一种2-氨基取代的含氮六元杂环类化合物的制备方法与流程
本发明涉及精细化工技术领域,具体涉及一种2-氨基取代的含氮六元杂环类化合物的制备方法。
背景技术:
2-氨基取代的含氮六元杂环类化合物在化工领域具有重要应用,尤其是2-氨基吡啶及其衍生物是药物和农用化学品分子中重要的结构之一,广泛存在于天然产物、药物以及发光材料和各种精细化学品的合成应用中。现有技术中,制备2-氨基吡啶的方法包括:1)金属催化卤代吡啶胺化制备法,如通过铜催化2-卤代吡啶的胺化是最常用的方法(adv.syn.catal.,2013,355,627-631;chemcommun.,2010,46,925-927)、钯催化2-溴吡啶和(me3si)2nhli的胺化得到2-氨基吡啶产物(产率为88%,catallett.,2017,147,204-214),这类方法具有收率高,反应条件温和等优点,然而却存在经济成本高等缺陷;2)氨解制备法,如由2-氟-吡啶和氨的氨解制备,该方法是在高温下使用氨气作为反应原料,因此,需要使用高压和密封的反应容器(j.org.chem.,1988,53,2740-2744.);3)氰基水解法或霍夫曼降解法(bioorg.med.chem.lett,2010,20,2512-2515),然而,这类方法需要使用大量氧化剂和有毒的溴素,且难以控制反应性,不适合大规模生产;4)催化还原法,如用钯催化2-硝基吡啶的氢化还原中获得,但这方法需要使用高压氢气(org.lett.,2015,17,941–3943);5)其他方法,如通过吡啶化合物和氨基钠的chichibabin反应制备2-氨基吡啶虽然具有非常高的原子经济性,然而,该方法的底物适用性不高(chem.rev.,1937,20,413–487;zhurnalrusskagofiziko-khimicheskagoobshchestva,1914,46,1216-1236)且存在许多副反应和需要严格无水无氧反应条件。
因此,尽管现有技术中存在多种合成2-氨基吡啶的方法,但继续发展温和、反应可控、高化学选择性且经济成本低的2-氨基吡啶合成方法是具有重要意义和应用前景。
技术实现要素:
本发明所要解决的技术问题是:提供一种经济效益高且产率高的2-氨基取代的含氮六元杂环类化合物的制备方法。
为了解决上述技术问题,本发明采用的技术方案为:一种2-氨基取代的含氮六元杂环类化合物的制备方法,所述制备方法包括以下步骤:将2-氟取代的含氮六元杂环类化合物与盐酸脒盐类化合物混合后,在碱性物质的作用下反应得到2-氨基取代的含氮六元杂环类化合物。
优选地,所述2-氨基取代的含氮六元杂环类化合物为2-氨基吡啶类化合物、2-氨基嘧啶类化合物或2-氨基吡嗪类化合物。
优选地,所述2-氟取代的含氮六元杂环类化合物的结构通式如式(i)所示:
进一步地,所述盐酸脒盐类化合物的结构通式如式(ii)所示:
进一步地,所述碱性物质包括无机碱和/或有机碱;优选地,所述碱性物质包括氢氧化钾、氢氧化钠、氢氧化锂、叔丁醇钠和/或甲醇钠。
进一步地,所述2-氟取代的含氮六元杂环类化合物与盐酸脒盐的摩尔比为1:(1~2),优选为1:1.2。
进一步地,所述碱性物质的用量为所述2-氟取代的含氮六元杂环类化合物用量的1~3倍;优选地,所述碱性物质的用量为所述2-氟取代的含氮六元杂环类化合物用量的2倍。
进一步地,反应温度为100~150℃,反应时间为24~36小时;优选为130℃,反应24h。
进一步地,所述反应在溶液中进行,溶剂包括有机溶剂和无机溶剂,所述有机溶剂选自氯苯、二乙二醇二甲醚、n-吡咯烷酮、二甲基亚砜(dimethylsulfoxide,dmso)、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲苯、二甲苯和1,4-二氧六环中的至少一种;有机溶剂优选为二甲基亚砜。
进一步地,所述制备方法还包括反应结束后进行提纯处理操作,所述提纯处理操作为反应结束后加入乙酸乙酯淬灭反应,加入饱和食盐水洗涤,分离出有机相;
再用乙酸乙酯萃取分离出有机相后剩余的水相,将萃取后的溶液与所述有机相合并,加入无水硫酸钠干燥,经减压蒸馏去除溶剂包括有机溶剂和无机溶剂,所述有机溶剂后,再经柱层析得到纯化后的2-氨基取代的含氮六元杂环类化合物。
本发明的有益效果在于:本发明方案反应条件温和,操作步骤少;采用的催化剂廉价易得,排放的废弃物少,原子效益高,经济环保;底物适应性广、收率高且化学选择性好,易于规模化生产。本发明制备方法无需过渡金属催化,以2-氟取代的含氮六元杂环类化合物为原料,廉价安全的盐酸脒盐为胺源,在碱性物质的作用下,2-氟取代的含氮六元杂环类化合物与盐酸脒盐类化合物通过亲核反应、水解反应制得2-氨基取代的含氮六元杂环类化合物。
附图说明
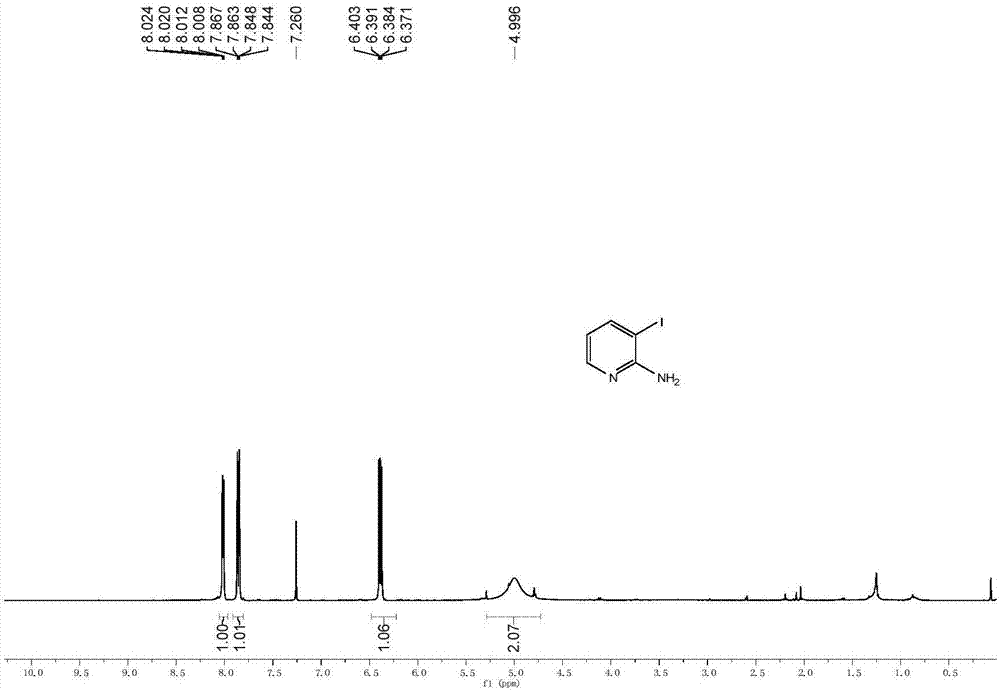
图1为本发明实施例1制得的产物的核磁共振氢谱;
图2为本发明实施例1制得的产物的核磁共振碳谱。
具体实施方式
为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式并配合附图予以说明。
本发明制备方法的反应原理如下:
上式中,a1和a2为n或ch;r1为单个或多个取代基,所述r1选自氢、卤素、烷基、烷氧基、硝基、芳香基、醛基、酯基、羧基中的至少一种;r2选自烷基或芳香基。
本发明实施例一为3-碘-2-氨基吡啶的制备:在25毫升的反应管中加入2-氟-3-碘吡啶(1mmol,223.0mg),乙脒盐酸盐(1.2mmol,113.4mg),naoh(2.5mmol,100mg),h2o(0.5ml)和二甲亚砜(2.5ml)。反应在130℃下进行,反应时间为24小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物3-碘吡啶-2-胺,产率为95%。
通过气相色谱-质谱联用仪(gaschromatography-massspectrometry,gc-ms)和核磁共振谱仪(nuclearmagneticresonancespectrometer,nmr)对制得的产物进行定性分析。
其中,gc-ms采用电子轰击离子(electronimpactionization,ei)源,电离电压为70ev,测得m/z.220,127,93,66。
1hnmr(400mhz,cdcl3),如图1所示,δ8.02(dd,j=4.8,1.5hz,1h),7.86(dd,j=7.7,1.6hz,1h),6.39(dd,j=7.7,4.9hz,1h),5.00(s,2h);
13cnmr(100mhz,cdcl3),如图2所示,δ157.5,147.8,147.1,115.3,77.7。
本发明实施例二为6-碘-2-氨基吡啶的制备:在25毫升的反应管中加入2-氟-6-碘吡啶(1mmol),乙脒盐酸盐(1.3mmol),koh(2.5mmol),h2o(0.5ml)和二甲亚砜(2.5ml)。反应在130℃下进行,反应时间为24小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为93%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.220,127,93,66。
1hnmr(600mhz,cdcl3)δ7.10–6.92(m,2h),6.41(d,j=7.6hz,1h),4.64(s,2h);
13cnmr(150mhz,cdcl3)δ158.5,138.9,124.3,115.7,107.3。
本发明实施例三为3-氯-5-氟-2-氨基吡啶的制备:在25毫升的反应管中加入2-氟-3-氯-5-氟-吡啶(1mmol),叔丁脒盐酸盐(1mmol),甲醇钠(2mmol),h2o(0.5ml)和n,n-二甲基甲酰胺(2.5ml)。反应在140℃下进行,反应时间为30小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为87%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.146,119,111,84。
1hnmr(400mhz,cdcl3)δ7.89(d,j=2.6hz,1h),7.34(dd,j=7.5,2.6hz,1h),4.81(s,2h);
13cnmr(100mhz,cdcl3)δ153.9(s,1c),151.5(d,jc-f=12.02hz,1c),133.2(d,jc-f=24.04hz,1c),125.0(d,jc-f=22.83hz,1c),144.4(s,1c)。
本发明实施例四为3-硝基-2-氨基吡啶的制备:在25毫升的反应管中加入2-氟-3-硝基-吡啶(1mmol),丙脒盐酸盐(2mmol),叔丁醇钠(3mmol),h2o(0.5ml)和n-吡咯烷酮(2.5ml)。反应在100℃下进行,反应时间为36小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为92%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.139,122,93,66。
1hnmr(600mhz,乙腈(acetone))δ8.51-8.23(m,2h),7.46(s,2h),6.80(dd,j=8.3,4.5hz,1h);
13cnmr(150mhz,acetone)δ156.9,154.9,135.6,128.6,113.7。
本发明实施例五为4-甲基-5-硝基-2-氨基吡啶的制备:在25毫升的反应管中加入2-氟-4-甲基-5硝基-吡啶(1mmol),苯脒盐酸盐(2mmol),叔丁醇钠(3mmol),h2o(0.5ml)和甲苯(2.5ml)。反应在130℃下进行,反应时间为28小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为95%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z153,136,80。
1hnmr(600mhz,cdcl3+d6-dmso)δ8.76(s,1h),7.29(s,2h),6.31(s,1h),2.47(s,3h);
13cnmr(150mhz,cdcl3+d6-dmso)δ160.5,146.5,142.2,133.6,106.9,19.2。
本发明实施例六为4-氟-5-氯-2-氨基嘧啶的制备:在25毫升的反应管中加入2-氟-4-氟-5-氯-嘧啶(1mmol),苯脒盐酸盐(2mmol),叔丁醇钠(3mmol),h2o(0.5ml)和甲苯(2.5ml)。反应在130℃下进行,反应时间为28小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为91%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.147,120,114,40。
1hnmr(600mhz,acetone)δ8.13(s,1h),7.16(d,j=180.3hz,2h);
13cnmr(150mhz,acetone)δ163.5(d,jc-f=18.6hz,1c),162.1(d,jc-f=210.65hz,1c),156.5(d,jc-f=57.08hz,1c),111.2(d,jc-f=6.49hz,1c)。
本发明实施例七为6-溴-2-氨基-吡啶的制备:在25毫升的反应管中加入2-氟-6-溴-吡啶(1mmol),戊脒盐酸盐(2mmol),叔丁醇钠(3mmol),h2o(0.5ml)和二乙二醇二甲醚(2.5ml)。反应在150℃下进行,反应时间为24小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为93%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.174,172,93,63。
1hnmr(600mhz,cdcl3)δ7.21(t,j=7.8hz,1h),6.74(d,j=7.5hz,1h),6.37(d,j=8.1hz,1h),4.93(s,2h);
13cnmr(151mhz,cdcl3)δ158.73,139.76,139.70,116.57,106.77。
本发明实施例八为3-溴-2-氨基-吡啶的制备:在25毫升的反应管中加入2-氟-3-溴-吡啶(1mmol),戊脒盐酸盐(1.2mmol),叔丁醇钠(3mmol),h2o(0.5ml)二乙二醇二甲醚(2.5ml)。反应在130℃下进行,反应时间为24小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为89%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.174,172,145,93。
1hnmr(600mhz,cdcl3)δ7.99(d,j=4.8hz,1h),7.68–7.56(m,1h),6.51(dd,j=7.7,4.9hz,1h),5.11(s,2h);
13cnmr(150mhz,cdcl3)δ155.60,146.88,140.26,114.82,104.38。
本发明实施例九为5-溴-2-氨基-吡啶的制备:在25毫升的反应管中加入2-氟-5-溴-吡啶(1mmol),乙脒盐酸盐(1.2mmol),氢氧化钠(3mmol),h2o(0.5ml)二甲基亚砜(2.5ml)。反应在130℃下进行,反应时间为24小时,待反应完成后,冷却至室温。
加入乙酸乙酯10ml淬灭反应,加入6ml饱和食盐水洗涤,分出有机相,再用乙酸乙酯萃取水相3次(每次乙酸乙酯用量为6ml)合并有机相,加入无水硫酸钠干燥,经减压蒸馏除去溶剂包括有机溶剂和无机溶剂,所述有机溶剂,再经柱层析分离得到目标产物,产率为86%。
通过gc-ms和nmr进行定性表征:
gc-ms(ei,70ev)m/z.172,145,93,66。
1hnmr(600mhz,acetone)δ7.85(d,j=2.4hz,1h),7.36(dd,j=8.8,2.5hz,1h),6.40(dd,j=8.8,0.5hz,1h),5.52(s,2h);
13cnmr(150mhz,acetone)δ158.72,148.30,139.33,109.90,106.06。
本发明实施例十为6-氯-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-6-氯-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为79%;定性表征数据如下:
gc-ms(ei,70ev)m/z.128,101,93,66。1hnmr(600mhz,acetone)δ7.38(t,j=7.8hz,1h),6.54(d,j=7.5hz,1h),6.48(d,j=8.1hz,1h),5.83(s,2h).13cnmr(150mhz,acetone)δ160.95,149.83,140.64,111.92,107.08。
本发明实施例十一为5-氯-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-5-氯-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为84%;定性表征数据如下:
gc-ms(ei,70ev)m/z.128,101,93,66。1hnmr(600mhz,acetone)δ7.77(d,j=3hz,1h),7.25(dd,j=8.8,2.7hz,1h),6.43(dd,j=8.8,0.6hz,1h),5.49(s,2h);13cnmr(150mhz,acetone)δ158.53,146.00,136.75,118.73,109.20。
本发明实施例十二为3-氟-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-3-氟-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为81%;定性表征数据如下:
gc-ms(ei,70ev)m/z.112,85,57。
1hnmr(400mhz,cdcl3)δ7.82(d,j=5.0hz,1h),7.17(ddd,j=10.9,7.9,1.3hz,1h),6.59(ddd,j=8.3,5.0,3.5hz,1h),4.78(s,2h);
13cnmr(100mhz,cdcl3)δ148.53(d,jc-f=12.52hz,1c),146.91(d,jc-f=252.5hz,1c),142.84(d,jc-f=5.96hz,1c),121.39(d,jc-f=15.35hz,1c),113.79(d,jc-f=1.72hz,1c)。
本发明实施例十三为3,5-二溴-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-3,5-二溴-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为83%;定性表征数据如下:
gc-ms(ei,70ev)m/z.254,252,249,170。
1hnmr(600mhz,acetone)δ8.03(d,j=2.4hz,1h),7.88(d,j=1.8hz,1h),5.96(s,2h);
13cnmr(150mhz,acetone)δ156.33,148.40,142.31,106.14,104.48。
本发明实施例十四为6-三氟甲基-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-6-三氟甲基-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为82%;定性表征数据如下:
gc-ms(ei,70ev)m/z.162,143,135,115,93,66。
1hnmr(600mhz,acetone)δ7.46(t,j=7.8hz,1h),6.80(d,j=7.2hz,1h),6.65(d,j=8.4hz,1h),5.84(s,2h);
13cnmr(150mhz,acetone)δ160.049(s,1c),145.82(dd,jc-f=66.59,66.44hz,1c),138.23(s,1c),122.00(d,jc-f=273.31hz,1c),111.756(s,1c),108.38(dd,jc-f=6.80,6.80hz,1c)。
本发明实施例十五为4-三氟甲基-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-4-三氟甲基-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为61%;定性表征数据如下:
gc-ms(ei,70ev)m/z.162,143,135,116。
1hnmr(600mhz,acetone)δ8.17(d,j=4.8hz,1h),6.81(s,1h),6.77(d,j=4.8hz,1h),5.94(s,2h);
13cnmr(150mhz,acetone)δ161.31(s,1c),150.68(s,1c),139.56(dd,jc-f=65.53,65.69hz,1c),124.36(dd,jc-f=544.36,544.51hz,1c),108.03(t,jc-f=3.322hz,1c),104.25(t,jc-f=4.228hz,1c)。
本发明实施例十六为3-氯-5-三氟甲基-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-3-氯-5-三氟甲基-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为94%;定性表征数据如下:
gc-ms(ei,70ev)m/z.196,177,169,161,141。
1hnmr(151mhz,acetone)δ16.99(d,j=1.8hz,1h),13.80(d,j=3.9hz,1h),3.66(s,2h);
13cnmr(150mhz,acetone)δ158.18(s,1c),144.10(dd,jc-f=8.91,8.76hz,1c),133.41(dd,jc-f=.80,6.80hz,1c),124.07(dd,jc-f=540.28,540.28hz,1c),115.59(ddjc-f=66.44,66.59hz,1c),113.48(s,1c)。
本发明实施例十七为5-硝基-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-5-硝基-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为98%;定性表征数据如下:
gc-ms(ei,70ev)m/z.139,109,93,66。
1hnmr(600mhz,acetone)δ8.91(d,j=2.8hz,1h),8.20(dd,j=9.2,2.8hz,1h),6.89(s,2h),6.73-6.63(m,1h)。
13cnmr(150mhz,acetone)δ163.81,147.11,136.11,133.11,107.47。
本发明实施例十八为5-硝基-6-甲基-2-氨基-吡啶的制备:其与实施例一的区别仅在于:以2-氟-5-硝基-6-甲基-吡啶替代2-氟-5-溴-吡啶,制得的目标产物的产率为97%;定性表征数据如下:
gc-ms(ei,70ev)m/z153,136,80.1hnmr(600mhz,cdcl3)δ7.63(d,j=9.1hz,1h),6.90(s,2h),5.92(d,j=9.1hz,1h),2.15(s,3h);13cnmr(150mhz,cdcl3)δ151.71,146.56,125.22,124.74,96.21,15.49.
本发明实施例十九为2-氨基吡嗪的制备:其与实施例一的区别仅在于:以2-氟-吡嗪替代2-氟-5-溴-吡啶,制得的目标产物的产率为52%;定性表征数据如下:
gc-ms(ei,70ev)m/z.95,68,41。
1hnmr(600mhz,acetone)δ7.98(d,j=1.2hz,1h),7.89(dd,j=2.4,2.4hz,1h),7.73(d,j=2.7hz,1h),5.84(s,2h);
13cnmr(150mhz,acetone)δ156.97,142.74,133.5,133.4。
与现有技术相比,本发明方案通过无过渡金属催化的方式,以2-氟取代的含氮六元杂环类化合物为原料,以脒盐酸盐作为胺源,使用氢氧化钠等碱性物质作为催化剂进行催化反应,相对于产物来说,原料成本低廉;本发明方案既无需使用价格高昂且难以回收的过渡金属催化剂、氢气等易燃易爆的危险性气体,也无需使用有毒的溴素及氨基锂等难以工业化的化工原料,本发明方案在较简单的工业条件下即可实现生产,安全环保;本发明的合成2-氨基吡啶方法具有转化率和收率高,工艺流程短,反应规模易于扩大,产物分离较简单,成本低,适于工业化生产优势。
综上所述,本发明提供的一种2-氨基取代的含氮六元杂环类化合物的制备方法,该方法合成条件简单、反应步骤少、反应条件温和、所用催化剂廉价、废弃物排放少且官能团耐受性好。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!