吡啶并咪唑利福霉素衍生物的抗菌剂的制作方法
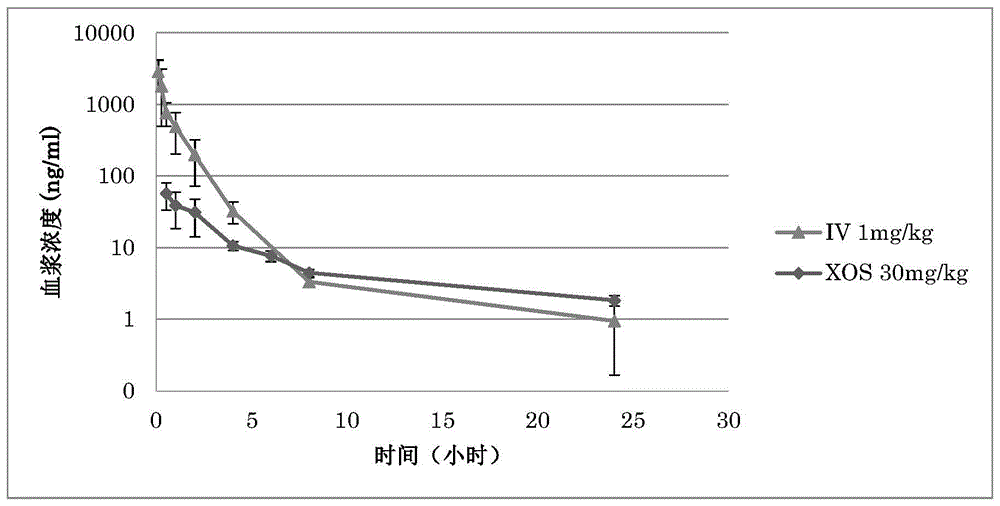
本发明涉及新型吡啶并咪唑利福霉素,其特征在于具有高选择性的抗菌活性和口服途径的低吸收。
背景技术:
:吡啶并(1’,2’:1,2)咪唑并(5,4-c)利福霉素是从利福霉素s或利福霉素o起合成而得到的抗生素,其特征在于,一方面口服吸收率低,另一方面对革兰氏阳性和革兰氏阴性细菌均具有良好的抗菌活性。这类抗生素的祖先是利福昔明,它是在70年代末由marchi等人合成的(us4341785a)。目前,这种抗生素在欧洲和美国市场上,在治疗人类胃肠道疾病方面特别成功,例如克隆氏症、憩室病、肠易激、门体分流性氨血症等;以及特别是在美国,预防旅行者的腹泻。该抗生素还在兽医市场上用于生殖器细菌性疾病和奶牛乳腺炎疾病。从1988年利福昔明进入意大利市场的那一年起,从生物学和化学的角度继续研究这种抗生素。一方面,有许多配方技术专利,另一方面,由于不同的溶解度与多晶型物和溶剂化物相关,要求保护多晶型物或假多晶型物(也称为溶剂化物)的专利具有根本的重要性,不同的生物利用度和毒性似乎是相对应的:在us7045620和wo2006094662专利中,分别要求保护多晶型物α、β、γ、ε和δ。基于对结晶粉末的部分x射线折光率测量,很难确定各种要求保护的产品是否确实是多晶型的。实际上,未能提供诸如化合物的熔点之类的数据,该数据可以突出多晶型形式与可能的溶剂化形式之间的差异。这些多晶型形式似乎具有不同的溶解度,似乎与不同的生物利用度和毒性有关(wo2006094662)。所有这些看起来非常混乱,而且,跟我们的需求相矛盾。实际上,为了发挥其活性,必须将抗生素溶解在介质中,在这种情况下,应溶解在肠液中,因此,如果抗生素更易溶,则应具有更高的活性,为此只要有较低剂量就足够。此外,胆汁酸的存在(胆汁酸改善利福昔明的抗菌作用。darkoh等人,antimicrob.agentschemother.:2010;54(9),3618-3624)会增加抗生素的溶解量,因此增加了产品的活性。但是,在这种情况下,如果吸收与溶解药物的量有关,则胆汁酸会增加所有多晶型形式的生物利用度,因此也会增加治疗中所用形式的可能的毒性。此外,在水性环境中,很难理解哪一种是稳定的多晶型形式,因为所述的各种形式,即α、β、γ、ε和δ,即使在固体状态中也可以通过吸收水分相互转化(wo2006094662)。可以看出,如有关门体分流性脑病的工作所述,利福昔明关键点之一的确与它的生物利用度有关(gastrointest.pharmacol.ther.:2012;3(4):62-67;chronicexposuretorifaximincauseshepaticsteatosisinpregnanexreceptor-humanizedmice.jiecheng,kristopherw.krausz,naokitanaka,andfrank,j.gonzalez,toxicol.sciences:2012;129(2),456-468),其应受其不溶性的限制,而取决于所用假多晶型或溶剂化形式、或者肠液中胆汁酸的存在,以及肠炎状态。进一步令人感兴趣的是抗生素对真核细菌菌群例如对大型生物群的活性。利福昔明对构成人肠真核菌群的细菌种类具有可评估的活性。例如,即使不是所有的物种都以相同的方式抑制,利福昔明对乳杆菌、双歧杆菌、肠球菌、肠杆菌等菌株也具有明显的抗菌活性。显然,能够拥有缺乏的抗生素、或任何对肠道大型生物群中的细菌物种具有高选择性的抗生素,将是一项进一步重要的创新,与低口服生物利用度相结合,将会代表产品的根本的治疗改善。实际上,如今,无论是否为局部用药,利福昔明与所有其他抗生素的治疗用途均与真核菌株补充剂相辅相成,这是因为,一方面使用抗生素可消除和/或减少病原菌在肠道中的存在,但同时导致形成大型菌群的真核菌株混合物的显著减少和改变。因此,考虑到这些抗生素除了偶尔用于旅行者的腹泻之外大多数是慢性的,拥有对真核生物菌株表现出良好选择性的抗生素,例如将其在肠道中的存在减少10个单个因素或最多100个因素,可以认为是一项根本的治疗创新。正是为了强调对真核生物菌株的选择性的重要性,我们记得此类抗生素的一个重要特征是在先前的治疗后三到四周可以重复治疗,这是因为耐药菌株的选择受到了利福霉素的特殊作用机理的很大的阻碍。这些实际上对dna依赖性细菌rna聚合酶具有活性,由于复制能力有限,因此在治疗期间可以选择为抗性的菌株实际上很容易由非抗性菌株控制。此外,与其他抗生素如青霉素等不同,对利福霉素的抗性不能通过质粒传播。显然,这些特征以及吡啶并咪唑利福霉素如利福昔明经口服途径吸收不良,使得这类抗生素对胃肠道疾病尤其是慢性疾病特别重要。很明显拥有一种如下抗生素具有重要意义:对可能与肠道定殖有关的致病菌株具有活性,对形成微生物群的真核菌株具有良好的选择性,并且同样重要的是显示出非常低的吸收性。如上所述,利福昔明表现出不同的吸收率,也取决于所用的溶剂化物,其吸收范围可以从0.5%到大于1%,并且肾脏排泄量约为0.36%(bajajj.s.,riggioo.,drugtherapy:rifaximin.hepatology2010;52:1484-148)。这种吸收与溶剂化物本身的溶解度有关,因此水分含量最低的形式吸收最多。无定形形式也非常易溶,在健康志愿者中其吸收量是利福昔明α的5-6倍(marzoa.,ismailis.,dig.liv.dis.2010;42(s2):s191-s192),因此认为可能有风险将抗性细菌选择至系统水平。显然,利福昔明α也确实存在这种与溶解度/吸收有关的风险,这是因为已证实(darkohc.,lichtenbergerl.m.,ajamin.,etal.antimicrob.agentschemother.2010;54:3618-24)利福昔明在胆汁酸存在下从在水中的0.6%的溶解度变为72.6%,即在低至20mm胆汁酸的存在下的溶解度的一百倍。由于利福昔明的高剂量给药,从旅行者腹泻的最低剂量400mg/天到憩室炎的800mg/天、到门体分流性氨血症的2400mg/天,甚至不考虑在胆汁酸存在下的溶解度,很明显即使是如利福昔明那样的有限吸收,也可能代表一个问题,即可能选择包括结核分枝杆菌的耐药菌菌株至系统水平,还可能与药物例如他汀类药物相互作用,它们在肝脏水平上具有共同的酶促介质。例如,关于利福昔明,据报道称在门体分流性氨血症的治疗期间,血液中钠和钾的浓度增加。此外,利福昔明对构成肠道菌群的细菌菌株也具有如上所述的显著活性,因此有必要干预真核细菌菌株的补充,以在每次抗生素给药后恢复肠道益生菌复合物。另外,利福昔明对肝pxr受体具有激动作用,并从中衍生出其抗炎性肠作用,利福昔明虽吸收程度不高但如何在具有这种人源化受体的小鼠中引起大量脂肪变性已被强调(cheng,kristopherw.krausz,naokitanaka,andfrank,j.gonzalez,toxicologicalsciences,129(2),456-468(2012))。鉴于这些结果,需要具有吡啶并咪唑结构的新型利福霉素衍生物,其特征在于具有高度选择性的抗菌活性和低的口服吸收率,例如比利福昔明低50%。技术实现要素:在利福霉素领域中具有抗生素活性的产品必须符合构象和化学要求,并且必须在亲脂性和亲水性之间具有适当的平衡(l.cellai,s.cerrini,a.segre,c.battistoni,g.cossu,g.mattogno,m.brufaniande.marchi.mol.pharm.1985,27:103-108)。因此,在本发明中,为了维持利福霉素sv上的吡啶基-咪唑结构,在吡啶核上引入了易于官能化的基团,因此随后可以得到具有非常不同的化学和物理化学特性的新衍生物。以此方式,发明人惊奇地发现了具有合适特征的分子,即:比利福昔明的肠吸收低至少50%,对致病菌株的良好的抗菌活性,以及对构成肠道大型生物群的细菌菌株的微不足道的活性且在任何情况下都低于利福昔明。在各种可能的化学结构中,发明人通过实验发现,在甲酰基的吡啶核的4’-或5’-位的引入,可提供4’-或5’-甲酰基-4-脱氧吡啶基[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv衍生物,该衍生物本身完全没有活性,但容易被转化为腙或肟或亚胺。两个位置4’-或5’-的醛基显著增加了位置8-的作为强吸电子基团的羟基的解离,并且因其穿过细菌壁的能力受阻而导致没有抗菌活性(p.bellomo,e.marchi,g.mascellani,j.med.chem,1981,24(11),1310-1314;m.f.dampier,h.w.whitlock,jr.j.am.chem.soc.1975,97:21,6254-6256)。然而,令人惊讶地,通过将羰基转化为腙、肟或亚胺,可以有利地改变其电子贡献。这些衍生物用不同的诱导/共聚作用特征化,通常是阳性,这些衍生物令人惊讶地显示出具有更可调和合适的生物学特性。由于甲酰基的反应性,利福霉素o与位置4-或5-带有甲酰基的2-氨基吡啶之间的反应是困难的,本发明人试图使用被保护为缩醛的2-氨基-4-甲酰基吡啶。该反应是可能的,但难以在任何情况下均能进行,这是因为受保护的2-氨基-4-甲酰基吡啶的获取并不容易,并且会导致试剂特别脏。因此,发明人猜测在具有进一步官能化基团的利福霉素sv上引入吡啶-咪唑基之后,可在缩合的吡啶核上得到甲酰基。出人意外地,在通过利福霉素o与2-氨基-4-羟甲基-吡啶的反应得到了高产率的4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv之后,可以通过在适当的溶剂中用二氧化锰氧化羟甲基来得到4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv,然后用抗坏血酸还原。本发明人能够验证由此得到的产物是稳定的并且可以进一步官能化为腙、肟或亚胺。随后,发明人能够令人惊讶地证实,通过其他方法,通过使利福霉素o或3-碘或3-溴利福霉素s与2-氨基-4/5-甲酰基吡啶的合适衍生物反应,得到4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的腙、肟或亚胺是可能的。通过使用这些合成路线,能够合成多种腙,其中4’-和5’-位置的具有良好的抗菌活性。在这些产物中,特别是4’-[((4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2,]咪唑并[5,4-c]利福霉素sv(以下为实施例3)对革兰氏阳性和革兰氏阴性菌株显示出良好的抗菌活性,并且令人惊讶地显示,与利福昔明在文献中显示和证实的相比,肠道吸收降低了7-8倍,即约15%。此外,该衍生物的更加显著特征是对被检验的真核菌株表现出的高选择性,所述真核菌株是肠道菌群的基本组成部分。附图说明图1示出在iv和口服给药后利福昔明(rfx)的血液水平的比较;图2示出在iv和口服给药后实施例3的化合物的血液水平的比较;图3示出实施例3的化合物和利福昔明对所测试的真核细菌菌株的作用。在柱上方报告了这些治疗的统计分析(*p<0.1;**p<0.05);图4示出实施例1中得到的化合物4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图5示出实施例1中得到的化合物4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图6示出实施例1中得到的化合物4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图7示出实施例2中得到的化合物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s的i.r.光谱;图8示出实施例2中得到的化合物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s在cdcl3中的1hnmr光谱;图9示出实施例2中得到的化合物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s在cdcl3中的13cnmr光谱;图10示出实施例2中得到的化合物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图11示出实施例2中得到的化合物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图12示出实施例2中得到的化合物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图13示出实施例3中得到的化合物4’-[(4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图14示出实施例3中得到的化合物4’-[(4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图15示出实施例3中得到的化合物4’-[((4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图16示出实施例4中得到的化合物4’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图17示出实施例4中得到的化合物4’-[((1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图18示出实施例4中得到的化合物4’-[((1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图19示出实施例5中得到的化合物4’-[((n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图20示出实施例5中得到的化合物4’-[((n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图21示出实施例5中得到的化合物4’-[((n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图22示出实施例6中得到的化合物4’-[(4-羧基酰氨基吡啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在meod中的1hnmr光谱;图23示出实施例6中得到的化合物4’-[(4-羧基酰氨基吡啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在meod中的13cnmr光谱;图24示出实施例7中得到的化合物5’-[((n,n-二甲基氨基)亚氨基甲基]-4-去氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图25示出实施例7中得到的化合物5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图26示出实施例7中得到的化合物5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图27示出实施例8中得到的化合物5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图28示出实施例8中得到的化合物5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图29示出实施例8中得到的化合物5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图30示出实施例9中得到的化合物5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的1hnmr光谱;图31示出实施例9中得到的化合物5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在cdcl3中的13cnmr光谱;图32示出实施例10中得到的化合物4’-[((4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s在cdcl3中的1hnmr光谱;图33示出实施例10中得到的化合物4’-[(4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s在cdcl3中的13cnmr光谱;图34示出实施例11中得到的化合物25-脱乙酰基-5’-[(1-哌啶基)亚氨基甲基]-4-去氧吡啶基[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的i.r.光谱;图35示出实施例11中得到的化合物25-脱乙酰基-5’-[(1-哌啶基)亚氨基甲基]-4-去氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在meod中的1hnmr光谱;图36示出实施例11中得到的化合物25-脱乙酰基-5’-[(1-哌啶基)亚氨基甲基]-4-去氧吡啶基[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在meod中的13cnmr光谱;图37示出实施例12中得到的化合物25-脱乙酰基-5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在meod中的1hnmr光谱;图38示出实施例12中得到的化合物25-脱乙酰基-5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv在meod中的13cnmr光谱。定义除非另有定义,否则本文中使用的所有本领域术语、符号和其他科学术语旨在具有本说明书所属领域的技术人员通常理解的含义。在某些情况下,为了清楚和/或以备参考,本文定义了具有通常理解的含义的术语。因此,本说明书中所包含的这样的定义不应被解释为代表相对于本领域通常所理解的实质性差异。本文中的术语“卤素”是指氟(f)、氯(cl)、溴(br)或碘(i)。本文中的术语“c1-c10烷基”是指含有1至10个碳原子的支链或直链烃。c1-c10烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、正己基。本文中的术语“芳基”是指芳香单碳环和多碳环系统,其中多碳环系统中的各个碳环可以通过单键来稠合或相连。合适的芳基包括但不限于苯基、萘基和联苯基。本文中的术语“芳氧基”是指o-芳基,其中“芳基”如上所定义。本文中的术语“烷氧基”是指o-烷基,其中“烷基”如上所定义。本文中的术语“杂环”是指4元、5元、6元、7元或8元单环,其饱和或不饱和,并且由碳原子和一个或多个选自n、o和s的杂原子组成,其中,氮和硫杂原子可任选被氧化,氮杂原子可任选被季铵化。杂环可以连接到任何杂原子或碳原子上,条件是该连接转化为形成稳定的结构。该术语还包括任何上述杂环与芳基或另一个杂环稠合而成的任何双环系统。当杂环为芳香族杂环时,可以定义为“杂芳香族环”。本文中的术语“不饱和环”是指部分或完全不饱和的环。例如,不饱和c6单环是指环己烯、环己二烯和苯。本文中的术语“取代”是指定义的(或未定义的)取代基的单取代或多取代,其程度是化学上允许这样的单个或多个取代。术语“生理上可接受的辅料”是指没有其自身任何药理作用的物质,并且当施用于哺乳动物时、优选施用于人类时,不会产生不良反应。生理上可接受的辅料在本领域中是众所周知的,例如,在《药用辅料手册》(handbookofpharmaceuticalexcipients)、第六版2009中进行了描述,该文献通过引用并入本文。术语“药学上可接受的盐或衍生物”是指具有盐化化合物的生物学效力和特性并且当施用于哺乳动物时、优选施用于人类时不产生不良反应的那些盐或衍生物。药学上可接受的盐可以是无机或有机盐。药学上可接受的盐的实例包括但不限于:碳酸盐、盐酸盐、氢溴酸盐、硫酸盐、硫酸氢盐、柠檬酸盐、马来酸盐、富马酸盐、三氟乙酸盐、2-萘磺酸盐和对甲苯磺酸盐。关于药学上可接受的盐的其他信息可以在《药用盐手册》(handbookofpharmaceuticalsalts),p.stahl,c.wermuth,wiley-vch,127-133,2008中找到,通过引用并入本文。术语“包括”、“具有”、“具备”和“包含”应被解释为开放式术语(即,意思是“包含但不限于”),并且应被视为对诸如“基本上由......组成”、“基本上由......构成”、“由......组成”或“由......构成”之类的术语的支持。术语“基本上由......组成”、“基本上由……构成”应理解为半封闭式术语,意味着不包括任何实质上影响本发明新颖特征的成分。术语“由......组成”、“由……构成”应解释为封闭式术语。具体实施方式发明人通过实验发现,具有吡啶-咪唑结构的利福霉素sv的腙显示出高选择性的抗菌活性并且口服途径的吸收差。根据第一方面,本发明涉及式(i)的化合物或其药学上可接受的盐。其中,r和r1可以是h、条件是:若则r1=h和r2=ch3co-或h;若则r=h和r2=ch3co-或h;若则r1=h和r2=ch3co-或h;以及若则r=h和r2=ch3co-或h;其中,r3和r4相同或不同,并且选自:氢,任选地被一个或多个选自氨烷基、烷氧基、苯氧基或磺基的取代基取代的直链或支链的c1-c10烷基,以及任选地被c1-c4烷基或烷氧基、卤素、氨基、硝基单取代或双取代的芳基;或者r3和r4与吡啶核的两个连续碳原子一起可以形成任选被c1-c4烷基取代的苯环,或任选被c1-c4烷基取代的5或6元杂环,r5选自:氢,羟基,任选地被一个或多个选自氨烷基、烷氧基、苯氧基或磺基的取代基取代的直链或支链的c1-c10烷基,以及任选地被c1-c4烷基或烷氧基、卤素、氨基、硝基单取代或双取代的芳基。一类优选的化合物包括式(i)的化合物,其中r和r1可以是h或其中,任选地被c1-c4烷基或烷氧基、卤素、氨基、硝基单取代或双取代的芳基选自苯基和苄基。另一类更优选的化合物包括式(i)的化合物,其中r和r1可以是h或其中,5或6元杂环选自吡咯烷,哌啶,哌嗪和吗啉。特别优选式(i)的以下化合物或其药学上可接受的盐:4”-[(4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例3);4’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例4);4’-[((n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例5);4’-[(4-羧基酰胺基吡啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例6);5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例7);5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例8);5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例9);4’-[(-4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s(实施例10);25-脱乙酰基-5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例11);25-脱乙酰基-5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(实施例12);4’-[(n-吗啉基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;5’-[(n-吗啉基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-4’-[(n-吗啉基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-5’-[(n-吗啉基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;4’-[((n-丙基,n-丁基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;5’-[(n-丙基,n-丁基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-4’-[((n-丙基,n-丁基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-5’-[((n-丙基,n-丁基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;4’-[(n,n-二戊基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;5’-[(n,n-二戊基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-4’-[(n,n-二戊基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-5’-[(n,n-二戊基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;4’-[(4-乙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;5’-[(4-乙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-4’-[(4-乙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-5’-[(4-乙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;4’-[(4-丙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑[5,4-c]利福霉素sv;5’-[(4-丙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv;25-脱乙酰基-4’-[(4-丙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑[5,4-c]利福霉素sv;以及25-脱乙酰基-5’-[(4-丙基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv。根据第二方面,本发明涉及制备式(i)的化合物的第一方法,该方法包括以下步骤:-使利福霉素o与2-氨基-4-羟甲基吡啶反应,得到4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv;-在液相中、优选在非质子溶剂中,用合适的氧化剂、优选用二氧化锰,氧化4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv,得到4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv;-使4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv与式(ii)的肼nh2-n(r3)(r4)反应,以得到相应的腙,其中r3和r4如式(i)所定义。在根据本发明的第一方法的优选实施方式中,每摩尔利福霉素o使用约1至约6摩尔当量的2-氨基-4-羟基甲基吡啶,和/或,每摩尔4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv使用约1至约6摩尔当量的肼。根据第三方面,本发明涉及制备式(i)的化合物的第二种方法,包括以下步骤:-使式(ii)的肼nh2-n(r3)(r4)与2-氨基-4-甲酰基吡啶或2-氨基-5-甲酰基吡啶反应,以得到相应的腙,其中r3和r4如式(i)所定义;-使所得到的腙与利福霉素o、3-碘利福霉素s、3-溴利福霉素s或25-脱乙酰基-25-羟基利福霉素s反应。在本发明的第二方法的优选实施方式中,每摩尔利福霉素o或3-碘/溴利福霉素s使用约1至约6摩尔当量的所得到的腙。优选地,在根据本发明的方法中,所使用的溶剂是选自含有1-4个碳原子的脂肪烷醇的有机溶剂,其单独使用、或与水或与dmf混合使用,或者使其处于短链卤代溶剂中、优选在二氯甲烷中。优选地,在根据本发明的方法中,反应温度在10℃至溶剂沸点之间变化,优选不超过80℃,和/或反应时间在几分钟至24h之间变化。根据一个优选的实施方式,式(i)的化合物可以通过用合适的氧化剂、优选通过二氧化锰的处理来氧化。根据第四方面,本发明涉及产物4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv,其可作为中间体用于4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv的获取。产物优选通过1摩尔的利福霉素o与1至5摩尔的2-氨基-4-羟甲基吡啶的反应来获取。根据第五方面,本发明涉及产物4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv,其作为得到式(i)的化合物的中间体。产物优选从产物4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s或sv开始,通过在液相如锰二氧化锰中进行氧化来得到。根据第六方面,本发明涉及用作药物的式(i)的化合物。在一个优选的实施方式中,式(i)的化合物用于治疗和/或预防胃肠道疾病,所述胃肠道疾病优选选自由细菌性疾病、ibd、克隆氏症、憩室病和旅行者腹泻引起的疾病,或者用于皮肤、眼睛、阴道或牙齿的感染,优选用于需要长期治疗的疾病的治疗中。在另一个优选的实施方式中,式(i)的化合物用于治疗和/或预防干燥性乳腺炎、牛的阴道疾病、宠物的肠道疾病或畜牧业的生长素疗法,优选用于鸡、火鸡、鸭子或兔子。根据第七方面,本发明涉及药物组合物,其包含至少一种式(i)的化合物以及至少一种药学上可接受的辅料。优选地,上述制剂适合于局部给药,优选以速释或程序释放固体、液体或凝胶的剂型。一般综合方案本发明中描述的化合物可以使用以下方法制备。1°方法第一种方法包括在质子溶剂如甲基、乙基、丙醇等中,使利福霉素o与2-氨基-4-羟甲基吡啶反应,以得到4’-羟甲基吡啶并[1’,2’-1,2]咪唑(5,4-c)利福霉素sv,该质子溶剂可以单独使用,也可以与其他溶剂如乙腈、二恶烷等混合使用。根据方案1,在非质子溶剂如乙腈或二氯甲烷中,用二氧化锰将该化合物氧化为相应的羰基衍生物4’-甲酰基吡啶并[1’,2’-1,2]咪唑并(5,4-c)利福霉素s,接着用抗坏血酸还原为sv形式,然后与合适的肼、羟胺或胺反应:方案1其中r3和r4具有在式(i)中说明的相同含义。通过这种方法,也可以使用合适的试剂得到亚胺和肟。利用方案1,还可以例如从(2-氨基-5-吡啶)甲醇得到5’衍生物。2°方法根据方案2,第二种方法包括使2-氨基-4/5-羰基吡啶与适当的胺、肼或羟胺反应,得到加成产物,其随后与3-碘/溴利福霉素s(根据us4179438专利得到)或利福霉素o反应。方案2其中r3和r4具有与方案1相同的含义。使用这种合成方案,从3-碘/溴利福霉素s或利福霉素o和2-氨基-4/5-甲酰基吡啶的腙开始,可以得到4’/5’-甲酰基-4-脱氧吡啶[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的腙衍生物。特别地,约1至约6摩尔的腙用于每摩尔利福霉素o或3-碘/溴利福霉素s,其中,腙通过2-氨基-4-甲酰基吡啶或2-氨基-5-甲酰基吡啶与合适的被取代的肼的缩合而得到,取代基r3和r4如上所定义。得到的最终产物都是还原形式,即sv形式。然而,也可以使用合适的氧化剂,容易得到s形式的衍生物,即氧化形式,该氧化剂通常为二氧化锰,其通过过滤很容易消除(参见方案3)。方案3其中,r3和r4具有与方案1相同的含义。同样,从25-脱乙酰基利福霉素s开始,可以按照例如下面的合成方案(方案4)得到在本发明中考虑的位置25-处脱酰的所有衍生物。从3-卤代-25-脱乙酰基利福霉素s开始,使其与所需的2-氨基-4/5-甲酰基吡啶或与2-氨基-4-羟甲基利福霉素s反应,将缩合产物用合适的氧化剂氧化为25-脱乙酰基-4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv,并与所需的肼缩合(方案4)。方案4r3和r4具有与方案1相同的含义。在方案1-4中,使用选自含1-4个碳原子的脂族烷醇的有机溶剂,其可以单独使用,可以与水或与dmf混合使用,也可以使其处于短链卤代溶剂如二氯甲烷中。优选地,反应温度可以在10℃至溶剂沸点之间变化,但是不高于80℃,而反应时间可以在几分钟至24h之间变化。实施例a)按照方案1所示的方法得到的产品。实施例1.4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。在70℃下,将5.03g的利福霉素o(6.67mmol)悬浮在38ml的乙醇中;加入2.48g的2-氨基-4-羟基甲基吡啶(19.97mmol),并将该混合物在70℃下搅拌4h。将溶液调至室温,并用190ml的二氯甲烷稀释,将有机层用10%抗坏血酸洗涤两次,并用饱和氯化钠溶液洗涤一次,最后用蒸馏水洗涤。将干燥的有机层过滤并蒸发,将粗残留物通过1:5.5的乙醇/乙醚混合物结晶,得到3.60g的红橙色固体产物。实施例2.4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。将4.52g的4’-羟甲基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(5.64mmol)溶于32ml的二氯甲烷中,并将22.6g的二氧化锰在40℃下加入溶液中1小时。使反应混合物回到室温后,将二氧化锰滤出并吸收在二氯甲烷中。通过蒸发溶剂得到3.73g的4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s。然后将3.73g的4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s溶解在45ml乙腈中,用10ml的15%抗坏血酸剧烈搅拌30’。将反应混合物用200ml的二氯甲烷稀释并用蒸馏水洗涤几次,然后干燥,并且在蒸发溶剂之后,将反应粗产物用乙醚洗涤,得到3.03g的紫色固体产物。实施例2a.获取4’-[(alkyloxy-iminomethyl]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的一般方法。将根据实施例2得到的0.79g的4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(1mmol,1当量)溶于10%(w/v)乙腈中。加入o-烷基-羟胺(1.5-4当量),并在搅拌下放置60-120分钟,通过tlc监测反应。将反应混合物用乙酸乙酯(3倍体积)稀释,并用水洗涤几次。将有机层蒸发至干,并将残留物从异丙醇中结晶纯化,得到相应的4’-烷氧基-亚氨基甲基利福霉素(55-80%收率)。用这种方法,优选得到o-甲基、-乙基、-丙基、-异丙基、-丁基、-戊基羟胺肟。实施例2b.获取4’-[n-烷基亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的一般方法。将根据实施例2得到的0.79g的4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(1mmol,1当量)溶于20%(w/v)二氯甲烷和三氟乙酸催化溶液(5滴)。添加n-烷基胺(4-10当量),并在搅拌下放置2-6h之间的时间,通过tlc监测反应。将反应混合物用乙酸乙酯(5体积)稀释,并用0.1n的hcl洗涤几次,最后用水洗涤。将有机层蒸发至干,并将残留物通过柱色谱法纯化以得到相应的4’-n-烷基亚氨基甲基利福霉素(30-70%收率)。用这种方法,优选得到甲基、乙基、丙基、异丙基、丁基、戊基和己胺亚胺。实施例3.4’-[(4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。向根据实施例2获取且溶于30ml乙腈中的3.05g(3.79mmol)的4’-甲酰基-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv,加入2.5当量的1-氨基-4-甲基-哌嗪。将该溶液在室温下搅拌约1小时,然后用250ml的二氯甲烷稀释,并用10%抗坏血酸溶液洗涤,再用蒸馏水洗涤几次;蒸发溶剂,得到3.38g的粗产物,在硅胶上使用95∶5的二氯甲烷/甲醇混合物对其进行纯化,得到1.06g的产物。实施例4.4’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。向根据实施例2获取且溶于10ml乙腈的0.40g(0.5mmol)的还原产物,加入0.25g的1-氨基-哌啶(2.5mmol)。将溶液在室温下放置3.5小时,然后将其用100ml的乙酸乙酯稀释,先后用10%抗坏血酸溶液、ph5.2-5.3的氯化铵、饱和氯化钠溶液和蒸馏水洗涤。干燥有机层,蒸发溶剂后,在硅胶上使用95∶5的二氯甲烷/甲醇混合物对粗产物进行纯化,得到0.15g的产物。实施例5.4’-[((n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。向根据实施例2获取且溶于20ml乙腈中的0.55g(0.675mmol)的还原产物,加入0.203g的二甲基肼(3.38mmol),并将该溶液在室温下搅拌3.5小时。反应混合物用80ml的乙酸乙酯稀释,有机层用10%抗坏血酸洗涤两次,用饱和氯化钠溶液洗涤一次,并用蒸馏水洗涤两次。蒸发有机层,并在硅胶上使用95:5二氯甲烷/甲醇混合物对残留物进行纯化,得到0.25g的产物。实施例6.4’-[(4-羧基酰胺基吡啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。向根据实施例2获取且溶于7ml乙腈中的0.35g(0.44mmol)的还原产物,加入0.30g的异烟肼(2.19mmol)。将该溶液在室温下搅拌18小时,然后将其用80ml的乙酸乙酯稀释,并用饱和氯化铵溶液洗涤一次,并用水洗涤三次。蒸发有机层,并在硅胶上使用1:3:1的二氯甲烷/乙酸乙酯/甲醇混合物对残留物进行纯化。得到0.11g的产物。b)按照方案2中所示的方法得到的产品。实施例7.从3-碘-利福霉素s开始的5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。在室温下,使根据us4179438专利得到的0.20g的3-碘-利福霉素s(0.26mmol)与1.5当量的5-(n,n-二甲基氨基)-亚氨基甲基-2-氨基吡啶在二氯甲烷中(5%溶液)反应。使其反应20小时,用2倍体积的二氯甲烷稀释,并用10%抗坏血酸洗涤两次,用0.1m盐酸洗涤两次,用饱和氯化钠溶液洗涤一次,用蒸馏水洗涤两次。蒸发有机层,并在硅胶上使用9:1的二氯甲烷/甲醇混合物对残留物进行纯化,得到0.15g的产物。实施例8.从3-碘-利福霉素s开始的5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。在室温下,使根据us4179438专利得到的0.35g的3-碘-利福霉素s(0.45mmol)与1.5当量的5-(1-哌啶基)-亚氨基甲基-2-氨基吡啶在二氯甲烷(5%溶液)中反应。使其反应20小时,用2倍体积的二氯甲烷稀释,并用10%抗坏血酸洗涤两次,用0.1m盐酸洗涤两次,用饱和氯化钠溶液洗涤一次,用蒸馏水洗涤两次。蒸发有机层,并在硅胶上使用9:1的二氯甲烷/甲醇混合物对残留物进行纯化,得到0.13g的产物。实施例9.从利福霉素o开始的5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。将0.30g的利福霉素o(0.40mmol)溶解在30ml的乙醇中,并与2当量的5-(n,n-二甲基氨基)-亚氨基甲基-2-氨基吡啶反应。使其反应约10小时,然后加入80ml的二氯甲烷,并用10%抗坏血酸、0.1n盐酸和蒸馏水洗涤几次,然后干燥并蒸发至干。在硅胶上使用9:1的二氯甲烷/甲醇混合物对残留物进行纯化,得到0.13g的产物。通过以与实施例9相同的方式操作,还可以从相应的2-氨基吡啶开始得到所有的4-和5-位被取代的衍生物。c)按照方案3中所示的方法得到的产品。实施例10.4’-[(4-methyl-1-piperazinyl)iminomethyl]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素s的获取。将根据实施例3得到的0.50g的产物4’-[((-4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv(0.56mmol)溶于32ml的二氯甲烷中。将1.3g的二氧化锰加入到溶液中;使其在室温下反应30分钟,然后滤出二氧化锰,并用二氯甲烷吸收。通过蒸发溶剂得到0.45g的产物。用实施例10中使用的方法,可以得到所有的以还原形式合成的产物的氧化形式。d)按照方案4所示方法得到的产物。实施例11.25-脱乙酰基-5’-[(1-哌啶基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。在室温下,使根据us4179438专利得到的0.050g的3-碘-25-脱乙酰基利福霉素s(0.066mmol)与2.5当量的5-(1-哌啶基)-亚氨基甲基-2-氨基吡啶在二氯甲烷(1.25%溶液)反应。将反应在搅拌下放置12小时,并且产物在相同的溶剂中结晶。用乙酸乙酯洗涤以除去杂质并得到0.055g的产物。实施例12.25-脱乙酰基-5’-[(n,n-二甲基氨基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv的获取。在室温下,使根据us4179438专利得到的0.20g的3-碘-25-脱乙酰基利福霉素s(0.26mmol)与2.5当量的5-(n,n-二甲基氨基)-亚氨基甲基-2-氨基吡啶在二氯甲烷(3%溶液)中反应。将反应在搅拌下放置18小时,产物在相同溶剂中结晶,通过过滤得到0.09g的产物。实施例13.生物测试结果。表1示出实施例中列出的各种化合物相较于利福昔明(rfx)的最低抑菌浓度(mic),单位为μg/ml。mic使用系列稀释法计算,并表示为细菌菌株生长的最后浓度与测试菌株不再生长的第一浓度之间的范围。表1实施例菌株a菌株b菌株c菌株d18-162-48-164-82>64>64>64>6430.03-0.060.125-0.254-80.02-0.0440.03-0.060.062-0.1254-88-1650.03-0.060.062-0.12532-6432-646>648-16>6432-6470.03-0.060.062-0.12532-6432-6480.03-0.060.062-0.1254-88-169----10----118-164-832-6432-64128-164-8>64>64rfx0.25-0.50.125-0.252-44-8从表1可以看出,实施例3示出完全可以与rfx相比的抗菌活性,而其他实施例尽管具有非最佳的亲水性/亲脂性比,但仍具有微生物活性。实施例14.药代动力学测试结果。通过给药化合物3(4’-[(4-甲基-1-哌嗪基)亚氨基甲基]-4-脱氧吡啶并[1’,2’-1,2]咪唑并[5,4-c]利福霉素sv)和利福昔明(rfx)进行药代动力学研究,两者在大鼠中以相同剂量口服和胃肠外给药。通过静脉和口服两种化合物,并计算与肠胃外给药相比的动力学参数auc、cmax、tmax和口服生物利用度,计划了相较于rfx的衍生物3的药代动力学研究。-rfx和衍生物3的静脉给药:将两种产品分别溶于dmso,得到4mg/ml储备液。向0.250ml的该溶液中加入4.750ml的生理盐水,得到最终的0.2mg/ml(5%dmso,95%生理盐水)溶液。向每组3只大鼠给药5ml/kg,两种产品的剂量分别为1mg/kg。-rfx和衍生物3的口服给药:将两种产物溶解在dmso中,得到两种浓度为60mg/ml的溶液(储备液)。在搅拌下,向0.5ml的每种溶液中加入2.0ml的peg400和7.5ml的生理盐水溶液,得到标称浓度为3mg/ml的稳定的悬浮液。给动物给药等于30mg/kg剂量的体积。该研究使用了雄性斯普拉格道利(斯普拉格道利)大鼠,体重在250至300g之间。根据意大利法律,根据所批准的gmp协议,在实验前对动物进行饲养和观察。将大鼠分为三组:-第1组:i.v.治疗-第2组:用于血液采样的口服治疗-第3组:用于尿液和粪便采集的口服治疗第1组:在5、15、30、60、120、240、480、1440分钟,从尾静脉进行血液采样。第2组:在30、60、120、240、360、480、1440分钟,从尾静脉进行血液采样。第3组:在0-4h、4-8h、8-24h、24-48h、48-72h,在代谢笼中收集粪便和尿液。在实验结束时,也从该组大鼠中取样72h时的血液作为检查。用于分析各种血液、粪便和尿液样本的分析方法是基于以下方法开发:uflc岛津ac20与api3200三重四极杆absciex结合使用。注入容量:10μl色谱柱:gemini-nx5μc18110a(50x2.00mm),色谱柱t35℃。梯度:流量0.3ml/min流动相a:水中15mm醋酸铵流动相b:meoh分析范围:血浆1-1000ng/ml。尿液:1-500ng/ml。粪便:1-1000ng/ml。表2:色谱梯度时间(min)%b0.01300.5301.5983.598430530进行了esi阳性的mrn分析,根据表2监测了主峰产生的片段。msesi阳性结果。化合物母离子产品离子利福昔明786.3754.33897.2865.3样品准备血浆:用未经处理的大鼠血浆制备校准曲线和qc样品,将每种储备液分别加入到5μl至45μl的未经处理的大鼠血浆中。将所得的50μl血浆用含有内标(i.s.)的冷乙腈以50ng/ml的最终浓度稀释至100μl,并在5℃下以9000rpm的转速离心5分钟。将上清液转移至96孔板进行分析。使用来自治疗过的大鼠的50μl血浆类似地制备第1组和第2组的样品。尿液:用未经处理的大鼠尿液制备校准曲线和qc样品,将每种储液分别添加5μl到45μl未经处理的大鼠尿液中。将所得的50μl尿液用含有i.s.的冷乙腈以50ng/ml的最终浓度稀释至100μl,并在5℃下以9000rpm的转速运转离心5分钟。将上清液转移至96孔板进行分析。使用来自治疗过的大鼠的50μl血浆类似地制备第1组和第2组的样品。粪便:通过将1g的粪便悬浮于ph值为4.3的3ml的15mm醋酸铵缓冲液中来匀浆粪便样品。通过将5μl的每种储备液加入到45μl的均质化的未经处理的大鼠粪便中,用未经处理的大鼠粪便制备校准曲线和qc样品。将所得的50μl均质粪便用含i.s.的冷乙腈以50ng/ml的最终浓度稀释至200μl,并置于96孔板中。将样品以3000rpm的转速离心15分钟,并将上清液转移到干净的平板中进行分析。分析数据-药代动力学分析应用非房室分析,并针对每个受试者考虑以下药代动力学参数:cmax:最大血浆浓度clast:最后一次采样时的血浆浓度tmax:最大血浆浓度的时间auc0-last:从t0到最后可检测浓度的时间的aucaucinf:从时间0外推到无限时间的mrtinf:t1/2:使用analysttm6.1软件(appliedbiosystems)外推浓度数据。使用线性梯形法计算auc,并采用重量均匀性作为第一种方法。使用pksolver2.0软件、excel2007microsoftaddin对结果进行对数转换后,进行浓度/时间图。结果表3:在雄性大鼠中静脉内(iv)和口服(po)给药后3和利福昔明(rfx)的药代动力学参数。表4:以30mg/kg口服给药后大鼠尿液中3和利福昔明(rfx)的浓度。表5:以30mg/kg口服给药后大鼠粪便中3和利福昔明(rfx)的浓度。与利福昔明在文献中显示和证实的相比,本发明的实施例3显示肠吸收低7-8倍,即约15%。实施例15.作为对照,在斯普拉格道利大鼠中口服给药产品3和利福昔明后,检测到对双歧杆菌、肠球菌、肠杆菌和乳杆菌种的真核细菌菌株的抗菌活性。治疗作为对照,在适应期后,通过胃探针以50mg/kg剂量的3和利福昔明对两组6只约250g/大鼠的斯普拉格道利大鼠进行治疗5天。在t0和最后治疗后的第二天收集粪便。首先将两种产物以100mg/ml的浓度溶于dmso(储备液)中,然后在搅拌下向0.5ml的每种溶液中加入2.0mlpeg400和7.5ml生理盐水溶液。得到标称浓度为5mg/ml的稳定悬浮液。给药体积为10ml/kg、等于50mg/kg。方法该检测方法基于使用特定的商业试剂盒从每个粪便样品中分离总基因组dna的方法。在对dna进行定性定量评估后,将相同的dna样品存储在-20℃下。使用实时qpcr进行定量分析,并使用双歧杆菌、肠球菌、肠杆菌和乳杆菌物种特异的dna引物对。每次使用dna分离试剂盒(mobiolaboratories)商业试剂盒对250mg粪便样品进行dna分离。分离后,通过分光光度分析法评估dna样品。每个dna样品的定量范围为60-100ng/μl(蛋白质和糖含量很高),这是粪便来源的dna的典型特征。qpcr的定量分析每个样品被评估三次。对于每个dna样品和每个特定的dna引物对,分析提供的ct值与特定于细菌物种的dna数量相关,因此与粪便样品中存在的该物种的细菌细胞数量密切相关。为了进行相对定量,应用了“在泊松对数正态误差下的广义线性混合模型(glmm)”。使用mcmc.qpcrr软件包将ct值转换为细菌细胞数。对于转换,使用公式:细胞数=e(ct1-ct),其中e表示扩增效率,在当前情况下假设值为2。ct1表示检测单个目标分子所需的qpcr循环数。(在本研究中,ct1的值为39)。马尔可夫链蒙特卡罗算法(mcmc)用于估计时间对每个微生物属发育水平的影响。glmm方法用于验证每个细菌属在t0和t5的水平是否不同。此外,进行了统计分析以评估t0和t5值之间差异的显著性。结果图3表示针对两组中的每个大鼠的、每个属的t5/t0时的细菌细胞数之间的比率。如下表7-9分别示出ct值、从ct值得到的每毫克粪便的细胞数,最后表9示出针对每个产品的、每个细菌属的t0和t5时的细胞数平均值。表7:三个qpcr分析值的平均ct表8:从表7的ct值得出的每毫克粪便中的细胞数表6:从图3和表7-8-9可以看出,以5mg/kg剂量的产品3口服治疗5天后,双歧杆菌属细菌计数降低了10倍,但不引起其他属,即肠球菌、肠杆菌和乳杆菌的细胞计数的显著变化。相反,使用利福昔明在相同的口服剂量下连续5天治疗会引起肠球菌属的显著下降(约100倍),肠杆菌属的下降较轻(约10倍),而双歧杆菌和乳杆菌没有显著差异,尽管两个物种的下降都可以检测到。显然,产物3对肠球菌、肠杆菌选择性高于利福昔明,虽程度相对较轻但对乳杆菌属的选择性高于利福昔明,而对双歧杆菌的选择性与利福昔明相似。有利地,由于其不相关的吸收和在保持肠道真核细菌菌群基本不变方面的高选择性,产品3可用于治疗由于细菌的微生物而引起的所有胃肠道疾病,特别是在需要长期治疗的疾病中。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1