用于多靶标扩增的避免二聚体的多重聚合酶链式反应的制作方法
相关申请的交叉引用
本申请要求在2017年3月9日提交的题为“用于多靶标扩增的避免二聚体的多重聚合酶链式反应”的美国申请no.:62/469,309的优先权,其通过引用并入本文。
背景技术:
聚合酶链反应(pcr)已经使dna扩增的使用能够用于多种用途,包括分子诊断测试。已经开发了多重pcr方法来扩增样品中的多种核酸以能够检测和鉴定多种靶序列。使用多重pcr,必须选择将特异性结合不同靶序列的多个引物以允许扩增那些序列。然而,多个引物的使用已证明在常规的多重pcr方法学的情况下是有问题的,所述方法学需要优化引物组、温度条件和酶,以满足对于不同引物可能需要的不同条件。因此,需要精心的计划和测试以找到用于常规多重pcr方法的兼容的引物组。
与多重pcr相关的挑战由引物-二聚体的形成而加剧,当使用多个引物时经常产生引物-二聚体。引物-二聚体的形成可能产生一些靶序列非常有效地扩增,而其它靶序列非常低效地扩增或根本不能扩增的结果。这种不均匀扩增的可能性也使得精确地进行终点(end-point)定量分析是困难的至不可能的,并且需要大量的引物优化以确定哪些引物可以在特定的多重pcr测定中适当地组合。即使在以下方法中引物-二聚体的问题仍可能存在:在其中在早于使用共用引物进一步扩增的初始pcr循环期间使用基因特异性引物以富集靶序列的方法。尽管如此,目前的范例是引物的选择必须被优化以实现多重pcr的理想执行[例如,canzar等人,bioinformatics,2016,1-3(“canzar”)]。
因此,仍然需要一种方法,其能够使用多个引物扩增多个靶序列,同时避免引物-二聚体形成的负面影响,而不需要繁重且昂贵的引物优化。
发明概述
在一个实施方案中,本公开涉及一种方法,其包括以下步骤:使用反向引物混合物从含有至少一种靶序列的mrna中反转录cdna的至少一个第一条链,形成第一条链cdna;其中所述反向引物混合物含有至少一个反向引物,所述反向引物被配置以将反向共用(common)引物结合位点掺入cdna的每个第一条链中;选择每个第一条链cdna;使用正向引物混合物从所述cdna的至少一个第一条链中的每个中合成cdna的至少一个第二条链,形成至少一个第一条链:第二条链复合物;其中所述正向引物混合物含有至少一个正向引物,每个正向引物被配置以结合特定的cdna第一条链,并将正向共用引物结合位点掺入cdna的每个第二条链中;选择cdna的每个第二条链;选择每个第一条链:第二条链复合物;使用与至少一个反向共用引物结合位点结合的反向共用引物并使用与至少一个正向共用引物结合位点结合的正向共用引物扩增cdna链;和选择经扩增的cdna链。在某些实施方案中,所述方法进一步包括使用与至少一个反向共用引物结合位点结合的反向共用引物并使用与至少一个正向共用引物结合位点结合的正向共用引物扩增经扩增的cdna链的步骤。在所述方法的某些实施方案中,所述反向引物混合物包含至少一个反向引物,其中所述至少一个反向引物包含掺入到每个第一条cdna链中作为鉴定标志物的另外的核苷酸。在所述方法的某些实施方案中,所述正向引物混合物包含至少一个正向引物,其中所述至少一个正向引物包含掺入到每个第二cdna链中作为鉴定标志物的另外的核苷酸。在所述方法的某些实施方案中,每个选择包括使用磁珠从引物混合物中分离cdna链。在所述方法的某些实施方案中,每个选择包括通过柱纯化从引物混合物中分离cdna链。在所述方法的某些实施方案中,每个选择包括引物混合物的酶切割。在某些实施方案中,所述第一条链cdna包含第一条链cdna:rna复合物。在某些实施方案中,本公开涉及诊断受试者中疾病的存在的方法,所述方法包括:提供来自所述受试者的样品,所述样品怀疑含有疾病因子,其中所述疾病因子的特征在于靶序列;对样品中的核酸进行本段所述的方法;对经扩增的dna链测序;和检测来自所述疾病因子的靶序列。在某些实施方案中,本公开涉及产生受试者的免疫状态概况的方法,所述方法包括:对来自受试者的白细胞样品的核酸进行本段所述的方法;对经扩增的dna链测序;并鉴定和定量代表t细胞受体、抗体和mhc重排的一个或更多个dna序列以生成受试者的免疫状态概况。在某些实施方案中,所述mrna获自单个细胞。
在某些实施方案中,本公开涉及一种方法,其包括以下步骤:使用第一引物混合物从含有至少一个靶序列的基因组dna合成dna的至少一个第一条链,形成第一条链:dna复合物;其中所述第一引物混合物含有至少一个第一引物,每个第一引物被配置以结合特定的靶序列并将第一共用引物结合位点掺入dna的每个第一条链中;选择每个第一条链:dna复合物;使用第二引物混合物从dna的至少一个第一条链中的每个合成dna的至少一个第二条链,形成第一条链:第二条链复合物;其中所述第二引物混合物含有至少一个第二引物,每个第二引物被配置以结合特定的dna第一条链,并将第二共用引物结合位点掺入dna的每个第二条链中;选择每个第一条链:第二条链复合物;使用与至少一个第一共用引物结合位点结合的第一共用引物并使用与至少一个第二共用引物结合位点结合的第二共用引物扩增dna链;和选择经扩增的dna链。在某些实施方案中,所述基因组dna从单个细胞获得。在某些实施方案中,本公开涉及诊断受试者中疾病的存在的方法,所述方法包括:提供来自受试者的样品,所述样品怀疑含有疾病因子,其中所述疾病因子的特征在于靶序列;从样品中分离核酸;对分离的核酸进行本段所述的方法;对经扩增的dna链测序;并检测来自所述疾病因子的靶序列。在某些实施方案中,本公开涉及产生受试者的免疫状态概况的方法,所述方法包括:对来自受试者的白细胞样品的核酸进行本段所述的方法;对经扩增的dna链测序;并鉴定和定量代表t细胞受体、抗体和mhc重排的一个或更多个dna序列以生成受试者的免疫状态概况。
在所述方法的某些实施方案中:所述第一引物混合物为反向引物混合物,每个第一引物为反向引物,每个第一共用引物为反向共用引物,且每个第一共用引物结合位点为反向共用引物结合位点;且所述第二引物混合物为正向引物混合物,每个第二引物为正向引物,每个第二共用引物为正向共用引物,且每个第二共用引物结合位点为正向共用引物结合位点。在所述方法的某些实施方案中:所述第一引物混合物为正向引物混合物,每个第一引物为正向引物,每个第一共用引物为正向共用引物,且每个第一共用引物结合位点为正向共用引物结合位点;且所述第二引物混合物为反向引物混合物,每个第二引物为反向引物,每个第二共用引物为反向共用引物,且每个第二共用引物结合位点为反向共用引物结合位点。在所述方法的某些实施方案中:所述第一引物混合物为包含至少一个正向引物和至少一个反向引物的引物混合物,每个第一引物为正向或反向引物,每个第一共用引物为正向或反向共用引物,且每个第一共用引物结合位点为正向或反向共用引物结合位点;且所述第二引物混合物包含至少一个正向引物和至少一个反向引物,其中第一引物混合物中不包括第二引物混合物中的正向或反向引物,每个第二共用引物为正向或反向共用引物,且每个第二共用引物结合位点为正向或反向共用引物结合位点。在某些实施方案中,所述方法还包括使用与至少一个反向共用引物结合位点结合的反向共用引物并使用与至少一个正向共用引物结合位点结合的正向共用引物扩增经扩增的dna链的步骤。在所述方法的某些实施方案中,所述第一引物混合物包含含有另外的核苷酸的引物,所述另外的核苷酸作为鉴定标志物掺入到每个第一条dna链中。在所述方法的某些实施方案中,所述第二引物混合物包含含有另外的核苷酸的引物,所述另外的核苷酸作为鉴定标志物掺入到每个第二条dna链中。在所述方法的某些实施方案中,每个选择包括使用磁珠从引物混合物中分离dna链。在所述方法的某些实施方案中,每个选择包括通过柱纯化从引物混合物中分离dna链。在所述方法的某些实施方案中,每个选择包括引物混合物的酶切割。
附图的简要说明
参考以下附图可以更好理解本公开。附图的元件不必相对于彼此成比例,而重点在于清楚地说明本公开的原理。此外,遍及几个视图中相似的参考数字标出相应的部分。
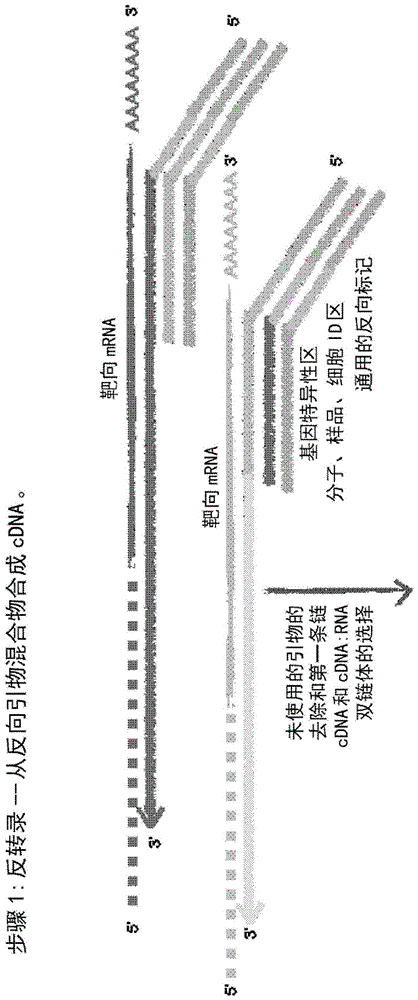
图1a-1c说明使用rna模板的dam-pcr方法学的示例性实施方案的步骤。图1a描述利用反转录的单循环第一条链cdna合成(第一条链标记(tagging))的步骤,未使用的反向引物混合物的去除和第一条链cdna的选择。图1b描述单循环第二条链dna合成(第二条链标记)的步骤,未使用的正向引物混合物的去除和第一条链:第二条链dna双链体的选择。图1c描述使用共用引物扩增靶标的步骤和重复选择和扩增的可选的步骤。
图2a-2c说明使用基因组dna(“gdna”)模板的dam-pcr方法学的示例性实施方案的步骤。图2a描述用第一引物混合物的第一条链dna合成(第一条链标记)的步骤,未使用的第一引物混合物的去除和第一条链:dna双链体的选择。图2b描述第二条链dna合成的步骤和未使用的第二引物混合物的去除。图2c描述使用共用引物扩增靶标的步骤和重复选择和扩增的可选的步骤。
图3a和3b说明引物-二聚体形成对产出(output)的影响。图3a是凝胶,其证明在单个细胞使用arm-pcr的引物-二聚体形成的普遍性和由于引物-二聚体形成,初级产物条带强度相应降低。凝胶上的百分比表明甚至在最终文库清除之后在对残余引物-二聚体产物测序上浪费的测序读数的百分比,并且通过分析每个细胞的数据的下一代测序数据获得。每个单独的泳道代表用覆盖α和βtcr基因座和另外的表型标志物的arm-pcr多重混合物扩增后的唯一的单个细胞。图3b是表格,其说明在pcr期间包括正向和反向引物的混合物中的引物对之间引物-二聚体形成的可能性,以及由于引物-二聚体形成而浪费的读数,所述读数可以占到用于样品的读数的多达31%。
图4a和4b说明具有少至一个碱基对重叠的引物-二聚体的形成。图4a是凝胶,其显示在几个循环条件下进行arm-pcr所导致的引物-二聚体的有意形成,所述循环条件包括在缺乏模板情况下的rt-单个细胞方案(scrt)、无rt单个细胞方案(scpcr)和ir-10-10方案(ir1010)。泳道1-3代表单核苷酸重叠的引物-二聚体形成,尽管使用该方案,但引物-二聚体依然形成于引物itrav_2和itrac之间。泳道4-6代表引物itrav_2和il-17f-ri-09之间4个核苷酸重叠的引物-二聚体形成,尽管使用该方案,但引物-二聚体依然形成。泳道6-9代表引物itrav_2和bcl6ri_md_11之间6个核苷酸重叠的引物-二聚体形成,尽管使用该方案,但引物-二聚体依然形成。泳道10-12代表引物itrav_40和bcl6ri_md_11之间6个核苷酸重叠的引物-二聚体形成,尽管使用该方案,但引物-二聚体依然形成。图4b说明某些引物-二聚体中的特异性重叠碱基对。还提供图3a和3b中证明的通过arm-pcr扩增的单个细胞的测序结果中引物-二聚体产物的等级,其中“第1高”代表ngs测序结果中最高等级引物-二聚体频率,其中“第2高”代表ngs测序结果中第二最丰富的引物-二聚体频率,等等。
图5是凝胶,其证明具有高引物-二聚体倾向的单对引物可以消除期望的产物条带的扩增。在描述的琼脂糖凝胶的第6泳道中,成功的pcr中包括扩增靶标il-10的4个引物。如泳道7所示,t-bet_fi的加入消除目的条带的扩增。同样,如泳道9所示,覆盖t-bet的4个引物和包括il-17a-fo的多重引物混合物成功扩增了靶标。然而,如泳道10显示加入一个破坏性引物,foxp3ri反向内部引物消除初级产物条带的扩增。
图6是凝胶,其证明调整退火温度不会去除引物-二聚体的影响。在范围从59.9℃至66℃的几个退火温度下测试已知形成引物-二聚体的4个不同的引物对以去除引物-二聚体的形成,均为完全无用。
图7a和7b是凝胶,其说明用arm-pcr对比dam-pcr对引物-二聚体形成的影响。图7a是凝胶,其证明dam-pcr使用珠子选择克服抑制的引物-二聚体的影响。泳道1-2是arm-pcr对照。泳道3-4是arm-pcr对照,其具有已知引起二聚化的一对引物的掺混(spike-in)。泳道5-6是没有引物-二聚体对掺混的单循环dam-pcr。泳道7-8是具有引物-二聚体对掺混的单循环dam-pcr。泳道9-10是具有没有引物-二聚体对掺混的线性扩增的dam-pcr。泳道11-12是具有引物-二聚体掺混的线性扩增的dam-pcr。泳道13是阴性对照。图7b是凝胶,其证明dam-pcr在用dam-pcr的共用正向和反向引物的第一轮扩增之后或在arm-pcr的rt-pcr的第一轮之后,使用转移而不是珠子选择来克服抑制的引物-二聚体的影响。泳道1-2是arm-pcr对照。泳道3-4是具有已知引起二聚化的一对引物的掺混的arm-pcr对照。泳道5-6是没有引物-二聚体对掺混的单循环dam-pcr。泳道7-8是具有引物-二聚体对掺混的单循环dam-pcr。泳道9-10是具有没有引物-二聚体对掺混的线性扩增的dam-pcr。泳道11-12是具有有引物-二聚体对掺混的线性扩增的dam-pcr。泳道13是阴性对照。
图8是比较使用dam-pcr对比arm-pcr的单个细胞扩增的凝胶。泳道1-6代表用dam-pcr扩增的单个细胞,而泳道7-14代表用arm-pcr扩增的单个细胞。dam-pcr扩增的细胞具有相似的终点强度,且缺乏引物-二聚体,而arm-pcr扩增的细胞在终点pcr中证明差异并且显示引物-二聚体扩增。
图9是凝胶,其证明去除未使用的引物的选择步骤的能力。使用标有“标准曲线”的泳道以评估第一条链标记和第二条链标记之间以及第二条链标记和扩增之间的引物遗留(carryover)量。泳道1和2代表磁珠选择步骤,其进行两次,并且当与标准曲线相比时,去除大于99.99%的未使用的引物。泳道3和4代表磁珠选择步骤,其进行一次,并且当与标准曲线相比时,去除大于99.9%的未使用的引物。
图10a-10b显示凝胶,其证明用gdna与不同的多重引物混合物的dam-pcr。图10a显示用于tcrβ基因座扩增的arm-pcr(泳道1-4)对比dam-pcr(泳道5-9)。图10b显示在两个gdna输入量下,用肿瘤多重面板的arm-pcr对比dam-pcr。泳道1-7代表200nggdna输入,对于泳道1-3进行arm-pcr,对于泳道4-7进行dam-pcr。泳道8-14代表540ng输入,对于泳道8-10进行arm-pcr,对于泳道11-14进行dam-pcr。
图11是描述使用被设计以覆盖两个外显子的引物的gdna的常规多重pcr的示意图。这种设计的类型在两个扩增子之间引入重叠以使基因在生物信息处理期间组装。由于需要生成重叠的另外的引物的兼容性,这种方法产生非靶标扩增,导致竞争性的较短pcr产物。
图12是描述gdna的dam-pcr以及与常规多重pcr相比dam-pcr相关的益处的示意图。特别地,在第一循环标记期间,可以使用一组引物覆盖较大的外显子产物。在第一引物混合物清除之后,第二引物混合物包括仅与它们各自的第一条链产物相互作用的内部引物。由于这些引物不参与任何扩增,并且在标记期间仅使用一次,因此没有产生竞争性的较短pcr产物。
图13是覆盖长的gdna基因靶标同时包括重叠部分(具体地覆盖hla靶标)的arm-pcr多重pcr策略和dam-pcr策略的琼脂糖凝胶比较。为了清楚起见,在琼脂糖凝胶上方提供图解以展示在琼脂糖凝胶中所引用的引物的位置。泳道1显示当arm-pcr仅用于扩增基因靶标a时的扩增模式。泳道2显示当arm-pcr仅用于扩增基因靶标b时的扩增模式。泳道3显示来自由于努力产生重叠区段的t2正向和t1反向的相互作用的一种潜在的脱靶扩增的扩增模式。泳道4显示t1正向引物和t2反向引物的长的扩增产物。泳道5-6显示从完全多重的混合物中进行的arm-pcr扩增。结果主要由不太理想的短的脱靶扩增产物占据主导地位。由于该短的产物与期望的产物竞争dna聚合酶活性,对于仅靶标a(从t1正向和t1反向产生),仅靶标b(从t2正向和t2反向产生)及靶标a和靶标b一起(从t1正向和t2反向产生)的基因靶标是模糊的且几乎不存在。泳道7显示当dam-pcr在第一循环标记期间用t1反向和在第二循环标记期间用t1正向用于仅扩增基因靶标a的扩增模式。泳道8显示当dam-pcr在第一循环标记期间用t2反向和在第二循环标记期间用t2正向用于仅扩增基因靶标b的扩增模式。泳道9-10显示完全多重dam-pcr策略的结果。在第一轮标记中,t1正向和t2反向用于生成更长的第一条链产物。在第二轮标记中,t1反向和t2正向与在第一条链标记过程中生成的第一条链产物独立地相互作用以合成第二条链。在第二条链标记、清除并用对第一轮靶标共用的一对引物扩增后,没有竞争性的短的产物,且可以实现期望的产物的扩增。
详细描述
本公开涉及用于扩增核酸的方法,所述方法避免与引物-二聚体形成相关的问题。本方法在本文中称为避免二聚体的多重聚合酶链式反应(dam-pcr)。本文公开的所述方法通常包括以下步骤:反转录dna(例如来自rna样品的cdna)的至少一个第一条链,其中dna的每个第一条链掺入反向共用引物结合位点;选择dna的每个第一条链;从dna的至少一个第一条链中的每个中合成dna的至少一个第二条链,其中cdna的每个第二条链掺入正向共用引物结合位点;选择cdna的每个第二条链;以及使用共用引物扩增dna链。或者,所述方法可以使用gdna模板进行。由于在扩增前dna链的选择和引物的去除,本文所述的方法避免引物-二聚体形成,并与常规的多重pcr方法相比,允许更高的灵敏度和效率。
如本文所用,“疾病”意指由所述因子引起的或与所述因子相关的感染、症状或病况。
如本文所用,“疾病因子”意指掺入核酸序列并在受试者中引起或促成疾病的任何生物体,所述生物体不论形式,包括但不限于细菌、癌细胞、病毒或寄生虫。
如本文所用,“第一引物混合物”意指包含被配置以与gdna结合的至少一个反向引物和/或至少一个正向引物的混合物。
如本文所用,“正向引物混合物”意指包含至少一个正向引物的混合物。
如本文所用,“鉴定标志物”意指用作标签以鉴定mrna或gdna的特定样品、核酸链或单个细胞来源的核苷酸序列。
如本文所用,“反向引物混合物”意指包含至少一个反向引物的混合物。
如本文所用,“样品”意指包含dna或rna的材料。
如本文所用,“第二引物混合物”意指包含被配置以与dna的至少一个第一条链结合的至少一个反向引物和/或至少一个正向引物的混合物。
如本文所用,“受试者”意指哺乳动物,优选人。
常规的多重pcr方法包括选择步骤(即,引物混合物的去除或dna链的分离),如果有的话,所述选择步骤仅在扩增子从pcr产生之后(例如,arm-pcr[wo/2009/124293]、tem-pcr[wo/2005/038039],其中的每一个进一步需要使用嵌套引物)。然而,如果在pcr完成后进行选择,申请人显示引物-二聚体将不仅有机会在最初的几个pcr循环中形成,引物-二聚体还将降低反应的敏感性,因为引物-二聚体将在pcr反应期间持续地与靶向序列竞争dna聚合酶活性。此外,由于分子量和电荷的相似性,扩增子和引物-二聚体将难以分离。如以下实施例中所证明的,在极端情况下,引物-二聚体的扩增占据反应的主导地位,完全消除靶序列的扩增。与产生更长的产物相比,dna聚合酶更擅长产生并结合至较短的扩增子,从而进一步加剧引物-二聚体的生成。在形成引物-二聚体但不完全抑制靶序列扩增的情况下,由于靶序列和引物-二聚体之间的竞争,期望的扩增子的量仍然减少,降低了反应的整体敏感性并生成产物,当产物被继续进行测序时,导致测序读数丢失和不必要的花费。
申请人主张目前多重优化的范例固有地有缺陷,因为引物-二聚体可以形成具有少至1bp的重叠。因此,预测引物-二聚体是不可能的,必须使用新的方法学解决。在此,申请人显示当在反转录之后而不是pcr之后进行选择时,从引物混合物中分离期望的dna链的效率(本文中称为“选择”)要好得多。如果使用相对长的寡核苷酸,诸如当制备下一代测序(ngs)-兼容的文库时,引物-二聚体的形成可以产生长度约为200bp的产物,使得从期望的产物条带中分离更加困难并且效率更低。然而,将选择步骤置于反转录之后意味着从单个引物分离约2kbrna:dna双链体(通常<80bp),这是对于实现来说简单得多的分离。在所述方法中第二条链合成后的额外的选择也比pcr后的选择更容易,因为没有形成引物-二聚体,因此,分离是在第一条链dna:dna双链体和较短的引物(<80bp)之间。总之,如果等到pcr之后从引物-二聚体分离或拯救扩增子,则已经失去对单个细胞应用至关重要的灵敏度。相反,通过防止反向引物混合物与正向引物混合物相互作用(例如,反转录和第二链cdna合成的分离)或通过防止第一引物混合物与第二引物混合物(如与gdna在一起)相互作用,并通过在任何pcr步骤之前去除反向引物或正向引物混合物(或去除如与gdna一起使用的第一引物混合物或第二引物混合物),消除了引物-二聚体形成的可能性,并且将反应的能量集中在所期望的产物的扩增上。
作为额外的支持,申请人主张单个细胞可以被认为是具有不同基因表达模式的动态模板。对于单个细胞,鉴于变化的模板数量的存在与否,不可能预测多重系统将如何响应。如果缺少某个基因,可能形成引物-二聚体,否则在模板存在下将不形成引物-二聚体,使得由于单个细胞水平上的表达的变化,引物设计是不可能的。
为了克服这些困难,申请人已经开发了一种在其中避免引物-二聚体的方法学,在本文中称为避免二聚体的多重聚合酶链式反应(dam-pcr)体,其是针对rna的多步骤多重反转录(rt)pcr或针对gdna的多步骤多重pcr。反转录步骤与第二条链dna合成和使用通用的引物的pcr扩增分开进行。同样对于gdna,链标记是通过第一引物混合物进行的,与用第二引物混合物进行的第二条链标记分开的。通过在每一步骤选择合成的dna,并用一个标记和延伸循环来标记第一条链cdna或gdna,避免了形成引物-二聚体的倾向,并且将dna聚合酶活性集中于扩增感兴趣的靶标而不是引物-二聚体,大大提高了扩增灵敏度。使用rna模板的本公开的方法的一个示例性实施方案显示在图1a-1c,使用gdna模板的本公开的方法的一个示例性实施方案显示在图2a-2c。
在反转录过程中,使用一个或更多个含有3′基因特异性和5′通用的结合位点两者的反向引物用于第一条链cdna合成。反转录后,引物-二聚体减轻的一个关键是非常精确地选择第一条链cdna:rna复合物(“第一条链:rna复合物”)并从该初级产物条带有效去除序列长度(通常<80bp)的寡核苷酸。如果通过用不同的寡核苷酸标记每个rna来定量rna,这是特别重要的,因为独特(uniquely)标记的引物的遗留将损害精确定量经标记的核酸种类的能力。反转录后,进行选择的步骤以去除未使用的反向引物。“选择”步骤从未使用的引物混合物中分离dna链(例如溶液中的第一cdna链:rna复合物或单链第一条链cdna),并且可以是基于磁珠(例如,固相可逆固定(spri)珠子)的、基于链霉亲和素-生物素珠子的、酶的、基于柱的,通过凝胶纯化或其它物理的、化学的或生物化学的方法,以主动选择dna链,或相反地,以去除未使用的引物和任何dna聚合酶。在这些实施例中,申请人证明去除超过99.99%的未使用的引物的选择效率。
一旦第一条链cdna合成完成,并且通过选择第一条链cdna有效地去除反转录引物,就使用正向引物混合物进行第二条链cdna合成,称为第二条链dna“标记”。退火后dna聚合酶激活的一个循环足以在不存在任何反向引物的情况下用正向引物混合物标记第一条链cdna产物。在该步骤中也可以引入单个核酸种类的标记,因为只进行一个循环,并且在pcr之前去除未使用的引物。此时,也可以进行有限的线性扩增循环(引物退火和延伸和/或在没有反向引物的情况下的等温扩增)以增加产量。然而,太多的另外的退火和延伸循环可能增加在正向混合物中的引物之间形成有害的引物-二聚体的风险,并且对于给定的多重系统可能需要经验上的确定。申请人证明,分别用于第一和第二条链标记的单个循环足以实现扩增,如通过从单个细胞扩增所证实的。在第二链dna合成后,进行第二选择步骤以去除未使用的正向引物混合物。选择步骤从未使用的引物混合物中分离dna链,并且可以是基于磁珠(例如,spri珠子)的、基于链霉亲和素-生物素珠子的、酶的、基于柱的,通过凝胶纯化或其它物理的、化学的或生物化学的方法,以主动选择dna链,或相反地,以去除未使用的引物和任何dna聚合酶。基本的构思与最初的选择相同,即去除引物的复杂性,使得引物-二聚体不能形成,所述引物-二聚体与初级产物和期望的产物竞争。
此时,“经标记的”dna在5′和3′端都含有通用的引物结合位点。通过该阶段去除所有的反向引物和正向引物标记混合物。通用的位点通常是测序接头或ngs选择平台所需的接头的一部分。然而,任何通用的经设计的位点将是适用的。使用一对对通用地“经标记的”第二条链cdna共用的引物进行pcr。这些引物完成指数扩增阶段。由于引物复杂性降低为双引物系统(其中3′末端不是反向互补的,并且其中引物对已经被筛选防止引物-二聚体形成),在该pcr期间引物-二聚体产生的倾向被消除。
在低输入的情况下,例如单个细胞,可能有必要进行第二轮pcr。在这种情况下,如上所述完成第一轮pcr的选择,并用新鲜的酶、缓冲液和dntp进行使用通用的引物对的两步pcr以富集第一轮产物。在所有情况下,进行最终的文库选择,并且所述文库直接准备用于汇集、预测序qc和使用本领域已知技术的测序。
可以对gdna进行修饰;然而,相同的全部原理适用于避免引物-二聚体的产生。使用单一循环变性和退火步骤,第一轮pcr仅用一组多重引物(第一引物混合物)进行。为了提高灵敏度,只要限制混合物之间引物-二聚体的形成,就可以进行线性扩增或等温扩增。如上所述纯化第一条链:dna复合物,并使用第二引物混合物在单个循环中进行第二链合成。为了提高灵敏度,可以进行线性扩增或等温扩增。在第二条链合成后,如上所述进行选择,并如rna过程中所述用一对通用的引物进行pcr。
本文公开的方法可用于检测来自病毒、细菌、真菌、植物和/或动物细胞的核酸的存在和存在的相对量,用于评价医学、环境、食品和其它样品,以鉴定那些样品中的微生物和其它因子。
本文所述方法的一个优点是,与常规多重pcr方法不同,避免了引物-二聚体的形成,这大大简化了引物组的选择,否则引物组将产生能够影响靶序列扩增的引物-二聚体。本文所述方法的另一个优点是避免引物-二聚体的形成导致了下至单个细胞的高度灵敏的多重pcr。使用所公开的方法,可在单个细胞水平上靶向rna。进一步地,通过减少浪费的读数的数量,否则浪费的读数将由于引物-二聚体的存在而发生,每个靶序列的每个读数的成本显著降低。该方法的另一个优点,尤其是涉及用gdna覆盖大基因区段,是可以从重叠扩增子中获得数据,而不会不利地引入降低灵敏度的较短的扩增子副产物。如果使用常规多重pcr的相似策略,这些副产物是不可避免的。dam-pcr方法允许策略性地设计第一和第二引物混合物,允许更长的覆盖,同时限制某些引物之间的相互作用,所述相互作用产生竞争dna聚合酶活性的短扩增子产物。
实施例
材料和方法:dam-pcr和arm-pcr设置
覆盖t细胞受体α基因座、β基因座和另外的表型标志物的引物在相同混合物中或在正向和反向混合物中是多重的,这取决于pcr策略。arm-pcr混合物由相同混合物中的218个正向引物和68个反向引物组成,覆盖247个或更多个靶标。靶标是根据参考序列定义的,但是由于重排的tcr基因座的可变性,对于大多数rna样品,给定样品中靶序列的实际数量通常以千计。对于单个细胞,根据细胞表型,在任何位置可能存在5-10个或更多个靶标。对于dam-pcr,正向混合物与反向混合物分开处理,并且排除与arm-pcr的嵌套部分相关的外部引物,总共107个正向引物和32个反向引物。含有通用的标记的内部引物是混合物所共有的,并包括已知引起引物-二聚体形成的引物对。在两个引物组中,illumina双索引的兼容的测序引物b的一部分与每个正向内部引物连接,而illumina双索引的测序共用引物a的一部分和6个核苷酸的样品条形码序列连接至反向内部引物。在某些实验中,在样品条形码附近包括标记单个核酸种类的20个随机核苷酸的索引。
对于arm-pcr方法,使用来自onesteprt-pcr试剂盒(qiagen,valencia,ca)的286个引物的嵌套引物混合物和试剂,从总rna样品反转录cdna。对于arm-pcr,第一轮rt-pcr(称为“rt-pcr1”或“pcr1”)在以下条件进行:50℃,60分钟;95℃,15分钟;94℃,30秒,60℃,5分钟,72℃,30秒,10个循环;94℃,30秒,72℃,3分钟,10个循环;在72℃,5分钟,4℃保持。在rt-pcr1后,进行0.7×spriselect珠子选择(beckmancoulter,brea,ca),并使用promegagotaqg2热启动pcr混合物(promega,madison,wisconsin)从珠子洗脱核酸产物。用一组公共的引物进行第二轮pcr(称为“pcr2”),所述公共的引物在以下条件下完成illumina接头序列:95℃,3分钟;94℃,30秒,72℃,90秒,30个循环;72℃,5分钟,4℃保持。
对于dam-pcr,使用qiagenonesteprt-pcr试剂盒在反向引物混合物存在下在50℃进行逆反转录步骤240分钟,通过进行两次0.7×spri珠子选择从反向引物混合物分离第一条链cdna。反转录后,在bioradc1000热循环仪中使用promegagotaqg2hotstartdna聚合酶在标记的一个循环或在线性扩增策略中仅使用正向引物混合物(无反向引物)进行第二条链dna合成,针对所述标记的一个循环在以下条件进行:95℃,3分钟初始变性和热启动;60-65℃,退火,每度变化3分钟;和72℃,10分钟延长,以及最终4℃保持;针对线性扩增策略在以下条件下进行:初始变性和热启动95℃,3分钟,随后6个循环的退火和延伸;60℃,5分钟退火,72℃,1分钟延伸以及最后4℃保持。在第二条链dna合成后,第二条链dna:dna双链体使用0.7×spri珠子选择两次从正向引物混合物分离。使用promegagotaqg2热启动pcr混合物从珠子洗脱dna产物,所述pcr混合物含有一对针对在反向和正向引发步骤期间引入的部分接头序列共用的引物。使用与arm-pcr方法的总循环相等的二十个循环的pcr,用通用的引物对扩增cdna:95℃,3分钟;94℃,30秒,72℃,6分钟,10个循环;94℃,30秒,72℃,3分钟,10个循环;72℃,5分钟,并4℃保持。对于dam-pcr,如前所述进行另外的spri珠子选择,并在新鲜酶、缓冲液和dntp存在下进行第二次pcr:95℃,3分钟;94℃,30秒,72℃,90秒,30个循环;72℃,5分钟,并4℃保持。
在第二次pcr反应(即,在arm-pcr的情况下的pcr2)之后,将10μlpcr产物在2.5%琼脂糖凝胶上电泳以评估扩增成功。使用0.7×spri珠子选择以在测序前选择针对arm-pcr和dam-pcr产物的文库。在25μl无核酸酶的水中从珠子上洗脱最终的文库,并用nanodrop(thermoscientific,carlsbad,ca)测量。将等摩尔量的每个文库合并用于测序,除了用引物-二聚体掺混产生的文库。由于文库的可用性,这些文库以一半的量合并。用qubit定量法对合并的文库进行定量,并用生物分析仪进行评估。然后用kappaqpcr定量文库,用10%phix掺混稀释至8pm,并用illuminamiseqv2,500循环试剂盒使用250个配对末端读数测序。类似的策略用于所描述的gdna模板,除了去除了的反转录步骤外。当设计如讨论中所述的第一和第二引物混合物时,使用策略性引物设计以避免短产物的产生。
引物-二聚体的形成对ngs产出的影响:导致对感兴趣的基因的敏感性丧失、浪费的读数,以及因此增加的测序费用
如图3a中的琼脂糖凝胶所示,传统的arm-pcr与优化的多重混合物一起使用来扩增单个细胞。可使用本领域已知的技术分离单个细胞。arm-pcr是嵌套的多重rtpcr,其中产物被“拯救”,例如,在rt-pcr之后,可以从完成的第一扩增反应中取少量样品以提供用于第二扩增的扩增子。
扩增后,将代表每个细胞的文库合并,并在illuminamiseqv2500循环试剂盒上使用250个配对末端读数在测序之前进行另外一轮选择。分析原始数据以确定相对于成功的靶标读数的引物-二聚体的证据。发现引物-二聚体序列占据的读数的百分比与初级产物条带的条带强度成反比(图3b)。例如,对于样品bb-s73,初级产物条带是强的,并且该细胞的测序读数中仅有1%被引物-二聚体占据,而对于样品xh-s28,产物测序读数相对较少,并且89%的该细胞数据由引物-二聚体占据。甚至相对强的产物(诸如样品bb-s25)条带数据被25%的引物-二聚体占据。
与其它方法(诸如rnaseq)相比,靶向测序方法的益处是需要较少的测序深度来覆盖感兴趣的基因,因为感兴趣的基因被特异性靶向和扩增。当分析单个细胞时,如果需要25倍以上的读数来实现与靶向序列方法相同的覆盖,则成本可能非常迅速地变得过高,特别是当每个单个细胞被作为样品处理时。在所述实验中对引物-二聚体的序列的分析揭示多达31%的总测序读数被引物-二聚体占据,浪费了有价值的测序资源,除了在覆盖给定细胞的感兴趣的基因中的灵敏度的损失之外,还导致了过度的成本。然而,如果可以实现与最强扩增子条带相等的扩增,则引物-二聚体废物基本上被消除,同时提供降低测序成本以及增加感兴趣的基因的灵敏度和覆盖的益处。
引物-二聚体的ngs揭示1bp重叠足以形成引物-二聚体,使得不可能通过单独设计除去
为了去除引物-二聚体,申请人通过在不存在模板的情况下用多重混合物进行arm-pcr有目的地生成引物-二聚体(图4a),并使用下一代测序(ngs)技术对所得扩增子进行测序,以突出可能需要重新设计的引物序列。令人惊讶地,在数据中超过几百个不同的相互作用对是明显的,少至1bp的重叠产生二聚产物(图4b)。事实上,1bp重叠引物-二聚体是最常观察到的引物-二聚体对之一。一些潜在的配对基于碱基对互补性和每个引物的3′末端是可预测的,但是1bp重叠是不可能预测的且因此不可能进行周围设计,再次显示目前多重pcr的“优化”引物的范例(例如canzar)的谬误。
然而,对引物-二聚体的单个的克隆的克隆和测序不能提供理解问题的完整程度和可能的相互作用的绝对数量的能力。其在物理观察用ngs的引物-二聚体条带的序列之前是不清楚的,所述ngs不可能“设计排除(out-design)”引物-二聚体。前面的结果(图3a)证明即使在“成功的”多重pcr中,仍然有显著的引物-二聚体产生。该引物-二聚体产物在整个pcr中与感兴趣的条带竞争,大大降低了pcr的灵敏度,并且损害了样品的感兴趣的基因的覆盖。在ngs之前,引物-二聚体效应的程度的观察是不能获得的。总之,这两个结果揭示了使用本文公开的dam-pcr的pcr替代方法,是解决多重pcr中的兼容性和敏感性问题的唯一可能的解决方案。
单一引物对可能对多重pcr造成的损伤的证据
从图3a中,如上所述,引物-二聚体的形成对测序产量有影响是明显的,即使扩增被认为是相对“成功”的。换句话说,扩增产生了感兴趣的条带,但测序结果证明了伴随着感兴趣的产物的显著量的引物-二聚体。然而,申请人也能在极端情况下证明在多重pcr混合物中根除期望的产物条带的扩增的引物对的实例。如图5所示,向含有il-10的引物的混合物中加入t-bet正向引物,导致在向所述混合物中加入该单一引物之前存在的期望的产物条带的消除。类似地,将foxp3反向引物加入t-bet正向和反向引物的混合物中类似地导致扩增的初级产物条带的丧失。
申请人证明,单一引物对可以消除感兴趣的靶标的扩增。根据本公开的启示,本公开显示在pcr期间引物-二聚体与期望的产物竞争dna聚合酶活性,许多多重pcr失败可被认为是“成功的”(虽然是不期望的)引物-二聚体扩增产物,代表了理解多重pcr的范例转变。一旦这一点显而易见,本公开就证明最好的pcr方法是避免初级产物条带和潜在引物-二聚体之间对dna聚合酶活性的竞争,如用目前公开的dam-pcr方法学实现的。
提高退火温度不足以克服引物-二聚体倾向
最常尝试的去除引物-二聚体形成的方法之一是减少pcr循环和提高退火温度,构思是引物-二聚体形成在较高温度下将被抑制。在单个细胞扩增的情况下,高循环数是必要的以实现扩增,因为起始拷贝数低,所以如果要保持灵敏度,循环减少不是可行的选择。在图6中,申请人证明将退火温度调整至混合物中的引物将不再结合靶标模板的点不足以去除引物-二聚体的形成,证明简单地提高退火温度将不能去除竞争。在各种退火温度下测试形成引物-二聚体的已知引物对。如果引物-二聚体产生可以通过提高退火温度而减少,那么引物-二聚体条带的强度应该随着温度的升高而降低。相反,在所有情况下,尽管退火温度提高,引物-二聚体产物的条带强度相对不变。在最高的允许的退火温度下,测试对中的两对仅有轻微的降低。
具有引物-二聚体倾向的引物对的有目的掺混以在arm-pcr和dam-pcr之间进行比较
dam-pcr能够克服引物-二聚体对扩增的抑制作用。利用多重混合物进行arm-pcr与dam-pcr实验的比较,所述多重混合物含有来自cd3+t细胞和脾的混合物的150ngrna。在不存在和存在已知引起引物-二聚体形成的另外的引物对的情况下,通过加入正向和反向内部引物(为了更好的比较,没有外部引物)的引物混合物进行arm-pcr实验。对于dam-pcr,在反转录步骤期间加入与arm-pcr扩增中使用的相同的反向引物混合物。使用磁珠选择第一条链cdna,并用与arm-pcr实验一起使用的相同的正向引物混合物进行第二条链标记。还进行了单循环dam-pcr(1个循环热变性、退火和延伸)和线性扩增dam-pcr(热变性后接着6个循环退火和延伸)的比较。使用磁珠选择经标记的dam-pcr文库,并用一对共用的引物扩增文库20个循环。在用该对共用引物标记和一轮扩增后,使用2μl的dam-pcr文库用于第一pcr扩增子至第二pcr反应的转移实验(图7b),用于与相似的转移arm-pcr试验直接比较。使用磁珠选择剩余的dam-pcr文库,并在存在新鲜酶和缓冲液的情况下使用相同的通用的引物对将其扩增30个以上循环。扩增循环的总数与arm-pcr方案相等,其包括20个循环扩增的rt-pcr步骤和30个循环的第二pcr。
图7a和7b中的结果清楚地证明,避免引物-二聚体的dam-pcr策略导致高度灵敏的扩增,而不论是否存在高二聚化潜力的引物对。在技术上,dam-pcr中违规(offending)引物对从不接触,因此,从没有机会二聚化。因此,标记步骤在两个独立的步骤中进行,并且通过pcr的扩增实际上是用一对共用引物(而不是多重混合物)进行的。多重的引物的唯一(only)部分分别是正向和反向组(或第一引物混合物和第二引物混合物和gdna在一起)。然而,由于每组(正向混合物和反向混合物)仅使用一次,因此潜在的混合内部引物-二聚体对从不允许累积。图7a和7b中的所有扩增包括技术复制。图7a代表第一pcr反应和第二pcr反应之间的珠子选择,而图7b代表第一pcr反应和第二pcr反应之间的2μl转移。如图7a和7b所示,当在已知的破坏性的引物对存在下进行arm-pcr时,期望的产物的扩增效率降低。在图7b中,对于pcr1和pcr2之间的转移的情况,该效果是显著的,这消除了初级产物条带。图7a和7b中,对于相同的模板rna,dam-pcr方法导致非常强的初级产物条带并且没有引物-二聚体产生是清楚的,特别是在图7a中,其中没有引物遗留的机会(如用2μl转移)。对于dam-pcr,单循环和线性扩增方法在琼脂糖凝胶上没有明显差异,表明单循环标记是可行的方法。
dam-pcr和单个细胞扩增
单个细胞是可论证的最具挑战性的模板中的一些,因为靶标的拷贝数低,并且细胞与细胞之间的表型变异使得多重混合物与模板的每次相互作用是动态的。在没有模板的情况下,某些在整体大块测量(ensemblebulkmeasurement)中未展示引物-二聚体形成的高度倾向的引物对由于缺乏靶标而开始二聚化。这增加了一层复杂性,因为高度多重化的引物混合物与变化的低拷贝模板的相互作用不能被预测或周围设计。在图8中,申请人证明尽管在单个细胞水平上基因表达的变化,但在单个细胞水平上dam-pcr产生没有引物-二聚体形成的具有强条带强度的扩增子,而arm-pcr证明单个细胞的动力学性质导致感兴趣的靶标的不同程度的扩增,该扩增具有增加的拖尾和引物-二聚体形成,其中没有明确避免引物-二聚体形成。这些负面的副作用通过导致测序结果中感兴趣的靶标的信号丢失(dropout)而减少了单个细胞的产出,必须增加每个细胞专用的测序读数以提供足够的基因覆盖,并且导致对无用的引物-二聚体进行昂贵的测序,最终显著增加单个细胞分析的成本。
选择后引物的遗留
为了证明本文公开的方法导致在步骤之间未使用的引物的几乎100%去除,申请人进行了实验,在所述试验中申请人通过将2pmol反向引物原液从0.5%连续稀释至0来制作标准曲线。这些稀释液用作参考以测量两种类型的清除方法中残留的引物遗留。稀释液模拟了在反转录和第二条链合成之间将遇到0.5%至0%的反向引物遗留的情况。将已知引物-二聚体倾向的正向引物加入到每个连续稀释的混合物中,并对混合物进行pcr以间接显现“残留的”反向引物的百分比。在测试样品中,在四个测试(包括技术重复)中的每一个包括2pmol的反向引物。这些混合物进行两种不同的spri珠子清除的方法。为了检测清除后的残留引物,将2pmol用于连续稀释样品的相同正向引物加入到测试样品的所选择的产物中。在这种情况下,清除后的残留反向引物是潜在扩增的仅有来源。对测试样品进行与标准稀释曲线相同的pcr。这样,如图9所示,通过与标准曲线比较,测试样品的扩增的条带强度可以直接表明清除测试中残留的反向引物的百分比。结果显示残留的引物在凝胶上未检测到,因此针对ces选择,具有小于0.01%的遗留(或大于99.99%的去除),而0.7×spri选择具有0.10%的遗留(或大于99.9%的去除)。
dam-pcr对于gdna的应用
使用覆盖不同靶标的三种分开的多重混合物用dam-pcr扩增gdna。一种靶标混合物用针对v基因区段的正向引物和在j区的反向引物覆盖人tcrβ基因座的可变区,这使得能够从gdna中获得tcrβ基因座的vdj重排。重排可变区和c-基因之间的内含子缺口连同测序长度的限制使得需要将反向引物放置在该位置以覆盖gdna,需要13个反向引物以覆盖该基因区段和超过33个正向引物。作为比较,该混合物也与arm-pcr策略一起使用。如图10a所示,当应用于gdna时,dam-pcr扩增产生扩增子条带,而arm-pcr产生拖尾,可能是由于位点外(offsite)背景扩增。当同时使用正向和反向引物混合物时(如典型的多重pcr策略),引物可以结合在重排中未使用的位点,并在最初的几个循环中产生与感兴趣的重排信号竞争性的产物。这些位点外反应用dam-pcr减少,因为在两者中的任一个方向只允许进行一个标记循环,并且在步骤之间只选择最初感兴趣的靶标。
为了证明dam-pcr也作用于除了适应性免疫细胞的其它靶标,还将覆盖在癌恶性肿瘤(肿瘤组;图10b)中通常突变的基因靶标的多重混合物和用于检测个体的人白细胞抗原型(hla-型)的引物混合物应用于gdna。
对于gdna,在每个混合物中包括哪些引物具有更大的灵活性。不必仅包括正向混合物或仅包括反向混合物。因此,我们称引物混合物为第一混合物和第二混合物。当设计引物混合物时可以应用该策略,以允许通过重叠扩增子产物的更长的测序覆盖,同时减少背景,所述背景对扩增过程和测序都是有害的。利用rna合成第一条链cdna的方向性需要首先使用反向混合物。当使用gdna作为模板时,引物混合物不必分为有义链和反义链混合物。重叠部分可以用引物设计,以便于生物信息学上测序后对较大基因产物的下游重组。通常,正如典型的多重pcr一样,如果允许整个引物混合物在相同的混合物中相互作用,则将产生脱靶扩增,产生将与感兴趣的产物竞争性的背景,从而在测序仪上产生如图11所示的昂贵的背景。对于dam-pcr,当试图覆盖更大的基因靶标时,引物混合策略能够以减少的背景进行靶向测序。在第一引物混合物中,如图12中所说明的,可以使用覆盖更大的靶标的一对引物。由于链是独立处理的,当将下一组引物或第二引物混合物施加到第二条链上时,它们将仅与各自的第一条链产物兼容。现在,用dam-pcr策略可以完全避免产生较短的无用产物的引物的相互作用。
图13提供应用于来自gdna的hla靶标的arm-pcr和dam-pcr的实例。泳道1和2分别显示当使用arm-pcr扩增基因靶标a或靶标b时的扩增模式。泳道3显示t2正向和t1反向相互作用的扩增模式。这只是为了证明该模式,但是当所有四个引物在相同pcr混合物中多重的时,该产物是可以产生的脱靶产物。泳道4显示t1正向引物和t2反向引物的长扩增产物。在这种特定情况下,这种产物也是不太理想的,因为由于序列长度的限制,该产物不能被目前使用的ngs平台覆盖。然而,能够覆盖更长产物的ngs平台可以对这种产物测序。泳道5-6显示从完全多重的混合物中进行的arm-pcr扩增。结果主要由不太理想的短的脱靶产物占据主导地位。由于该短的产物与期望的产物竞争dna聚合酶活性,对于仅有靶标a(从t1正向和t1反向产生),仅有靶标b(从t2正向和t2反向产生)及同时有靶标a和靶标b(从t1正向和t2反向产生)的基因靶标是模糊的且几乎不存在。泳道7显示当dam-pcr在第一循环标记期间仅用t1反向和在第二循环标记期间用t1正向用于扩增基因靶标a的扩增模式。泳道8显示当dam-pcr在第一循环标记期间仅用t2反向和在第二循环标记期间用t2正向用于扩增基因靶标b的扩增模式。泳道9-10显示完全多重dam-pcr策略的结果。在用第一引物混合物的第一轮标记中,t1正向和t2反向用于生成更长的第一条链产物。在用第二引物混合物的第二轮标记中,t1反向和t2正向与在第一条链标记过程中生成的第一条链产物独立地相互作用。在第二条链标记、清除并用一对引物扩增后,没有竞争性的短的产物,且可以实现期望的产物的扩增,所述一对引物具有对第一轮靶标共用的标签。如在泳道9-10的凝胶图像中明显的,产物条带是泳道1-2或泳道7-8中所代表的期望产物的总和。重要的是注意到标记的单个循环是关键的。如果允许引物t1正向和t2反向像正常pcr一样进行另外的循环,则可能产生短的竞争产物。然而,这些潜在有害的引物在用共用通用的引物进行任何真正的扩增之前在dam-pcr中去除。
除非另外明确说明或从文本中来看是清楚的,否则对单数的项目的引用应当理解为包括复数的项目,反之亦然。除非另有说明或从上下文中来看是清楚的,否则语法连接旨在表达连接的从句、句子、词等的任何和所有分离和连接组合。因此,术语“或”通常应当理解为意指“和/或”等等。
本文所述的系统和方法的各种实施方案是示例性的。用于本文所述的系统和方法的各种其它实施方案是可能的。
- 还没有人留言评论。精彩留言会获得点赞!