本发明中,所述柔性二胺单体和二酐单体反应的温度优选为100~120℃,进一步优选为110~115℃;反应时间优选为2~5h,进一步优选为3~4h。下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。实施例1:1,4-(3-氨基-5-三氟甲基环基氧基)苯的制备第一步反应:向装有机械搅拌装置的250ml三颈烧瓶中,加入2.7605g(10mmol)3-溴-5硝基-三氟环己烷、0.0200g(0.2mmol)二甲基铜锂、3.2582g(10mmol)碳酸铯、0.1169g(0.4mmol)乙二胺四乙酸,加入35mln-甲基吡络烷酮作为溶剂,在搅拌、充分通氩气的保护下,加入2.7528g(25mmol)的1,4-对苯二酚化合物,室温下搅拌30min,在搅拌、氩气保护下升温到回流温度210℃,tlc检测至原料点消失即为反应结束;体系冷却后出料于甲醇中,旋蒸得粗产品,100℃真空干燥12h,经石油醚/环己烷=2/1(体积比)的硅胶层析柱,得到1,4-(3-硝基-5-三氟甲基环基氧基)苯化合物;第二步反应:向装有机械搅拌装置的250ml三颈烧瓶中,加入第一步反应制备的二硝基化合物5.0039g(10mmol),加入40ml的1,4-二氧六环作为溶剂,加热到回流温度110℃,加入2g的硫化钠na2s,于回流状态下反应12h,tlc检测至原料点消失即为反应结束;趁热过滤反应液除去na2s,收集滤液并将其减压蒸馏至20ml,在氩气氛围下对浓缩液进行重结晶,得到3.4g1,4-(3-氨基-5-三氟甲基环基氧基)苯,具有下式所示结构:对实施例1制备得到的1,4-(3-氨基-5-三氟甲基环基氧基)苯进行核磁测试,测试结果如图1所示。实施例2:4,4-(3-氨基-5-三氟甲基环基氧基)联苯的制备第一步反应:向装有机械搅拌装置的250ml三颈烧瓶中,加入2.7605g(10mmol)3-溴-5-硝基三氟甲苯、0.0801g(0.8mmol)二甲基铜锂、3.2582g(10mmol)氟化铯、0.8766g(3mmol)乙二胺四乙酸,在搅拌、充分通氩气的保护下,加入1.9800g(15mmol)的4,4’-二羟基-联苯化合物,室温下搅拌30min,在搅拌、氩气保护下,升温到回流温度220℃,体系成为熔融反应,tlc检测至原料点消失即为反应结束;体系冷却后出料于甲醇中,旋蒸的粗产品,100℃真空干燥12h,经石油醚/环己烷=2/1(体积比)的硅胶层析柱,得到4,4-(3-硝基-5-三氟甲基环基氧基)联苯化合物;第二步反应:向装有机械搅拌装置的250ml三颈烧瓶中,加入第一步反应制备的二硝基化合物5.7649g(10mmol),加入75ml的1,4-二氧六环作为溶剂,加热到回流,加入2g的硫化钠na2s,将10.003g(200mmol)的质量分数为85%的水合肼缓慢滴加到反应体系中,于回流温度110℃下反应16h,tlc检测至原料点消失即为反应结束,趁热过滤反应液除去na2s,收集滤液并将其减压蒸馏至60ml,在氩气氛围下对浓缩液进行重结晶,得到4.8g的4,4’-(3-氨基-5-三氟甲基苯氧基)联苯化合物,具有下式所示结构:实施例3:1,4-(3-氨基-5-三氟甲基酯基)苯的制备第一步反应:向装有机械搅拌装置的250ml三颈烧瓶中,加入2.4117g(10mmol)3-硝基-5-(三氟甲基)环己基甲酸、2.7528g(25mmol)1,4-对苯二酚、5.0ml浓硫酸,混合均匀,加入沸石,在搅拌、冷凝、充分通氩气的保护下,保持缓慢回流温度120℃0.5h,待体系内反应物冷却,将回流装置改为蒸馏装置,蒸出生成的二硝基酯基化合物,直到馏出液体积约为反应物总体积的1/2,在馏出液中慢慢加入饱和碳酸钠溶液,并不断振荡,直至ph试纸不再显酸性,将混合液转入分液漏斗,分去下层水溶液,有机层依次用饱和食盐水、饱和氯化钙、水洗涤,分去下层液体,加入无水硫酸镁干燥,有机层旋蒸得粗产品,真空干燥粗产品,粗产品经重结晶处理,得到1,4-(3-硝基-5-三氟甲基酯基)苯的制备化合物;第二步反应:将得到的二硝基酯基化合物4.2539g(10mmol)加入到无水乙醇中,搅拌加热体系至回流温度85℃,加入2g的h2s水溶液,再缓慢滴加55ml质量分数为85%的水合肼溶液,与回流状态下反应,tlc检测至原料点消失即为反应结束,其氩气氛围下对浓缩液进行重结晶,60℃真空干燥得到3.2g的1,4-(3-硝基-5-三氟甲基酯基)苯的制备化合物,具有下式所示结构:实施例4:4,4’-(3-氨基-5-三氟甲基酯基)联苯的制备第一步反应:向装有机械搅拌装置的250ml三颈烧瓶中,加入2.4117g(10mmol)3-硝基-5-(三氟甲基)环己基甲酸、4.6553g(25mmol)4,4’-二羟基-联苯5.0ml浓硫酸,混合均匀,加入沸石,在搅拌、冷凝、充分通氩气的保护下,保持缓慢回流温度120℃0.5h,待体系内反应物冷却,将回流装置改为蒸馏装置,蒸出生成的二硝基酯基化合物,直到馏出液体积约为反应物总体积的1/2,在馏出液中慢慢加入饱和碳酸钠溶液,并不断振荡,直至ph试纸不再显酸性,将混合液转入分液漏斗,分去下层水溶液,有机层用饱和食盐水洗涤,再用饱和氯化钙洗涤,最后用水洗一次,分去下层液体,加入无水硫酸镁干燥,有机层旋蒸得粗产品,真空干燥粗产品,粗产品经过重结晶操作,得到4,4’-(3-氨基-5-三氟甲基酯基)联苯的制备化合物;第二步反应:将得到的二硝基酯基化合物5.7759g(10mmol)加入到1,4-二氧六环中,搅拌加热体系至回流温度110℃,加入2g的h2s水溶液,再缓慢滴加55ml质量分数为85%的水合肼溶液,与回流状态下反应,tlc检测至原料点消失即为反应结束,收集滤液并将其减压蒸馏至原反应液体积的1/4,最后在氩气氛围下对浓缩液进行重结晶,60℃真空干燥得到4.8g的4,4’-(3-氨基-5-三氟甲基酯基)联苯的制备化合物,得到的产物结构如下式:应用例1:1,4-(3-氨基-5-三氟甲基苯氧基)苯与4,4’-(六氟异丙烯)二酞酸酐,制备聚酰亚胺,具体过程如下:在装有氩气进出口、磁力搅拌子、温度计、冷凝管的50ml三颈烧瓶中,在氩气保护下,加入0.8809g(2mmol)的1,4-(3-氨基-5-三氟甲基苯氧基)苯,加入20ml的n,n-二甲基乙酰胺作为溶剂,缓慢加入0.8885g(2mmol)4,4’-(六氟异丙烯)二酞酸酐室温下搅拌6h,再加入4ml吡啶和8ml乙酸酐,加热至100℃反应2h,反应结束后冷却至室温,出料在乙醇中,乙醇回流洗三次,在真空烘箱80℃下烘干,得到1.4551g的1,4-联(3-氨基-5-三氟甲基苯氧基)苯六氟类聚酰亚胺,得到的产物结构如下式,标记为pi-1:对应用例1制备得到的聚酰亚胺进行核磁测试,测试结果如图2所示。应用例2:4,4’-(3-氨基-5-三氟甲基苯氧基)联苯与4,4’-(六氟异丙烯)二酞酸酐制备聚酰亚胺,具体过程如下:在装氩气进出口、磁力搅拌子、温度计、冷凝管的50ml的三颈烧瓶中,在氩气的保护下,加入1.0331g(2mmol)的4,4’-联(3-氨基-5-三氟甲基苯氧基)联苯、加入2ml的亚磷酸苯酯和1ml吡啶、0.2gcacl2、6ml的nmp作为溶剂,缓慢加入0.8885g(2mmol)4,4’-(六氟异丙基)二甲酸酐,加热到100℃反应3h,反应结束后冷却至室温,出料在乙醇中,抽滤,滤饼用乙醇、水、乙醇回流洗三次,在真空烘箱80℃下烘干,得到1.5005g的4,4’-二(3-氨基-5-三氟甲基苯氧基)联苯六氟类聚酰亚胺,得到的产物结构如下式,标记为pi-2:应用例3:1,4-(3-氨基-5-三氟甲基酯基)苯与4,4’-(六氟异丙烯)二酞酸酐制备聚酰亚胺,具体过程如下:在装氩气进出口、磁力搅拌子、温度计、冷凝管的50ml的三颈烧瓶中,在氩气的保护下,加入0.9929g(2mmol)的1,4(3)-(3-氨基-5-三氟甲基酯基)苯、加入2ml的亚磷酸苯酯和1ml吡啶、0.2gcacl2、6ml的nmp作为溶剂,缓慢加入0.8885g(2mmol)4,4’-(六氟异丙基)二甲酸酐,加热到100℃反应3h,反应结束后冷却至室温,出料在乙醇中,抽滤,滤饼用乙醇、水、乙醇回流洗三次,在真空烘箱80℃下烘干,得到1.5402g的1,4-(3-氨基-5-三氟甲基酯基)苯六氟类聚酰亚胺,得到的产物结构如下式,标记为pi-3:应用例4:4,4’-(3-氨基-5-三氟甲基酯基)联苯与4,4’-(六氟异丙烯)二酞酸酐制备聚酰亚胺,具体过程如下:在装氩气进出口、磁力搅拌子、温度计、冷凝管的50ml的三颈烧瓶中,在氩气的保护下,加入1.1451g(2mmol)的4,4’-(3-氨基-5-三氟甲基酯基)联苯、加入2ml的亚磷酸苯酯和1ml吡啶、0.2gcacl2、5ml的nmp作为溶剂,缓慢加入0.8885g(2mmol)4,4’-(六氟异丙基)二甲酸酐,加热到100℃反应3h,反应结束后冷却至室温,出料在乙醇中,抽滤,滤饼用乙醇、水、乙醇回流洗三次,在真空烘箱80℃下烘干,得到1.6085g的4,4’-(3-氨基-5-三氟甲基酯基)联环己烷六氟类聚酰亚胺,得到的产物结构如下式,标记为pi-4:对比例以二羟基-苯基类化合物为对比例,二羟基-苯基类化合物的结构如下式所示:对应用例和对比例的聚酰亚胺的溶解性进行测试,测试方法为:将聚酰亚胺分别溶解在nmp、dmac、dmf、dmso、thf和chcl3中,聚酰亚胺在不同溶剂中的浓度为10mg/ml。测试聚酰亚胺在不同溶剂中的溶解性能,++:室温全溶;+:加热全溶;+-:部分溶解;--:加热不溶。测试结果如表1所示。表1:应用例和对比例制备的聚酰亚胺在6种溶剂中的溶解性样品nmpdmacdmfdmsothfchcl3应用例1++++++++++++应用例2+++++++++++-应用例3++++++++++++应用例4+++++++++++-对比例+++++-+++++-由表1测试结果可知,本发明提供的柔性二胺单体制备得到的聚酰亚胺具有较好的溶解性。将应用例1~4制备得到的聚酰亚胺采用本领域常用方法制备成聚酰亚胺薄膜,对聚酰亚胺薄膜的透光率进行测试,测试方法为:采用日本岛津公司的uv2550型紫外可见分光光度计测定,扫描范围为250~800nm。测试结果如图1所示,由图1可以看出:聚酰亚胺薄膜在可见光范围内具有高的透过性,在可见光范围内平均透过率高达85%以上,截止波长300nm。另外,对应用例1~4制备得到的聚酰亚胺薄膜的热稳定性进行测试,测试结果如图2所示,由图2可以看出,聚酰亚胺薄膜具有良好的热稳定性,在氮气气氛下的5%热失重温度为480℃以上。对应用例制备得到的聚酰亚胺的力学性能进行测试,测试方法为:采用shimadzuag-i型万能试验机,在室温下测试,样品尺寸为20mm*3mm*0.02mm,拉伸速率为8mm/min。测试结果如表2所示。表2:应用例制备的聚酰亚胺的力学性能样品拉伸模量e(gpa)最大拉伸强度σmax(mpa)断裂伸长率εmax(%)应用例14.523014.5应用例25.122516.3应用例34.821017.2应用例45.021318.3由表2测试结果可知,本发明提供的柔性二胺单体制备得到的聚酰亚胺具有较好的力学性能,e(gpa)为4.5~5.1gpa,σmax(mpa)为210~230mpa,εmax(%)为14.5~18.3%。由以上实施例和应用例可知,由本发明提供的柔性二胺单体制备得到的聚酰亚胺具有较好的溶解性和力学性能,e(gpa)为4.5~5.1gpa,σmax(mpa)为210~230mpa,εmax(%)为14.5~18.3%,且聚酰亚胺薄膜的光透过率较高。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1