Delta-9,11-羟基黄体酮的合成方法与流程
本发明属于化合物合成领域,具体涉及一种delta-9,11-羟基黄体酮的合成方法。
背景技术:
delta-9,11-羟基黄体酮的传统合成方法为(合成路线如下所示):以奥式物为起始原料,经过上脱溴,然后发酵进行11位羟化,再消去11位羟基得到目标产品。但是该工艺的起始原料奥式物来自于黄姜提取物皂素,然后再经过多步化学合成才得到奥式物原料,在此过程中存在严重的环境污染以及三废特别是重金属铬难以处理问题,另外在11位羟基消去得到目标产品时也会存在较大的异构体杂质因而严重影响产品质量,在11位羟消去时,往往会引入11,12位双键异构体,该异构体含量最高可达10%左右,而且通常难以精制掉。
技术实现要素:
本发明提供一种delta-9,11-羟基黄体酮的合成方法,该方法以17-β-氰基为起始原料,经过硅醚化,溴甲基化,还原反应等简单的步骤得到目标产物。该起始原料17β-氰基来自于生物发酵产物9-ohad,生产环节绿色环保,而且其成本相对传统工艺极大降低,另外产品质量也比传统工艺大为提高。
所述delta-9,11-羟基黄体酮的合成方法,该方法包括以下步骤:
(1)硅醚反应:17-β-氰基物、氯甲基氯硅烷和催化剂在溶剂中进行硅醚反应,得到硅醚物;
(2)氯甲基化反应:上述硅醚物、lda和浓盐酸在溶剂中进行反应,反应完成后,调节体系ph值,得到氯甲基物;
(3)还原反应:上述氯甲基物、还原剂和浓盐酸在溶剂中进行反应,浓缩、析晶,得到delta-9,11-羟基黄体酮;
具体合成路线如下所示:
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(1)中所述溶剂可以选自二氯甲烷、四氢呋喃和乙腈等中的至少一种;例如,所述溶剂和17-β-氰基物的体积质量比为5~9:1,例如7:1。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(1)中所述催化剂可以选自咪唑、三乙胺和二异丙胺中的至少一种。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(1)中所述17-β-氰基物、催化剂和甲基氯硅烷的质量比为1:0.2~0.4:0.4~0.7,例如质量比为1:0.35:0.55。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(1)中所述氯甲基氯硅烷加入前控制反应体系温度-10~-5℃,加入过程中控制反应体系温度不超过0℃,加入完成后,保持温度-1~0℃,反应1~2小时。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(2)中所述溶剂可以选自四氢呋喃、2-甲基四氢呋喃和二氧六环中的至少一种。优选地,所述溶剂和硅醚物的体积质量比(ml/g)可以为1~2:1。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(2)中以每升溶剂计,所述lda的用量为1~5mol,所述lda以溶液形式滴加入反应体系中,滴加时反应体系温度控制在-60~-50℃,滴加完成后,保温反应1~2小时。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(2)中所述浓盐酸的用量为步骤(2)所述溶剂体积的2~3.5倍,浓盐酸加入后,于15-20℃保温反应4-5小时。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(2)中所述ph值为6-7。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(3)中所述溶剂可以选自甲醇、乙醇和乙腈中的至少一种,所述还原剂可以选自锌粉和铁粉中的至少一种。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(3)中所述溶剂、还原剂、氯甲基物和浓盐酸的用量比(ml/g/g/ml)为6-9:0.2-0.4:1:1。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(3)中所述浓盐酸加入时反应体系的温度为45-55℃,加入完成后保温反应9-10小时。
其中,上述delta-9,11-羟基黄体酮的合成方法,步骤(3)中析晶完成后,还可以进行精制。
与现有技术相比,本发明的有益效果:
以17-β-氰基为起始原料,经过硅醚化,溴甲基化,还原反应等简单的步骤得到目标产物。该起始原料17-β-氰基来自于生物发酵产物9-ohad,生产环节绿色环保,无重金属铬产生;而且其成本相对传统工艺极大降低,另外产品质量也比传统工艺大为提高,产品收率可达到90-95%,hplc纯度99.5%以上。
附图说明
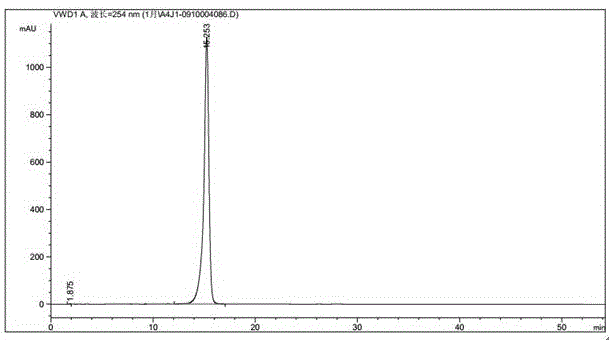
图1是本发明实施例1所得delta-9,11-羟基黄体酮的hplc谱图。
具体实施方式
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
实施例1
1、硅醚反应:
将700ml二氯甲烷,100g的17-β-氰基物,35g咪唑投入反应器中,搅拌溶清,氮气保护下(确保空气中水分不会因为反应体系温度降低而进入反应体系中)降温至-8℃,然后慢速滴加氯甲基氯硅烷55g,滴加过程控制反应体系温度不超过0℃,控温0℃,保温反应1.5小时,取样点板原料反应彻底,即可终止反应,然后负压浓缩至干,再加入10份水降温至0-5℃析晶5小时,抽滤,用水洗涤10-15分钟,抽干烘干,收率97%左右。
2、氯甲基化反应:
在反应釜中抽入150ml四氢呋喃,100g硅醚物,抽真空,再通氮气至常压,搅拌降温,于-60℃缓慢滴加2摩尔每升的lda溶液150ml,滴加完毕,保温反应1.5小时。
将440ml浓盐酸滴加到反应液中,于20℃下保温反应5小时。反应完毕后控制内温20℃下滴加由125g氢氧化钠和200g水配成的氢氧化钠溶液,调节体系ph为7,搅拌15分钟后,复测ph。于真空-0.04mpa到-0.09mpa下负压浓缩至温度升至75℃结束。加入水800g搅拌分散,于45°左右搅拌2小时。离心分离,将湿品放入真空干燥机中,于90℃,真空-0.07mpa到-0.09mpa下干燥12小时,收率95%。
3、还原反应:将800ml甲醇、25g锌粉、100g氯甲基物投入到反应器中,保温30℃下慢慢滴加100ml浓盐酸,滴加完毕搅拌下保温反应10小时,然后50℃下浓缩至干,加入1000g水5℃下水析析晶5小时,抽滤烘干。烘干后的粗品用400ml甲醇和二氯甲烷800ml的混合溶剂进行精制。
本实施例终产品收率90%,hplc纯度99.5%以上。
实施例2
1、硅醚反应:将700ml四氢呋喃,100g的17-β-氰基物和35g三乙胺投入反应器中,搅拌溶清,氮气保护下(确保空气中水分不会因为反应体系温度降低而进入反应体系中)降温至-10℃,然后慢速滴加氯甲基氯硅烷55g,滴加过程控制反应体系温度不超过0℃,控温0℃,保温反应1小时,取样点板原料反应彻底,即可终止反应,然后负压浓缩至干,再加入1000g水降温至3℃析晶5小时,抽滤,用水洗涤15分钟,抽干烘干,收率97%左右。
2、氯甲基化反应:在反应釜中抽入150ml2-甲基四氢呋喃,100g硅醚物。抽真空,再通氮气至常压,搅拌降温,于-60℃缓慢滴加2摩尔每升的lda溶液150ml,滴加完毕,保温反应1.5小时。
将440g浓盐酸滴加到反应液中,于18℃下保温反应4小时。反应完毕后控制内温20℃下滴加由125g氢氧化钠和200g水配成的氢氧化钠溶液,调节体系ph为6,搅拌15分钟后,复测ph。于真空-0.04mpa到-0.09mpa下负压浓缩至温度升至75℃结束。加入水800g搅拌分散,于50℃左右搅拌3小时。离心分离,将湿品放入真空干燥机中,于100℃,真空-0.07mpa到-0.09mpa下干燥12小时,收率95%。
3、还原反应:将800ml乙醇、25g铁粉和100g氯甲基物投入到反应器中,保温30°下慢慢滴加100ml浓盐酸,滴加完毕搅拌下保温反应10小时,然后50℃下浓缩至干,加入1000g水5℃下,水析析晶5小时,抽滤烘干。烘干后的粗品用400ml甲醇和二氯甲烷800ml的混合溶剂进行精制。
终产品收率90%,hplc纯度99.5%以上。
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!