用于人类呼吸道合胞病毒(HRSV)的诊断和疗法的制作方法
本发明涉及一种用于鉴定与传染病的严重程度有关的遗传变体的方法。本发明还涉及与例如在人类婴儿中的人类呼吸道合胞病毒(humanrespiratorysyncytialvirus,hrsv)感染的严重程度有关的一组遗传因素。根据本发明鉴定的遗传多态性(geneticpolymorphism)用于感染性疾病的诊断和患者分层,从而避免或减少感染期间致命事件的发生或选择最合适的治疗方法来治疗该疾病。
技术实现要素:
rsv感染是工业化国家中婴儿住院治疗的主要原因。一般在2岁以下发生的原发性rsv感染后,对rsv的免疫力仍不完全,并且可能发生再感染。此外,rsv可以在老年人中引起严重的疾病,并且通常在非大流行年份比a型流感具有更高的死亡率(falsey等,1995)。世卫组织估计的全球人口年感染率估计为6400万例,死亡率为160000;仅在美国,由于rsv感染,每年有85000至144000例婴儿住院治疗(http://www.who.int/vaccine_research/diseases/ari/en/index2.html,2009年更新)。rsv属于副粘病毒科肺病毒亚科肺炎病毒属;在人类中,有a和b两个亚组。除了人类rsv外,还有牛变体。人rsv的基因组大约长15200个核苷酸,是负义rna分子。rsv基因组编码11种已知蛋白:糖蛋白(g)、融合蛋白(f)、小疏水蛋白(smallhydrophobicprotein,sh)、核蛋白(n)、磷酸蛋白(p)、大蛋白(l)、基质蛋白(m)、m2orf-1蛋白(m2-1)、m2orf-2蛋白(m2-2)、非结构蛋白1(ns1)和非结构蛋白2(ns2)。g、f和sh是跨膜表面蛋白;n、p、l、m、m2-1是核衣壳相关蛋白,ns1和ns2是非结构蛋白。m2-2作为结构或非结构蛋白的状态尚不清楚。(hacking和hull,2002)。rsv亚组显示出在g、f、n和p蛋白的抗原特性上有所不同(ogra,2004)。
rsv感染后形成在血清和某些其他体液中可检测到的特异性igg和iga抗体。几项研究表明,抗体反应主要针对主要的rsv跨膜蛋白f和g;已知只有f和g特异性抗体具有体外rsv中和活性。对f蛋白的抗体反应通常在a和b亚组之间发生交叉反应,而对g蛋白的抗体反应则是亚组特异性的(orga,2004)。与f和g相反,跨膜蛋白sh被认为是非免疫原性的(gimenez等,1987;tsutsumi等,1989),并且在某些候选疫苗中,sh甚至被删除从而获得不可逆的减毒疫苗(karron等,2005)。宿主免疫应答似乎在该疾病的发病机理中起着重要的作用,从而使预防rsv感染的疫苗的开发变得复杂。早期用福尔马林灭活的rsv为儿童接种疫苗的尝试表明,与未接种疫苗的对照相比,接种疫苗的儿童在随后接触该病毒时会经历更严重的疾病(kapikian等,1969)。减毒活疫苗已经经过测试,但在临床研究中经常显示减毒过度或减毒不足(murata,2009)。
使用一种免疫原性蛋白质或免疫原性蛋白质的组合的亚单位疫苗被认为更安全,因为它们无法还原或突变为强毒病毒。已经开发出了基于纯化f蛋白的候选疫苗,并在啮齿动物、棉鼠和人类中进行了测试,结果证明是安全的,但仅具有中等免疫原性(falsey和walsh,1996;falsey和walsh,1997;groothuis等,1998)。同样,在第二阶段已停止使用f蛋白、g蛋白和m蛋白的混合物进行临床试验(adisinsight临床数据库)。一种替代方法包括将人rsvg蛋白的抗原结构域与链球菌g蛋白的白蛋白结合结构域的c末端进行重组遗传融合(bbg2na;power等,2001)。在健康志愿者中对bbg2na进行了直至iii期临床试验的研究,但由于在一些免疫志愿者中出现了意料不到的3型超敏反应副作用(紫癜),该试验不得不中止(meyer等,2008)。
最近的进展是使用嵌合重组病毒作为rsv抗原的载体。通过用来自hpibv-3的同源基因在bpiv-3基因组中替代f和hn基因,对3型嵌合重组牛/人副流感病毒(rb/hpiv-3)进行了工程改造。然后将所得的嵌合rb/hpiv-3菌株用于表达hrsvf和g基因(schmidt等,2002)。该疫苗目前正在临床研究中。
对于由rsv引起的严重疾病,只有有限的预防和治疗选择。最广泛使用的干预措施是基于源于小鼠单克隆抗体1129的人源化单克隆抗体的被动免疫预防(beeler和vanwykecoelingh,1989)。该抗体对rsvf蛋白具有特异性,可以中和a和b亚组病毒。重组人源化抗体1129被称为帕利珠单抗(palivizumab)(也称为synagis),用于对rsv感染后发生并发症风险高的婴儿进行预防性治疗。每月肌肉内施用该抗体,从而降低因早产、慢性肺病或血液动力学显著的先天性心脏病而处于危险之中的婴儿的rsv感染风险(bocchini等,2009)。一些研究报告了使用帕利珠单抗预防rsv的可接受的成本效益比(prescott等,2010)。
由于市场上没有批准的疫苗,因此对于安全和有效的rsv疫苗的开发和可用性仍存在未满足的需求。令人惊讶地,我们发现小疏水蛋白sh的胞外部分(胞外域,称为she)可以安全地用于抗rsv感染的疫苗接种,尤其是当它以低聚体(优选为五聚体)的形式存在于载体上时。此外,针对she的多克隆或单克隆抗体也可以分别预防性地或治疗性地用于rsv感染的预防或治疗。
因此,本发明的一个目的是提供诊断选择,以鉴定出严重的hrsv感染过程的风险,特别是在免疫受损的患者(例如非常年轻和年长的患者)中。本发明旨在为临床医生提供对致命病程的严重风险的早期检测,从而提供快速或预防性的治疗措施。
在第一方面,上述问题通过以下方法来解决,该方法用于评估对象对人类呼吸道合胞病毒(hrsv)感染的敏感性增加的风险,该方法包括以下步骤:在从所述对象中分离出的样品中确定至少一种选自表1的遗传多态性的存在,其指示该对象对hrsv感染的敏感性增加的风险。
本发明的另一方面涉及一种用于诊断对象对人类呼吸道合胞病毒(hrsv)感染的敏感性增加的方法,该方法包括以下步骤:在从所述对象中分离出的样品中确定至少一种选自表1的遗传多态性的存在,其指示该对象对hrsv感染的敏感性增加的风险。
本发明的另一方面涉及一种用于鉴定对象患有严重hrsv感染的风险增加的方法,该方法包括以下步骤:在从所述对象中分离出的样品中确定至少一种选自表1的遗传多态性的存在,其指示该对象对hrsv感染的敏感性增加的风险。
对于上述方法,优选根据本文公开的表1使用至少一种遗传多态性。然而,在本发明方面的某些实施方案中,使用本文公开表1的一种以上变体的组合可能是有利的。因此,本发明的方法优选涉及2、3、4、5、6、7或8或更多种遗传多态性。
术语“单核苷酸多态性(singlenucleotidepolymorphism)”或“snp”是指在人类群体中以明显的频率出现的遗传序列中的单核苷酸变异。人类基因组中有数百万个snp。最常见的是,这些变异是在基因之间的dna中发现的。当snp出现在基因内或基因附近的调控区中时,它们可能通过影响基因的功能而在疾病中发挥更直接的作用。snp对本领域技术人员来说是众所周知的,并且特别在ncbi数据库dbsnp(www.ncbi.nlm.nih.gov/snp/)中进行了描述。如本文所用,本发明所涉及的snp如表1所示。
通常,通过确定表1中公开的至少一种风险等位基因在纯合子或杂合子中的存在或不存在来进行本发明的方法。优选地,在上述定义的方法中,推论出如果对象对于表1中公开的至少一种风险等位基因是纯合的或杂合的,则所述对象快速发展hrsv感染的风险增加。更具体地,推论出对于风险等位基因是纯合的对象对hrsv的风险高于对于风险等位基因是杂合的对象。
在本发明的一些实施方案中,其中hrsv感染是hrsv的严重感染,优选与选自细支气管炎、肺炎、哮喘和复发性hrsv感染及其组合的一种或多种并发症相关。
根据本发明,从获自对象的样品中测试单核苷酸多态性的基因型。
样品是含有dna的样品,其优选例如选自组织样品或液体样品,例如头发样品、皮肤样品或口腔组织样品、刮擦或洗涤或生物流体样品,优选唾液、尿液或血液。通常从组织活检和尸检材料中获得组织提取物。可用于本发明的体液包括血液、尿液、唾液或任何其他人体分泌物或其衍生物。如本文所用,“血液”包括全血、血浆、血清、循环上皮细胞、血液的成分或任何衍生物。根据本发明,可以通过核酸测序、pcr分析或本领域已知的任何基因分型方法来确定风险等位基因的存在。此类方法的例子包括但不限于:化学测定法,例如等位基因特异性杂交、引物延伸、等位基因特异性寡核苷酸连接、测序、酶促切割、瓣核酸内切酶鉴别;以及检测方法,例如荧光、化学发光和质谱。
例如,可以优选在扩增后在rna或dna样品中检测所述遗传多态性的存在或不存在。例如,可以使用对多态性具有特异性的特定寡核苷酸引物,或者使得能够对含有多态性的区域进行扩增的特定寡核苷酸引物,对分离的rna进行偶联逆转录和扩增,例如通过聚合酶链反应(rt-pcr)进行逆转录和扩增。根据第一种选择,可以选择用于引物退火的条件以确保特异性逆转录(在适当的情况下)和扩增;因此,扩增产物的出现可诊断本发明多态性的存在。否则,可以将rna逆转录并扩增,或者可以扩增dna,然后可以通过与合适的探针杂交或通过直接测序或本领域已知的任何其他合适的方法,在扩增的序列中检测突变位点。例如,可以将从rna获得的cdna进行克隆并测序以对多态性进行基因分型(或鉴定等位基因)。实际上,许多用于基因型分析的策略可供选择。简而言之,可以测试核酸分子是否存在限制位点。当碱基多态性产生或消除限制酶的识别位点时,这允许对多态性进行简单的直接pcr基因分型。其他策略包括但不限于:直接测序,限制性片段长度多态性(rflp)分析;与等位基因特异性寡核苷酸(aso)杂交,后者是短的合成探针,在适当严格的杂交条件下仅与完全匹配的序列杂交;等位基因特异性pcr;使用诱变引物进行pcr;连接酶pcr,hot裂解;变性梯度凝胶电泳(dgge),温度变性梯度凝胶电泳(tgge),单链构象多态性(sscp)和变性高效液相色谱法(kuklin等,1997)。直接测序可以通过任何方法来完成,包括但不限于:化学测序,使用maxam-gilbert方法;通过酶测序,使用sanger方法;质谱测序;使用基于芯片的技术进行测序;以及实时定量pcr。优选地,首先使用特异性扩增引物通过聚合酶链反应(pcr)对来自对象的dna进行扩增。但是,还有其他几种方法可以使dna与pcr无关地进行研究,例如滚环扩增(rca)、invadertm分析、或寡核苷酸连接分析(ola)。ola可以用于揭示碱基多态性。根据该方法,构建了两个寡核苷酸,它们与靶核酸中的相邻序列杂交,连接位于多态性的位置。仅当它们与等位基因之一完美杂交时,dna连接酶才会共价连接这两个寡核苷酸。
因此,根据本发明的有用的核酸分子,特别是寡核苷酸探针或引物,包括与多态性的等位基因之一特异性杂交的核酸分子。
寡核苷酸探针或引物可以包含至少10、15、20或30个核苷酸。它们的长度可以短于400、300、200或100个核苷酸。
在一些实施方案中,本发明的方法由将会产生测试报告的实验室执行。测试报告因此将指示风险等位基因是否存在,并且优选地指示患者对于风险等位基因是杂合的还是纯合的。在一些实施方案中,测试结果将包括发展为肝纤维化的概率得分,其是从运行模型得出的,该模型包括针对所测试的本发明的一个或两个单核苷酸多态性确定的风险因子。为了计算分数,可以通过系数来考虑针对本发明的单核苷酸多态性确定的风险因素,该系数取决于所述单核苷酸多态性与另一个单核苷酸多态性相比在风险确定中的贡献。通常,用于计算分数的方法是基于对不同患者群体进行的统计研究。分数还可以包括其他各种患者参数(例如对象的年龄、性别、体重、饮酒量、hcv基因型、hiv感染)。在解释发展为肝纤维化的个体间变异性时,赋予每个参数的权重是基于其相对于其他参数的贡献。在一些实施方案中,测试报告可以因此由用于建立这种分数的计算机程序生成。
因此,本发明的另一方面涉及一种用于指示在对象中需要使用抗感染剂或抗感染疗法进行预防性治疗的方法,该方法包括以下步骤:在从所述对象中分离出的样品中确定至少一种选自表1的遗传多态性的存在,其指示所述预防性治疗或疗法的对象。
使用抗感染剂或抗感染疗法进行的预防性治疗优选涉及施用针对hrsv蛋白的抗体,例如靶向g、f或sh包膜蛋白的预防性抗体或病毒聚合酶,并且优选是使用帕利珠单抗的治疗;或者其中使用抗感染剂或抗感染疗法进行的所述预防性治疗涉及施用针对hrsv的疫苗。原则上,在这种情况下可以使用hrsv的任何治疗方法。在以上介绍中介绍了hrsv治疗或预防的一些优选方法。
“对象”优选是人类患者,例如人类婴儿,例如0至5岁的儿童、或0至3岁的儿童、或0至2岁的儿童、或0至1岁的儿童;和/或其中人类患者是免疫抑制的患者,例如非常年轻或年长的患者、或免疫受损的(成人)患者。
在一些优选的实施方案中,通过从样品中扩增或未能扩增扩增产物来鉴定多核苷酸的存在或不存在,其中扩增产物优选在分析之前用限制酶进行消化,和/或其中通过将核酸样品与作为可检测部分的引物标记进行杂交来鉴定snp。
还提供了一种计算机程序或计算机可读介质,其包含用于执行本公开中定义的方法的装置。
另一个方面涉及一种试剂盒,其包含用于检测含有选自表1的核苷酸的遗传多态性的身份的试剂。这种试剂盒优选用于本文公开的任何方法中。在一些实施方案中,试剂盒可以包含一个或多个核酸引物对,其对包含至少一种选自表1的多态性的区域的扩增具有特异性。
令人惊讶地发现,本文公开的一些遗传多态性位于与病毒感染功能相关的新基因中。这样的基因对于病毒感染可能是必需的,或者具有病毒抑制剂的活性。因此,本发明另一方面涉及化合物用于治疗hrsv感染的用途,其中该化合物选自(i)拮抗剂胞质多聚(a)结合蛋白1(poly(a)bindingproteincytoplasmic1,pabpc1)、干扰素诱导的蛋白激酶eif2ak2的蛋白激活剂(prkra)、芳基硫酸酯酶d(arsd)、“含有phd手指蛋白的mllt6”(mllt6)或ctd小磷酸酶2(ctdsp2);或者(ii)激动剂c末端结合蛋白2(ctbp2)、线粒体rna聚合酶(polrmt)、或跨膜蛋白259(tmem259)。基因是根据人类基因名称计划的命名法命名的:grayka,yatesb,sealrl,wrightmw,brufordea.genenames.org:thehgncresourcesin2015.nucleicacidsres.2015jan;43(databaseissue):d1079-85.doi:10.1093/nar/gku1071.pmid:25361968;hgncdatabase,hugogenenomenclaturecommittee(hgnc),embloutstation-hinxton,europeanbioinformaticsinstitute,wellcometrustgenomecampus,hinxton,cambridgeshire,cb101sd,ukwww.genenames.org。
在本发明的上下文中,“拮抗剂”优选是损害参考蛋白质或其mrna或基因的表达、稳定性和/或功能的化合物。最优选地,拮抗剂是相应因子的哺乳动物同源物的拮抗剂。如本文所用,术语“拮抗剂”是指影响蛋白质表达或活性的量或速率降低的物质。这种物质可以直接起作用,例如,通过与候选蛋白质结合并降低其表达或活性的数量或速率。拮抗剂还可以例如通过以下方法降低蛋白质表达或活性的量或速率:通过以减少或防止其与相互作用伴侣(例如受体或配体)相互作用的方式与参考因子结合;或通过与参考蛋白质结合并对其进行修饰,例如通过去除或添加部分;以及通过与其结合并降低其稳定性。拮抗剂也可以间接起作用,例如通过结合至调节分子或基因区域,从而调节调节蛋白或基因区域功能并影响蛋白表达或活性的量或速率的降低。
本发明的拮抗剂可以是例如天然或非天然存在的大分子,例如多肽、肽、拟肽、核酸、碳水化合物或脂质。拮抗剂还可以是抗体或其抗原结合片段,例如单克隆抗体、人源化抗体、嵌合抗体、微型抗体、双功能抗体、单链抗体(scfv)、可变区片段(fv或fd)、fab或f(ab)2。拮抗剂也可以是对相应蛋白质具有特异性的多克隆抗体。拮抗剂还可以是天然存在的大分子或小的有机或无机分子的部分或完全合成的衍生物、类似物或模拟物。
作为抗体的拮抗剂可以是例如这样的抗体,其与蛋白质结合并抑制与其受体结合,或改变调节表达或活性的分子的活性,从而使蛋白质表达或活性的量或速率降低。可用于本发明方法的抗体可以是天然存在的抗体,包括单克隆或多克隆抗体或其片段,或非天然存在的抗体,包括但不限于单链抗体、嵌合抗体、双功能抗体、互补决定区移植(cdr移植)抗体和人源化抗体或其抗原结合片段。
作为核酸的拮抗剂可以是例如反义核苷酸序列、rna分子、或适体序列。反义核苷酸序列可以结合细胞内的核苷酸序列,并调节候选蛋白的表达水平或调节控制候选蛋白的表达或活性的另一个基因的表达。类似地,rna分子,例如催化核酶,可以结合并改变基因或控制本发明候选物的表达或活性的其他基因的表达。适体是具有能够结合分子靶标的三维结构的核酸序列。
作为核酸的拮抗剂也可以是用于rna干扰方法的双链rna分子。rna干扰(rnai)是转录后rna降解导致序列特异性基因沉默的过程,该过程由与被沉默的基因序列同源的双链rna(dsrna)启动。用于rnai的合适的双链rna(dsrna)包含约21个连续核苷酸的有义和反义链,其与形成19个rna碱基对的待靶向基因相对应,在每个3'末端均保留两个核苷酸的突出端(elbashir等,nature411:494-498(2001);bass,nature411:428-429(2001);zamore,nat.struct.biol.8:746-750(2001))。大约25-30个核苷酸的dsrna也已成功用于rnai(karabinos等,proc.natl.acad.sci.usa98:7863-7868(2001)。dsrna可以体外合成,并通过本领域已知的方法引入细胞中。优选的反义干扰构建体是sirna和shrna或任何其他基于rna的抑制剂。还包括的是使用crispr/cas9介导的基因组编辑来破坏蛋白质表达的构建体和方法。这些方法和化合物是本领域公知的并且要求技术人员简单地设计和选择适当的遗传靶序列,这对于本文公开的所有靶基因都是已知的。
在其他方面,本发明涉及本文鉴定为抑制病毒感染的蛋白质的激动剂。当以激动剂化合物作为靶标时,这些因子是有用的。如本文所使用的,术语“激动剂”是指影响这种因子的表达或活性的量或速率的增加的物质。这样的物质可以直接作用,例如,通过结合蛋白质并增加表达或活性的量或速率。激动剂还可以例如通过以下方式增加表达或活性的量或速率:通过以增强或促进其与配体或受体的相互作用的方式与蛋白质结合;可以通过与蛋白质结合并对其进行修饰而影响激活,例如诱导构象变化、或部分的去除或添加;以及通过与蛋白质结合并增强其稳定性。激动剂还可以间接起作用,例如通过结合至调节分子或基因区域,从而调节调节蛋白或基因区域功能并影响表达或活性的量或速率的增加。
激动剂可以是例如天然或非天然存在的大分子,例如多肽、肽、拟肽、核酸、碳水化合物或脂质。激动剂还可以是抗体或其抗原结合片段,例如单克隆抗体、人源化抗体、嵌合抗体、微型抗体、双功能抗体、单链抗体(scfv)、可变区片段(fv或fd)、fab或f(ab)2。激动剂也可以是对作为本发明的hrsv抑制剂而公开的蛋白质具有特异性的多克隆抗体。激动剂还可以是天然存在的大分子或小的有机或无机分子的部分或完全合成的衍生物、类似物或模拟物。
可用于本发明方法的抗体可以是天然存在的抗体,包括单克隆或多克隆抗体或其片段,或非天然存在的抗体,包括但不限于单链抗体、嵌合抗体、双功能抗体、互补决定区移植(cdr移植)抗体和人源化抗体或其抗原结合片段。
根据本发明的激动剂也是表达任何hrsv抑制蛋白或其功能片段的表达构建体。术语“表达构建体”是指设计来转录rna的任何双链dna或双链rna,例如,包含至少一个可操作地连接至下游目的基因或编码区域的启动子的构建体(例如,编码蛋白质或任何目的rna的cdna或基因组dna片段)——特别是本文鉴定的hrsv抑制剂的基因或其片段。将表达构建体转染或转化成受体细胞使该细胞表达由表达构建体编码的rna或蛋白质。表达构建体可以是衍生自例如噬菌体、腺病毒、逆转录病毒、痘病毒、或疱疹病毒的基因工程质粒、病毒、或人工染色体、或下文“表达载体”下所述的其他实施方案。表达构建体可以在活细胞中复制,也可以通过合成制备。为了本申请的目的,术语“表达构建体”、“表达载体”、“载体”和“质粒”可互换使用,以一般性的说明性意义说明本发明的应用,而不意图将本发明限制至特定类型的表达构建体。此外,术语表达构建体或载体还意图包括这样的情况,其中用于测定的细胞已经内源地包含这种dna序列。
在另一方面,提供了一种用于鉴定遗传变体和传染病的关联的方法,该方法包括:对感染了传染病的患者的研究队列的整个外显子组进行测序,使用基因的预定子集的参考基因组数据在测序出的外显子组中进行变体调用(variantcalling),以及与参考队列比较研究队列中所述变体的等位基因频率,以及鉴定与传染病显著相关的遗传变体。
优选地,遗传变体是遗传多态性,优选地是单核苷酸多态性(snp)。
优选地,传染病是病毒病,例如是hsrv。
在本发明的上下文中,术语“参考队列”优选地属于外显子组聚集数据库(http://exac.broadinstitute.org/)的子队列。在选择参考队列时,技术人员应该选择与研究队列的遗传背景紧密匹配的群组。例如,如本情况中所述,如果研究队列属于某个年龄组或种族,则参考队列应在可能的范围内也符合这些标准。
在本发明的上下文中,基因的预定子集是受干扰素调节的基因。
进一步包括的是上述方法,其进一步包括验证鉴定出的关联的遗传变体的进一步步骤。
现在将在以下实施例中参考附图和序列进一步描述本发明,然而不被限于此。为了本发明的目的,本文引用的所有参考文献通过引用整体并入本文。在附图中:
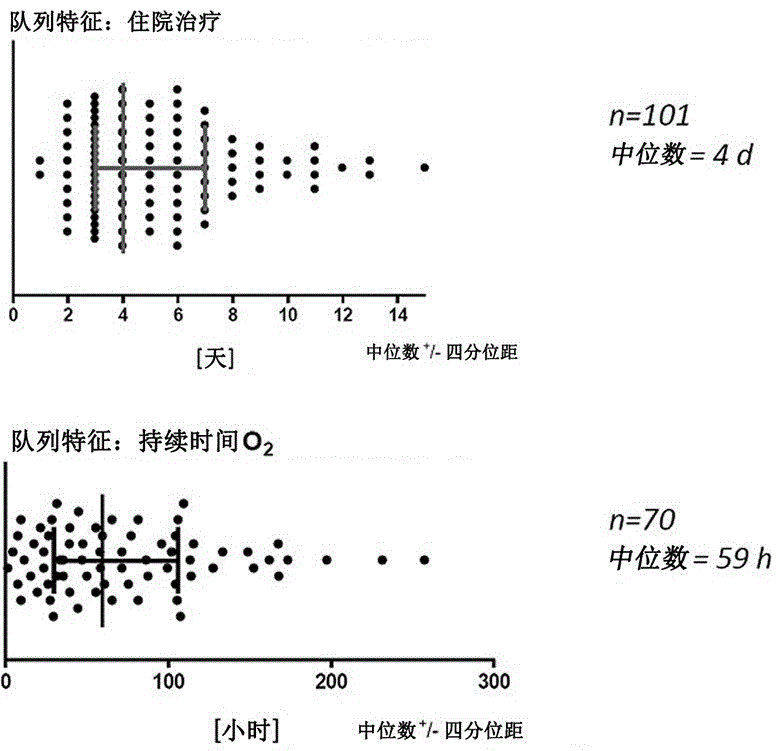
图1:示出了研究队列特征。
图2:示出了外显子组测序和数据处理、变体调用和过滤处理的工作流程。
图3:示出了测序和后续分析的工作流程。
图4:示出了snp的火山图。
图5:示出了snp的火山图。
图6:示出了snp的火山图。
图7:示出了sirna筛选的结果。左侧显示了如duprex等人发表的检测方法。右侧显示了eleouet等人的方法。
实施例
实施例1:外显子组测序和变体调用
研究队列的纳入标准是法定监护人的书面同意、年龄(0-5岁)和白种人。患者患有严重rsv感染,包括急性呼吸道感染、鼻拭子(pcr)中的rsv,需要住院治疗和低氧血症。排除标准为早产(<36ssw)、支气管肺发育不良、心脏缺陷或免疫缺陷。
图1显示了研究队列的特征。
使用truseqsbs试剂盒v3-hs在illuminahiseq2500上对文库进行测序(200个循环,配对末端运行),每个单个外显子组平均读取12,5x106个读数(平均覆盖率:50x)。在通过fastqc工具进行修剪之前和之后确定fastq文件的质量。通过trimgalore!(默认设置)进一步操作原始读数,从而裁剪人工illumina适配子序列并裁剪/删除质量差的序列读数。使用bwa对齐工具将修剪后的fastq文件与人类参考基因组hg19对齐,然后使用markduplicates(picard工具)标记重复的读数(pcr产物)。
对两个患者子集进行变体调用:101例严重rsv感染的儿童和70例非常严重rsv感染(低氧血症)的儿童。遵循其最佳实践指南使用基因组分析工具包(gatk)。具体而言,在“-ercgvcf”模式中在单独的样本上运行haplotypecaller。随后,使用genotypecaller以hg19人类基因组构建作为参考,进行联合变体调用。使用variantrecalibrator对单核苷酸变体和插入缺失的分数进行重新校准,并且仅将通过99.9%部分阈值的变体考虑用于下游分析。
分析了直接受干扰素调节和/或参与病原体感应和干扰素信号传导的总共5142个基因的变体。在以下数据库中鉴定出干扰素调节基因(irg):(i)interferome数据库(截止值至少是2倍调节,p值<0.05;总共4777个基因),以及(ii)原代人肺细胞中irg的内部数据库(lauberc等人,genesimmun.2015年9月;16(6):414-21;704个基因)。最后,发明人使用ingenuity通路分析(ipa)来鉴定涉及病原体感测和ifn信号传导的基因。为此,在基因和化学列表中使用了以下运算符:重组干扰素、干扰素α、干扰素β、干扰素γ、干扰素信号转导;在通路和毒物列表中使用了:jak1、jak2和tyk2在干扰素信号传导中的作用,pkr在干扰素诱导和抗病毒反应中的作用,胞质模式识别受体对irf的激活,模式识别受体在细菌和病毒识别中的作用;并且在疾病和功能列表中使用了:rsv。在分子的所有表格中,仅考虑具有人类entrez数的分子。通过ipa总共提取了194个基因。如venn图所示,通过所有三种方法总共鉴定出35个基因,最终根据选择标准将总共5142个基因纳入了分析。
使用snpeff对对这些基因所发现的变体进行注释。使用自定义perl脚本,为每个变体添加由美国人和非芬兰人欧洲人组成的exac子队列中的次要等位基因频率。我们的队列和exac子队列(freq_diff_exac)之间的次要等位基因频率差异用于对变体进行排名。使用fisher精确检验和cochran-armitage检验趋势(在r中实现),使用exac和我们的队列中的杂合度和纯合度计数来计算变体与严重rsv感染的关联的显著性。使用combinedannotationdependentdepletion(cadd)评分评估变体的有害影响。根据不同的度量对基因进行排名,包括基因所有变体的freq_diff_exac值之和以及所有非同义变体的freq_diff_exac值之和与基因的所有静默变体的freq_diff_exac值之和的对数比。
对于在等位基因频率上显示出显著变化的变体进行筛选,发现共有346个snv显示出显著的关联。在这些中,218个是编码snv而128个是非编码snv。这些snv总共影响了144个基因。编码snv影响了84个基因,非编码snv影响了87个基因,27个基因同时包含编码和非编码snv。
在图3中提供了对受影响基因进行功能跟进的策略。总共144个基因受到显著相关的snv的影响。84个基因带有编码snv,这些基因中的44个在原代人气道上皮细胞中表达mrna(hae;rpkm>1)。符合这些纳入标准(受snv影响并以rpkm>1在hae中表达mrna)的6个hla基因被排除在功能跟进之外,并以红色突出显示。剩余的38个候选基因被列入用于a549细胞(通常用于rsv感染研究的人肺癌细胞系)中的sirna沉默,此时基因在这些a549细胞中以rpkm>1表达mrna(29个候选基因满足此要求)。如果未检测到相应基因的mrna或rpkm低于1,则将该基因在a549中列入异位表达(9个候选基因)。
图4显示了火山图,突出显示了holm校正后的p值<0.05的所有编码snp和非编码snp。每个snv都根据其p值(y轴)和相对于exac队列的频率差异(x轴)进行绘图。颜色代码指示所标识的snv是编码的(红色/橙色)还是未编码的(深/浅蓝色)。hardyweinberg平衡(hwe)中的snv用深色绘制,非hwe中的snv用浅色绘制。图5显示的火山图突出显示了所有非编码snv。图6显示的火山图突出显示了所有编码snv。图7显示的火山图突出显示了所有snv。包围了先前已经在体外或体内作为抗病毒效应物进行了研究的snv靶向基因。
以下的表1提供了鉴定出的遗传多态性的结果:
示出的是变体的染色体位置、其数据库标识符以及计算出的显著性(p值)。
实施例2:用于hrsv感染因子的rna干扰筛选
用egfp报告病毒(rsv-b-05)和f-荧光素酶报告病毒(rsv-a-long)进行sirna筛选的结果如图7所示。报告了三个生物学重复的平均得分。aln01是针对rsv的对照sirna。绿色箭头标记了候选rsv依赖因子。敲除这些基因可减少rsv感染。红色箭头标记了候选rsv限制因子。敲除这些基因可增强rsv感染。结果,基因pabpc1、prrka、arsd、mllt6和ctdsp2被鉴定为hrsv感染的依赖因子,而ctbp2、polrmt和tmem259是hrsv感染的限制因子。
参考文献
andrewss.(2010).fastqc:aqualitycontroltoolforhighthroughputsequencedata.availableonlineat:http://www.bioinformatics.babraham.ac.uk/projects/fastqckruegerf.(2012).trimgalore!:awrappertoolaroundcutadaptandfastqctoconsistentlyapplyqualityandadaptertrimmingtofastqfiles,withsomeextrafunctionalityformspi-digestedrrbs-type(reducedrepresentationbisufite-seq)libraries.availableonlineathttp://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
alecwysoker,kathleentibbetts,timfennell(2013):picardtoolsversion1.90.availableonlineathttp://picard.sourceforge.net
mckennaa,hannam,bankse,sivachenkoa,cibulskisk,kernytskya,garimellak,altshulerd,gabriels,dalym,depristoma(2010)thegenomeanalysistoolkit:amapreduceframeworkforanalyzingnext-generationdnasequencingdata.genomere-search20:1297-303
cingolani,p.andplatts,a.andcoon,m.andnguyen,t.andwang,l.andland,s.j.andlu,x.andruden,d.m.(2012)aprogramforannotatingandpredictingtheeffectsofsinglenucleotidepolymorphisms,snpeff:snpsinthegenomeofdrosophilamelanogasterstrainw1118;iso-2;iso-3.fly6(2):80-92.pmid:22728672
lekmetal.(2016)analysisofprotein-codinggeneticvariationin60,706humans.nature536(7616):285-91.doi:10.1038/nature19057.
kircherm,wittendm,jainp,o'roakbj,coopergm,shendurej(2014).ageneralframeworkforestimatingtherelativepathogenicityofhumangeneticvariants.natgenet.doi:10.1038/ng.2892.pubmedpmid:24487276.
lih.,handsakerb.,wysokera.,fennellt.,ruanj.,homern.,marthg.,abecasisg.,durbinr.and1000genomeprojectdataprocessingsubgroup(2009)thesequencealignment/map(sam)formatandsamtools.bioinformatics,25,2078-9.[pmid:19505943]
- 还没有人留言评论。精彩留言会获得点赞!